
A Brief Overview of Molecular Biology in Pituitary Adenomas with a Focus on Aggressive Lesions


Abstract
1. Introduction
2. A Brief Overview of Molecular Biology
2.1. Inherited Pituitary Tumors
2.2. Intracellular Signaling Pathways
2.3. Epigenetics
2.4. Somatotroph Adenomas
2.5. Lactotroph Adenomas
2.6. Corticotroph Adenomas
2.7. Thyrotroph Adenomas
2.8. Gonadotroph Adenomas
2.9. Null Cell Adenomas
2.10. Landscape of Molecular Events in Pituitary Tumor Apoplexy
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Melmed, S. Pituitary-Tumor Endocrinopathies. N. Engl. J. Med. 2020, 382, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.C.; Wang, E.J.; Aghi, M.K. Recent advancements in the molecular biology of pituitary adenomas. Expert Rev. Endocrinol. Metab. 2022, 17, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Beckers, A. The Epidemiology of Pituitary Adenomas. Endocrinol. Metab. Clin. N. Am. 2020, 49, 347–355. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26. [Google Scholar] [CrossRef]
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrange, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inoshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (PitNET): An International Pituitary Pathology Club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef]
- Sathyakumar, R.; Chacko, G. Newer Concepts in the Classification of Pituitary Adenomas. Neurol. India 2020, 68, S7–S12. [Google Scholar] [CrossRef]
- Ho, K.K.Y.; Fleseriu, M.; Wass, J.; van der Lely, A.; Barkan, A.; Giustina, A.; Casanueva, F.F.; Heaney, A.P.; Biermasz, N.; Strasburger, C.; et al. A tale of pituitary adenomas: To NET or not to NET: Pituitary Society position statement. Pituitary 2019, 22, 569–573. [Google Scholar] [CrossRef]
- Liu, X.; Wang, R.; Li, M.; Chen, G. Pituitary adenoma or pituitary neuroendocrine tumor: A narrative review of controversy and perspective. Transl. Cancer Res. 2021, 10, 1916–1920. [Google Scholar] [CrossRef]
- Ho, K.K.Y.; Fleseriu, M.; Wass, J.; Katznelson, L.; Raverot, G.; Little, A.S.; Castaño, J.P.; Reincke, M.; Lopes, M.B.; Kaiser, U.B.; et al. A proposed clinical classification for pituitary neoplasms to guide therapy and prognosis. Lancet Diabetes Endocrinol. 2024, 12, 209–214. [Google Scholar] [CrossRef]
- Lake, M.G.; Krook, L.S.; Cruz, S.V. Pituitary adenomas: An overview. Am. Fam. Physician 2013, 88, 319–327. [Google Scholar] [PubMed]
- Zhan, X.; Desiderio, D.M. Editorial: Molecular Network Study of Pituitary Adenomas. Front. Endocrinol. 2020, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Zhan, X. The crucial role of multiomic approach in cancer research and clinically relevant outcomes. EPMA J. 2018, 9, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Wang, X.; Zhan, X. Multi-parameter systematic strategies for predictive, preventive and personalised medicine in cancer. EPMA J. 2013, 4, 2. [Google Scholar] [CrossRef]
- Cheng, T.; Zhan, X. Pattern recognition for predictive, preventive, and personalized medicine in cancer. EPMA J. 2017, 8, 51–60. [Google Scholar] [CrossRef]
- Chang, M.; Yang, C.; Bao, X.; Wang, R. Genetic and Epigenetic Causes of Pituitary Adenomas. Front. Endocrinol. 2020, 11, 596554. [Google Scholar] [CrossRef]
- Dai, C.; Kang, J.; Liu, X.; Yao, Y.; Wang, H.; Wang, R. How to Classify and Define Pituitary Tumors: Recent Advances and Current Controversies. Front. Endocrinol. 2021, 12, 604644. [Google Scholar] [CrossRef]
- Knosp, E.; Steiner, E.; Kitz, K.; Matula, C. Pituitary adenomas with invasion of the cavernous sinus space: A magnetic resonance imaging classification compared with surgical findings. Neurosurgery 1993, 33, 610–618. [Google Scholar] [CrossRef]
- Kasuki, L.; Raverot, G. Definition and diagnosis of aggressive pituitary tumors. Rev. Endocr. Metab. Disord. 2020, 21, 203–208. [Google Scholar] [CrossRef]
- Inomoto, C.; Tahara, S.; Oyama, K.; Kimura, M.; Matsuno, A.; Teramoto, A.; Osamura, R.Y. Molecular, functional, and histopathological classification of the pituitary neuroendocrine neoplasms. Brain Tumor Pathol. 2021, 38, 183–188. [Google Scholar] [CrossRef]
- Raverot, G.; Burman, P.; McCormack, A.; Heaney, A.; Petersenn, S.; Popovic, V.; Trouillas, J.; Dekkers, O.M. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur. J. Endocrinol. 2018, 178, G1–G24. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dai, C.; Feng, M.; Li, M.; Chen, G.; Wang, R. Diagnosis and treatment of refractory pituitary adenomas: A narrative review. Gland Surgery 2021, 10, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- García-Guzmán, B.; Portocarrero-Ortiz, L.; Dorantes-Argandar, A.A.; Mercado, M. Hereditary pituitary tumor syndromes: Genetic and clinical aspects. Rev. Investig. Clin. 2020, 72, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, V.; Daly, A.F.; Zacharieva, S.; Beckers, A. Clinical and Molecular Update on Genetic Causes of Pituitary Adenomas. Horm. Metab. Res. 2020, 52, 553–561. [Google Scholar] [CrossRef]
- Barry, S.; Korbonits, M. Update on the Genetics of Pituitary Tumors. Endocrinol. Metab. Clin. N. Am. 2020, 49, 433–452. [Google Scholar] [CrossRef]
- Cakir, M.; Grossman, A.B. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin. Ther. Targets 2009, 13, 1121–1134. [Google Scholar] [CrossRef]
- Dworakowska, D.; Wlodek, E.; Leontiou, C.A.; Igreja, S.; Cakir, M.; Teng, M.; Prodromou, N.; Góth, M.I.; Grozinsky-Glasberg, S.; Gueorguiev, M.; et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr. Relat. Cancer 2009, 16, 1329–1338. [Google Scholar] [CrossRef]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [CrossRef]
- Khan, K.; Gogonea, V.; Fox, P.L. Aminoacyl-tRNA synthetases of the multi-tRNA synthetase complex and their role in tumorigenesis. Transl. Oncol. 2022, 19, 101392. [Google Scholar] [CrossRef]
- Lamb, L.S.; Sim, H.W.; McCormack, A.I. Exploring the Role of Novel Medical Therapies for Aggressive Pituitary Tumors: A Review of the Literature-“Are We There Yet?”. Cancers 2020, 12, 308. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.S.; Wang, E.L.; Xu, W.F.; Yamada, S.; Yoshimoto, K.; Qian, Z.R.; Shi, L.; Liu, L.L.; Li, X.H. Overexpression of DNA (Cytosine-5)-Methyltransferase 1 (DNMT1) And DNA (Cytosine-5)-Methyltransferase 3A (DNMT3A) Is Associated with Aggressive Behavior and Hypermethylation of Tumor Suppressor Genes in Human Pituitary Adenomas. Med. Sci. Monit. 2018, 24, 4841–4850. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, S. Epigenetic control in pituitary tumors. Endocr. J. 2008, 55, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Usui, H.; Morii, K.; Tanaka, R.; Tamura, T.; Washiyama, K.; Ichikawa, T.; Kumanishi, T. cDNA cloning and mRNA expression analysis of the human neuronatin. High level expression in human pituitary gland and pituitary adenomas. J. Mol. Neurosci. 1997, 9, 55–60. [Google Scholar] [CrossRef]
- Revill, K.; Dudley, K.J.; Clayton, R.N.; McNicol, A.M.; Farrell, W.E. Loss of neuronatin expression is associated with promoter hypermethylation in pituitary adenoma. Endocr. Relat. Cancer 2009, 16, 537–548. [Google Scholar] [CrossRef]
- Simpson, D.J.; Clayton, R.N.; Farrell, W.E. Preferential loss of Death Associated Protein kinase expression in invasive pituitary tumours is associated with either CpG island methylation or homozygous deletion. Oncogene 2002, 21, 1217–1224. [Google Scholar] [CrossRef]
- Herrgott, G.A.; Asmaro, K.P.; Wells, M.; Sabedot, T.S.; Malta, T.M.; Mosella, M.S.; Nelson, K.; Scarpace, L.; Barnholtz-Sloan, J.S.; Sloan, A.E.; et al. Detection of tumor-specific DNA methylation markers in the blood of patients with pituitary neuroendocrine tumors. Neuro Oncol. 2022, 24, 1126–1139. [Google Scholar] [CrossRef]
- Kiseljak-Vassiliades, K.; Carlson, N.E.; Borges, M.T.; Kleinschmidt-DeMasters, B.K.; Lillehei, K.O.; Kerr, J.M.; Wierman, M.E. Growth hormone tumor histological subtypes predict response to surgical and medical therapy. Endocrine 2015, 49, 231–241. [Google Scholar] [CrossRef]
- Ogedegbe, O.J.; Cheema, A.Y.; Khan, M.A.; Junaid, S.Z.S.; Erebo, J.K.; Ayirebi-Acquah, E.; Okpara, J.; Bofah, D.; Okon, J.G.; Munir, M.; et al. A Comprehensive Review of Four Clinical Practice Guidelines of Acromegaly. Cureus 2022, 14, e28722. [Google Scholar] [CrossRef]
- Syro, L.V.; Rotondo, F.; Ramirez, A.; Di Ieva, A.; Sav, M.A.; Restrepo, L.M.; Serna, C.A.; Kovacs, K. Progress in the Diagnosis and Classification of Pituitary Adenomas. Front. Endocrinol. 2015, 6, 97. [Google Scholar] [CrossRef]
- Burcea, I.F.; Năstase, V.N.; Poiană, C. Pituitary transcription factors in the immunohistochemical and molecular diagnosis of pituitary tumours—A systematic review. Endokrynol. Pol. 2021, 72, 53–63. [Google Scholar] [CrossRef]
- Faltermeier, C.M.; Magill, S.T.; Blevins, L.S., Jr.; Aghi, M.K. Molecular Biology of Pituitary Adenomas. Neurosurg. Clin. N. Am. 2019, 30, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Hage, M.; Viengchareun, S.; Brunet, E.; Villa, C.; Pineau, D.; Bouligand, J.; Teglas, J.-P.; Adam, C.; Parker, F.; Lombès, M.; et al. Genomic Alterations and Complex Subclonal Architecture in Sporadic GH-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2018, 103, 1929–1939. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.J.; Reitman, Z.J.; Ma, Z.Y.; Chen, J.H.; Zhang, Q.L.; Shou, X.F.; Huang, C.X.; Wang, Y.F.; Li, S.Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Välimäki, N.; Demir, H.; Pitkänen, E.; Kaasinen, E.; Karppinen, A.; Kivipelto, L.; Schalin-Jäntti, C.; Aaltonen, L.A.; Karhu, A. Whole-Genome Sequencing of Growth Hormone (GH)-Secreting Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2015, 100, 3918–3927. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allolio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef]
- Lecoq, A.L.; Kamenický, P.; Guiochon-Mantel, A.; Chanson, P. Genetic mutations in sporadic pituitary adenomas—What to screen for? Nat. Rev. Endocrinol. 2015, 11, 43–54. [Google Scholar] [CrossRef]
- Tuominen, I.; Heliövaara, E.; Raitila, A.; Rautiainen, M.R.; Mehine, M.; Katainen, R.; Donner, I.; Aittomäki, V.; Lehtonen, H.J.; Ahlsten, M.; et al. AIP inactivation leads to pituitary tumorigenesis through defective Gαi-cAMP signaling. Oncogene 2015, 34, 1174–1184. [Google Scholar] [CrossRef]
- Bogusławska, A.; Korbonits, M. Genetics of Acromegaly and Gigantism. J. Clin. Med. 2021, 10, 1377. [Google Scholar] [CrossRef]
- Pejhan, S.; VanUum, S.; Duggal, N.; Rotenberg, B.; Mayich, M.; Hammond, R.; Zhang, Q. Somatotroph Adenoma with Dual Transcription Factor Expression. Can. J. Neurol. Sci./J. Can. Sci. Neurol. 2021, 48, S10. [Google Scholar] [CrossRef]
- Hunter, J.A.; Skelly, R.H.; Aylwin, S.J.; Geddes, J.F.; Evanson, J.; Besser, G.M.; Monson, J.P.; Burrin, J.M. The relationship between pituitary tumour transforming gene (PTTG) expression and in vitro hormone and vascular endothelial growth factor (VEGF) secretion from human pituitary adenomas. Eur. J. Endocrinol. 2003, 148, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, V.; Zhou, C.; Ben-Shlomo, A.; Zonis, S.; Tani, Y.; Ren, S.G.; Melmed, S. Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3331–E3339. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Lopes, M.B. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2017, 28, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hernandez, K.; Ezzat, S.; Asa, S.L.; Mete, Ö. Clinical Implications of Accurate Subtyping of Pituitary Adenomas: Perspectives from the Treating Physician. Turk Patoloji Derg. 2015, 31 (Suppl. S1), 4–17. [Google Scholar] [CrossRef]
- Trouillas, J.; Delgrange, E.; Wierinckx, A.; Vasiljevic, A.; Jouanneau, E.; Burman, P.; Raverot, G. Clinical, Pathological, and Molecular Factors of Aggressiveness in Lactotroph Tumours. Neuroendocrinology 2019, 109, 70–76. [Google Scholar] [CrossRef]
- Wierinckx, A.; Delgrange, E.; Bertolino, P.; François, P.; Chanson, P.; Jouanneau, E.; Lachuer, J.; Trouillas, J.; Raverot, G. Sex-Related Differences in Lactotroph Tumor Aggressiveness Are Associated With a Specific Gene-Expression Signature and Genome Instability. Front. Endocrinol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Lim, J.S.; Ku, C.R.; Lee, M.K.; Kim, T.S.; Kim, S.H.; Lee, E.J. A case of fugitive acromegaly, initially presented as invasive prolactinoma. Endocrine 2010, 38, 1–5. [Google Scholar] [CrossRef]
- Shimon, I. Giant Prolactinomas. Neuroendocrinology 2018, 109, 51–56. [Google Scholar] [CrossRef]
- Molitch, M.E. Diagnosis and Treatment of Pituitary Adenomas: A Review. JAMA 2017, 317, 516–524. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Tohti, M.; Hu, Y.; Wang, S.; Li, W.; Lu, Z.; Ma, C. The expression profile of Dopamine D2 receptor, MGMT and VEGF in different histological subtypes of pituitary adenomas: A study of 197 cases and indications for the medical therapy. J. Exp. Clin. Cancer Res. 2014, 33, 56. [Google Scholar] [CrossRef]
- Marques, N.V.; Kasuki, L.; Coelho, M.C.; Lima, C.H.A.; Wildemberg, L.E.; Gadelha, M.R. Frequency of familial pituitary adenoma syndromes among patients with functioning pituitary adenomas in a reference outpatient clinic. J. Endocrinol. Investig. 2017, 40, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Wildemberg, L.E.; Fialho, C.; Gadelha, M.R. Prolactinomas. La Presse Médicale 2021, 50, 104080. [Google Scholar] [CrossRef] [PubMed]
- Lemelin, A.; Lapoirie, M.; Abeillon, J.; Lasolle, H.; Giraud, S.; Philouze, P.; Ceruse, P.; Raverot, G.; Vighetto, A.; Borson-Chazot, F. Pheochromocytoma, paragangliomas, and pituitary adenoma: An unusual association in a patient with an SDHD mutation. Case report. Medicine 2019, 98, e16594. [Google Scholar] [CrossRef] [PubMed]
- Cotton, E.; Ray, D. DICER1 mutation and pituitary prolactinoma. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Igreja, S.; Chahal, H.S.; King, P.; Bolger, G.B.; Srirangalingam, U.; Guasti, L.; Chapple, J.P.; Trivellin, G.; Gueorguiev, M.; Guegan, K.; et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum. Mutat. 2010, 31, 950–960. [Google Scholar] [CrossRef]
- Araujo, P.B.; Kasuki, L.; de Azeredo Lima, C.H.; Ogino, L.; Camacho, A.H.S.; Chimelli, L.; Korbonits, M.; Gadelha, M.R. AIP mutations in Brazilian patients with sporadic pituitary adenomas: A single-center evaluation. Endocr. Connect. 2017, 6, 914–925. [Google Scholar] [CrossRef]
- Raverot, G.r.; Wierinckx, A.; Dantony, E.; Auger, C.; Chapas, G.; Villeneuve, L.; Brue, T.; Figarella-Branger, D.; Roy, P.; Jouanneau, E.; et al. Prognostic Factors in Prolactin Pituitary Tumors: Clinical, Histological, and Molecular Data from a Series of 94 Patients with a Long Postoperative Follow-Up. J. Clin. Endocrinol. Metab. 2010, 95, 1708–1716. [Google Scholar] [CrossRef]
- Roche, M.; Wierinckx, A.; Croze, S.; Rey, C.; Legras-Lachuer, C.; Morel, A.P.; Fusco, A.; Raverot, G.; Trouillas, J.; Lachuer, J. Deregulation of miR-183 and KIAA0101 in Aggressive and Malignant Pituitary Tumors. Front. Med. 2015, 2, 54. [Google Scholar] [CrossRef]
- Wierinckx, A.; Roche, M.; Raverot, G.; Legras-Lachuer, C.; Croze, S.; Nazaret, N.; Rey, C.; Auger, C.; Jouanneau, E.; Chanson, P.; et al. Integrated genomic profiling identifies loss of chromosome 11p impacting transcriptomic activity in aggressive pituitary PRL tumors. Brain Pathol. 2011, 21, 533–543. [Google Scholar] [CrossRef]
- Finelli, P.; Pierantoni, G.M.; Giardino, D.; Losa, M.; Rodeschini, O.; Fedele, M.; Valtorta, E.; Mortini, P.; Croce, C.M.; Larizza, L.; et al. The High Mobility Group A2 gene is amplified and overexpressed in human prolactinomas. Cancer Res. 2002, 62, 2398–2405. [Google Scholar]
- Guignat, L.; Assie, G.; Bertagna, X.; Bertherat, J. Adénome corticotrope. La Presse Médicale 2009, 38, 125–132. [Google Scholar] [CrossRef] [PubMed]
- George, D.H.; Scheithauer, B.W.; Kovacs, K.; Horvath, E.; Young, W.F., Jr.; Lloyd, R.V.; Meyer, F.B. Crooke’s cell adenoma of the pituitary: An aggressive variant of corticotroph adenoma. Am. J. Surg. Pathol. 2003, 27, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; Wagner, J.R.; Pekmezci, M.; Hiniker, A.; Chang, E.F.; Kunwar, S.; Blevins, L.; Aghi, M.K. A comprehensive long-term retrospective analysis of silent corticotrophic adenomas vs hormone-negative adenomas. Neurosurgery 2013, 73, 8–17, discussion 17–18. [Google Scholar] [CrossRef] [PubMed]
- Laws, E.R., Jr.; Penn, D.L.; Repetti, C.S. Advances and controversies in the classification and grading of pituitary tumors. J. Endocrinol. Investig. 2019, 42, 129–135. [Google Scholar] [CrossRef]
- Yamamoto, M.; Nakao, T.; Ogawa, W.; Fukuoka, H. Aggressive Cushing’s Disease: Molecular Pathology and Its Therapeutic Approach. Front. Endocrinol. 2021, 12, 650791. [Google Scholar] [CrossRef]
- Liu, X.; Feng, M.; Dai, C.; Bao, X.; Deng, K.; Yao, Y.; Wang, R. Expression of EGFR in Pituitary Corticotroph Adenomas and Its Relationship With Tumor Behavior. Front. Endocrinol. 2019, 10, 785. [Google Scholar] [CrossRef]
- Miao, H.; Liu, Y.; Lu, L.; Gong, F.; Wang, L.; Duan, L.; Yao, Y.; Wang, R.; Chen, S.; Mao, X.; et al. Effect of 3 NR3C1 Mutations in the Pathogenesis of Pituitary ACTH Adenoma. Endocrinology 2021, 162, bqab167. [Google Scholar] [CrossRef]
- Abraham, A.P.; Pai, R.; Beno, D.L.; Chacko, G.; Asha, H.S.; Rajaratnam, S.; Kapoor, N.; Thomas, N.; Chacko, A.G. USP8, USP48, and BRAF mutations differ in their genotype-phenotype correlation in Asian Indian patients with Cushing’s disease. Endocrine 2022, 75, 549–559. [Google Scholar] [CrossRef]
- Chasseloup, F.; Pankratz, N.; Lane, J.; Faucz, F.R.; Keil, M.F.; Chittiboina, P.; Kay, D.M.; Hussein Tayeb, T.; Stratakis, C.A.; Mills, J.L.; et al. Germline CDKN1B Loss-of-Function Variants Cause Pediatric Cushing’s Disease With or Without an MEN4 Phenotype. J. Clin. Endocrinol. Metab. 2020, 105, 1983–2005. [Google Scholar] [CrossRef]
- Du, L.; Bergsneider, M.; Mirsadraei, L.; Young, S.H.; Jonker, J.W.; Downes, M.; Yong, W.H.; Evans, R.M.; Heaney, A.P. Evidence for orphan nuclear receptor TR4 in the etiology of Cushing disease. Proc. Natl. Acad. Sci. USA 2013, 110, 8555–8560. [Google Scholar] [CrossRef]
- Riebold, M.; Kozany, C.; Freiburger, L.; Sattler, M.; Buchfelder, M.; Hausch, F.; Stalla, G.K.; Paez-Pereda, M. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat. Med. 2015, 21, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Garbicz, F.; Mehlich, D.; Rak, B.; Sajjad, E.; Maksymowicz, M.; Paskal, W.; Zieliński, G.; Włodarski, P.K. Increased expression of the microRNA 106b~25 cluster and its host gene MCM7 in corticotroph pituitary adenomas is associated with tumor invasion and Crooke’s cell morphology. Pituitary 2017, 20, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Casar-Borota, O.; Boldt, H.B.; Engström, B.E.; Andersen, M.S.; Baussart, B.; Bengtsson, D.; Berinder, K.; Ekman, B.; Feldt-Rasmussen, U.; Höybye, C.; et al. Corticotroph Aggressive Pituitary Tumors and Carcinomas Frequently Harbor ATRX Mutations. J. Clin. Endocrinol. Metab. 2021, 106, 1183–1194. [Google Scholar] [CrossRef]
- Beck-Peccoz, P.; Persani, L.; Lania, A. Thyrotropin-Secreting Pituitary Adenomas. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Pereira, B.D.; Raimundo, L.; Mete, O.; Oliveira, A.; Portugal, J.; Asa, S.L. Monomorphous Plurihormonal Pituitary Adenoma of Pit-1 Lineage in a Giant Adolescent with Central Hyperthyroidism. Endocr. Pathol. 2016, 27, 25–33. [Google Scholar] [CrossRef]
- Sanno, N.; Teramoto, A.; Osamura, R.Y. Thyrotropin-secreting pituitary adenomas. Clinical and biological heterogeneity and current treatment. J. Neurooncol. 2001, 54, 179–186. [Google Scholar] [CrossRef]
- Asteria, C.; Anagni, M.; Persani, L.; Beck-Peccoz, P. Loss of heterozygosity of the MEN1 gene in a large series of TSH-secreting pituitary adenomas. J. Endocrinol. Investig. 2001, 24, 796–801. [Google Scholar] [CrossRef]
- Dong, Q.; Brucker-Davis, F.; Weintraub, B.D.; Smallridge, R.C.; Carr, F.E.; Battey, J.; Spiegel, A.M.; Shenker, A. Screening of candidate oncogenes in human thyrotroph tumors: Absence of activating mutations of the G alpha q, G alpha 11, G alpha s, or thyrotropin-releasing hormone receptor genes. J. Clin. Endocrinol. Metab. 1996, 81, 1134–1140. [Google Scholar] [CrossRef]
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliövaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J. Clin. Endocrinol. Metab. 2010, 95, E373–E383. [Google Scholar] [CrossRef]
- Barlier, A.; Vanbellinghen, J.F.; Daly, A.F.; Silvy, M.; Jaffrain-Rea, M.L.; Trouillas, J.; Tamagno, G.; Cazabat, L.; Bours, V.; Brue, T.; et al. Mutations in the aryl hydrocarbon receptor interacting protein gene are not highly prevalent among subjects with sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. 2007, 92, 1952–1955. [Google Scholar] [CrossRef]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell 2020, 37, 123–134.e125. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Sarlis, N.J.; Krishnan, J.; Feng, X.; Refetoff, S.; Zhang, M.Q.; Oldfield, E.H.; Yen, P.M. Aberrant alternative splicing of thyroid hormone receptor in a TSH-secreting pituitary tumor is a mechanism for hormone resistance. Mol. Endocrinol. 2001, 15, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lian, W.; Xing, B.; Feng, M.; Ma, W.B. Functioning gonadotroph adenoma. Chin. Med. J. 2019, 132, 1003–1004. [Google Scholar] [CrossRef] [PubMed]
- Cote, D.J.; Smith, T.R.; Sandler, C.N.; Gupta, T.; Bale, T.A.; Bi, W.L.; Dunn, I.F.; De Girolami, U.; Woodmansee, W.W.; Kaiser, U.B.; et al. Functional Gonadotroph Adenomas: Case Series and Report of Literature. Neurosurgery 2016, 79, 823–831. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.L.; Gejman, R.; Ansell, P.J.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef]
- Ntali, G.; Capatina, C.; Grossman, A.; Karavitaki, N. Clinical review: Functioning gonadotroph adenomas. J. Clin. Endocrinol. Metab. 2014, 99, 4423–4433. [Google Scholar] [CrossRef]
- Lachkhem, A.; Yahi, A.; Kablia, S.; Derriche, A.; Staifi, A.; Derradji, H. Null cell adenoma: Case report. Endocrine Abstracts 2023, 90, EP867. [Google Scholar] [CrossRef]
- Singh, V.; Gupta, K.; Salunke, P.; Dhandapani, S.S. Null Cell Adenoma of the Pituitary: Pseudo-rosettes Say It Best When Immunohistochemistry Says Nothing At All! Head Neck Pathol. 2019, 13, 677–680. [Google Scholar] [CrossRef]
- Newey, P.J.; Nesbit, M.A.; Rimmer, A.J.; Head, R.A.; Gorvin, C.M.; Attar, M.; Gregory, L.; Wass, J.A.H.; Buck, D.; Karavitaki, N.; et al. Whole-Exome Sequencing Studies of Nonfunctioning Pituitary Adenomas. J. Clin. Endocrinol. Metab. 2013, 98, E796–E800. [Google Scholar] [CrossRef]
- Bahar, A.; Bicknell, J.E.; Simpson, D.J.; Clayton, R.N.; Farrell, W.E. Loss of expression of the growth inhibitory gene GADD45γ, in human pituitary adenomas, is associated with CpG island methylation. Oncogene 2004, 23, 936–944. [Google Scholar] [CrossRef]
- Simpson, D.J.; Bicknell, J.E.; McNicol, A.M.; Clayton, R.N.; Farrell, W.E. Hypermethylation of the p16/CDKN2A/MTS1 gene and loss of protein expression is associated with nonfunctional pituitary adenomas but not somatotrophinomas. Genes Chromosomes Cancer 1999, 24, 328–336. [Google Scholar] [CrossRef]
- Butz, H.; Likó, I.; Czirják, S.; Igaz, P.; Korbonits, M.; Rácz, K.; Patócs, A. MicroRNA profile indicates downregulation of the TGFβ pathway in sporadic non-functioning pituitary adenomas. Pituitary 2011, 14, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Biagetti, B.; Simò, R. Pituitary Apoplexy: Risk Factors and Underlying Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 8721. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Dutta, P. Landscape of Molecular Events in Pituitary Apoplexy. Front. Endocrinol. 2018, 9, 107. [Google Scholar] [CrossRef]
- Xiao, Z.; Liu, Q.; Mao, F.; Wu, J.; Lei, T. TNF-α-induced VEGF and MMP-9 expression promotes hemorrhagic transformation in pituitary adenomas. Int. J. Mol. Sci. 2011, 12, 4165–4179. [Google Scholar] [CrossRef]
- Xiao, Z.; Liu, Q.; Zhao, B.; Wu, J.; Lei, T. Hypoxia induces hemorrhagic transformation in pituitary adenomas via the HIF-1α signaling pathway. Oncol. Rep. 2011, 26, 1457–1464. [Google Scholar] [CrossRef]
- Shan, B.; Gerez, J.; Haedo, M.; Fuertes, M.; Theodoropoulou, M.; Buchfelder, M.; Losa, M.; Stalla, G.K.; Arzt, E.; Renner, U. RSUME is implicated in HIF-1-induced VEGF-A production in pituitary tumour cells. Endocr. Relat. Cancer 2012, 19, 13–27. [Google Scholar] [CrossRef]
- Mou, C.; Han, T.; Zhao, H.; Wang, S.; Qu, Y. Clinical features and immunohistochemical changes of pituitary apoplexy. J. Clin. Neurosci. 2009, 16, 64–68. [Google Scholar] [CrossRef]
- Gültekin, G.D.; Çabuk, B.; Vural, Ç.; Ceylan, S. Matrix metalloproteinase-9 and tissue inhibitor of matrix metalloproteinase-2: Prognostic biological markers in invasive prolactinomas. J. Clin. Neurosci. 2015, 22, 1282–1287. [Google Scholar] [CrossRef]
- Drummond, J.B.; Ribeiro-Oliveira, A., Jr.; Soares, B.S. Non-Functioning Pituitary Adenomas. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
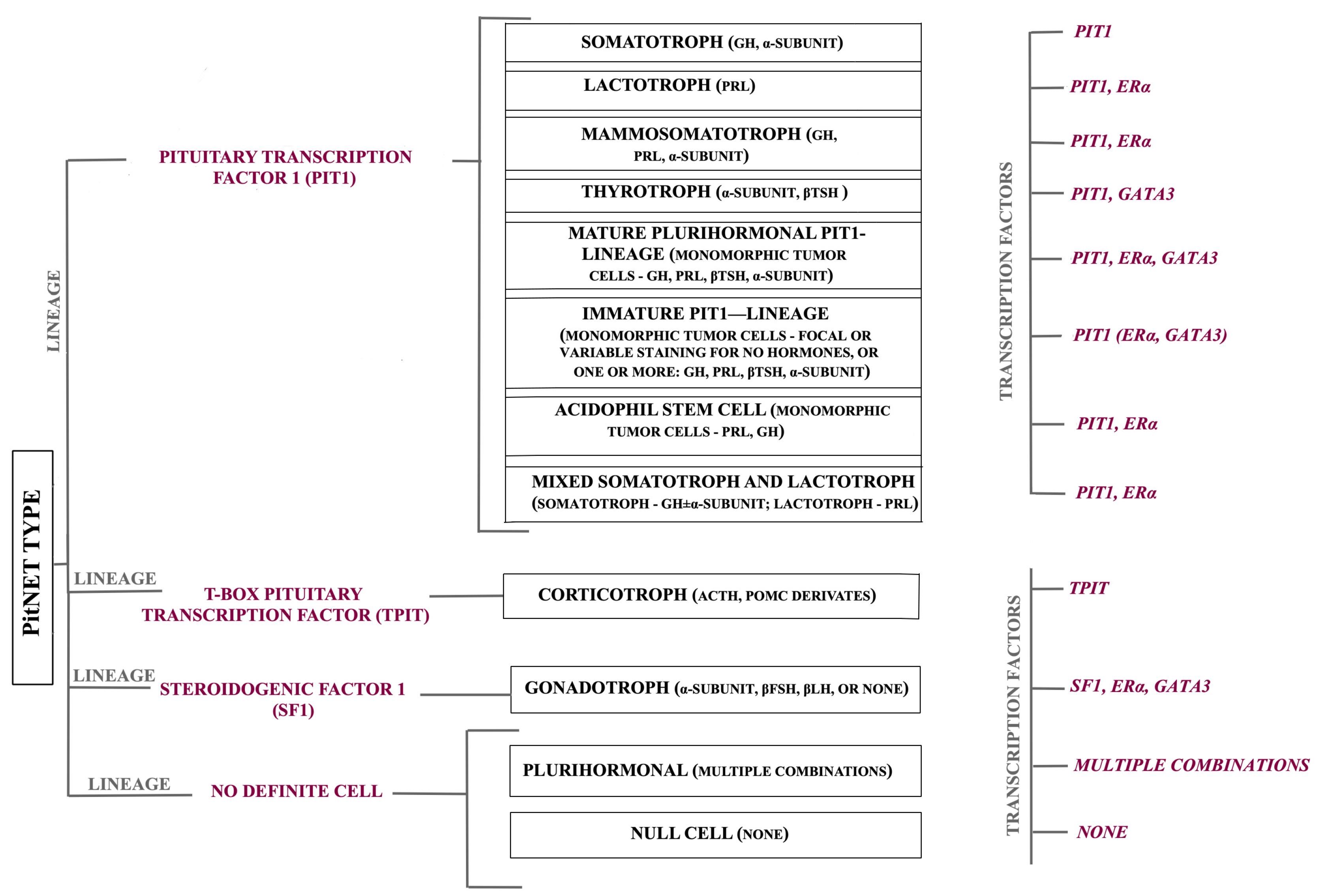
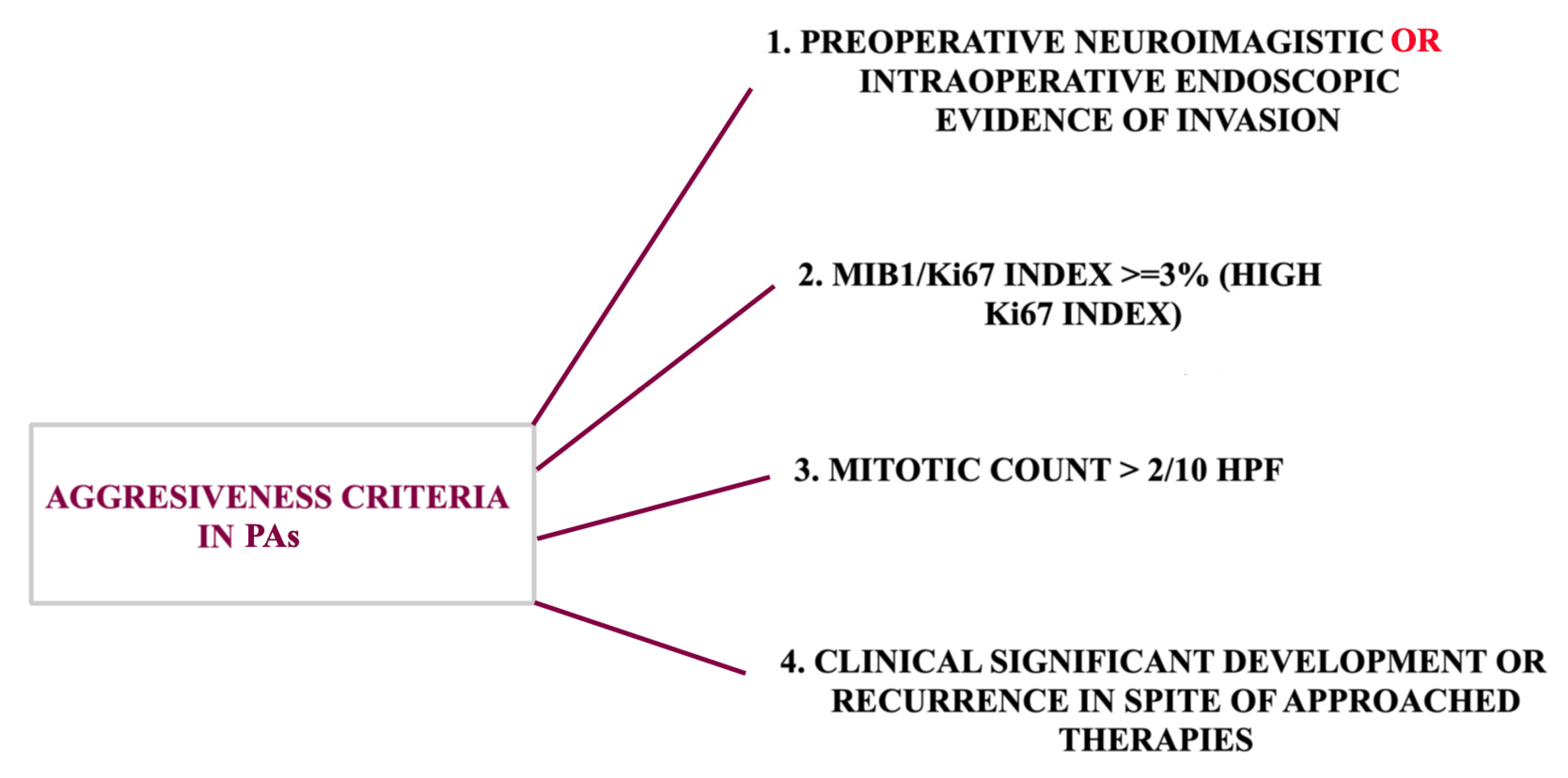

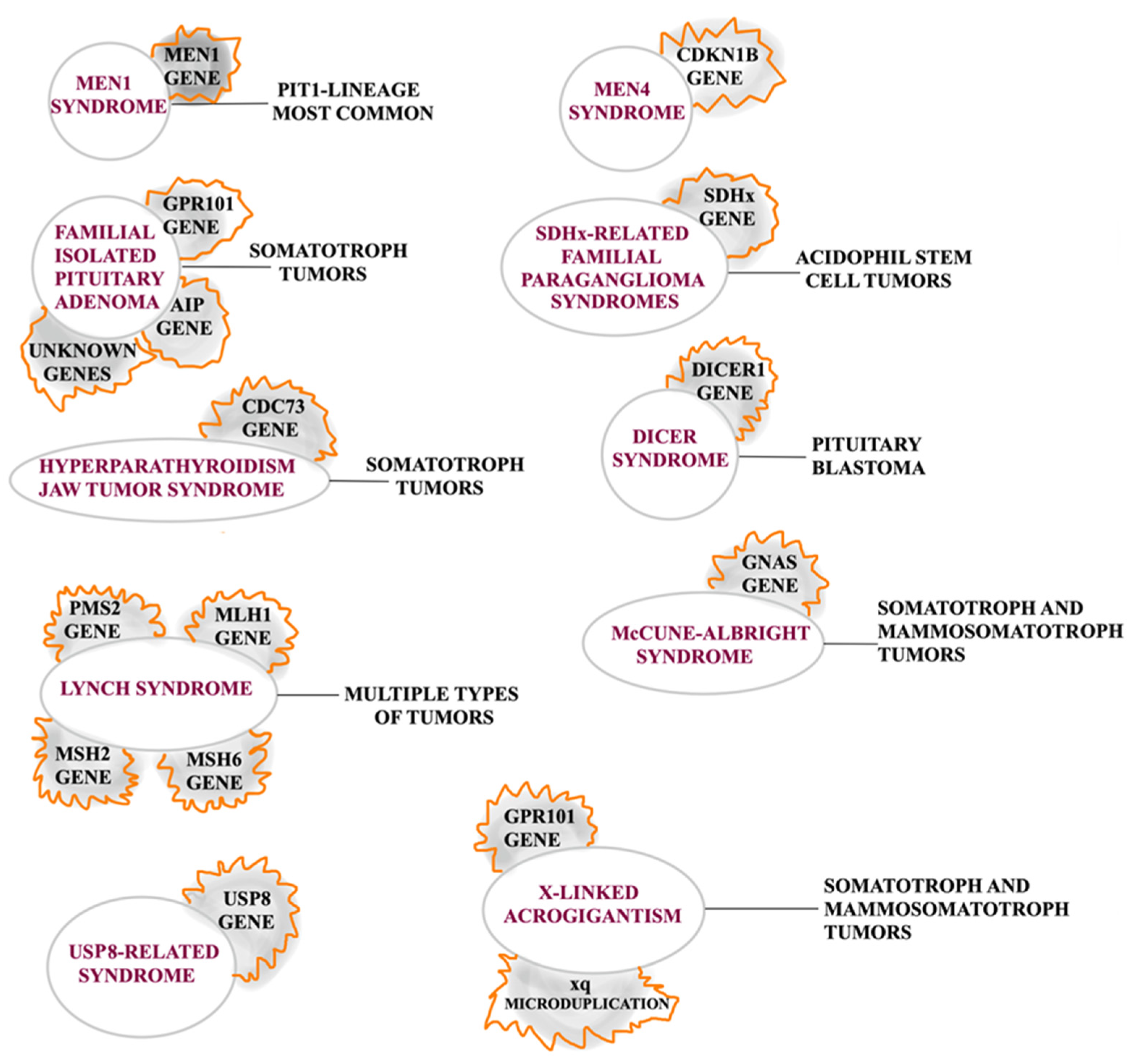

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aggressive PAs | Refractory PAs | |
|---|---|---|
| Resistance to standard therapies | Yes | Yes |
| Cerebral/spinal/distant metastasis | No | No |
| Tumor invasion | Proven on neuroimaging results | Proven intraoperatively and on neuroimaging results |
| Growth velocity | Not necessarily required | >2%/month |
| Recurrence | Not necessarily required | In less than 6 months after surgical removal |
| Ki67 index | Not necessarily required | >3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tataranu, L.G. A Brief Overview of Molecular Biology in Pituitary Adenomas with a Focus on Aggressive Lesions. Int. J. Mol. Sci. 2025, 26, 3717. https://doi.org/10.3390/ijms26083717
Tataranu LG. A Brief Overview of Molecular Biology in Pituitary Adenomas with a Focus on Aggressive Lesions. International Journal of Molecular Sciences. 2025; 26(8):3717. https://doi.org/10.3390/ijms26083717
Chicago/Turabian StyleTataranu, Ligia Gabriela. 2025. "A Brief Overview of Molecular Biology in Pituitary Adenomas with a Focus on Aggressive Lesions" International Journal of Molecular Sciences 26, no. 8: 3717. https://doi.org/10.3390/ijms26083717
APA StyleTataranu, L. G. (2025). A Brief Overview of Molecular Biology in Pituitary Adenomas with a Focus on Aggressive Lesions. International Journal of Molecular Sciences, 26(8), 3717. https://doi.org/10.3390/ijms26083717