
Development and Validation of a Prognostic Model for Lung Adenocarcinoma Based on CAF-Related Genes: Unveiling the Role of COX6A1 in Cancer Progression and CAF Infiltration


Abstract
1. Introduction
2. Results
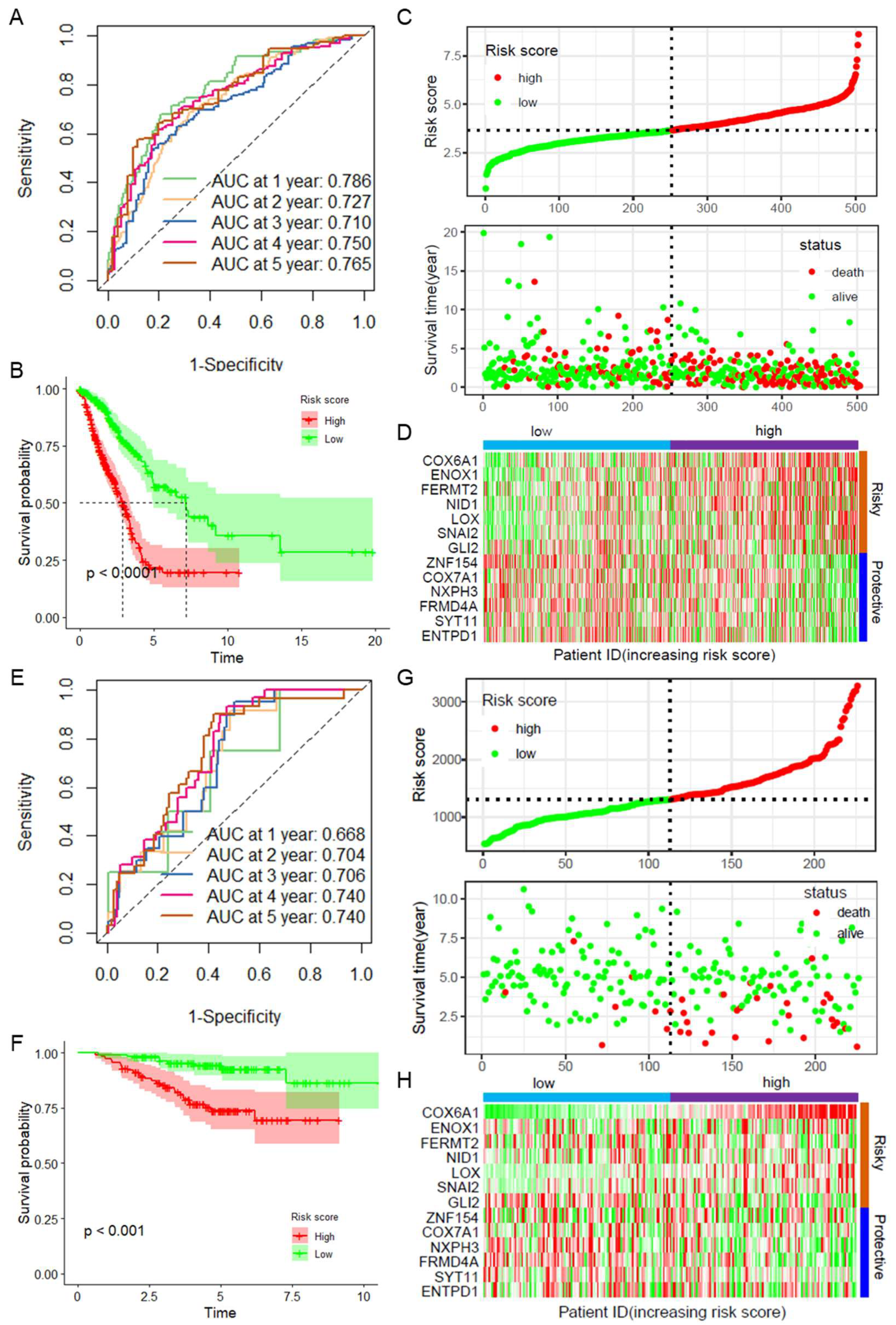
2.1. Construction and Validation of the Prognostic Model Based on CAFRGs
2.2. Construction and Validation of a Prognostic Model Based on CAF-Related Genes
2.3. Validation of the CAFRGs Risk Model
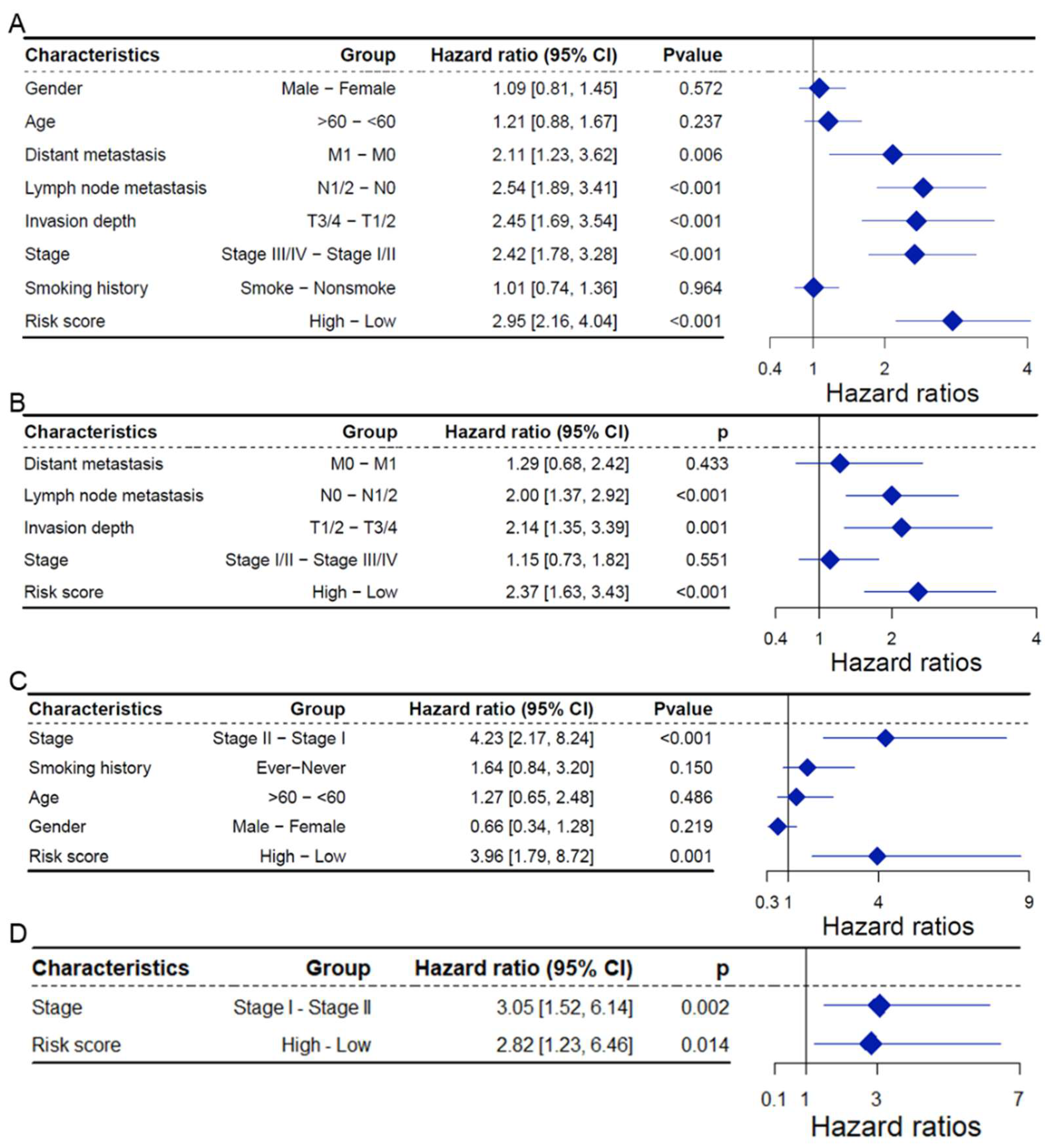
2.4. CAFRG Risk Score as an Independent Prognostic Factor
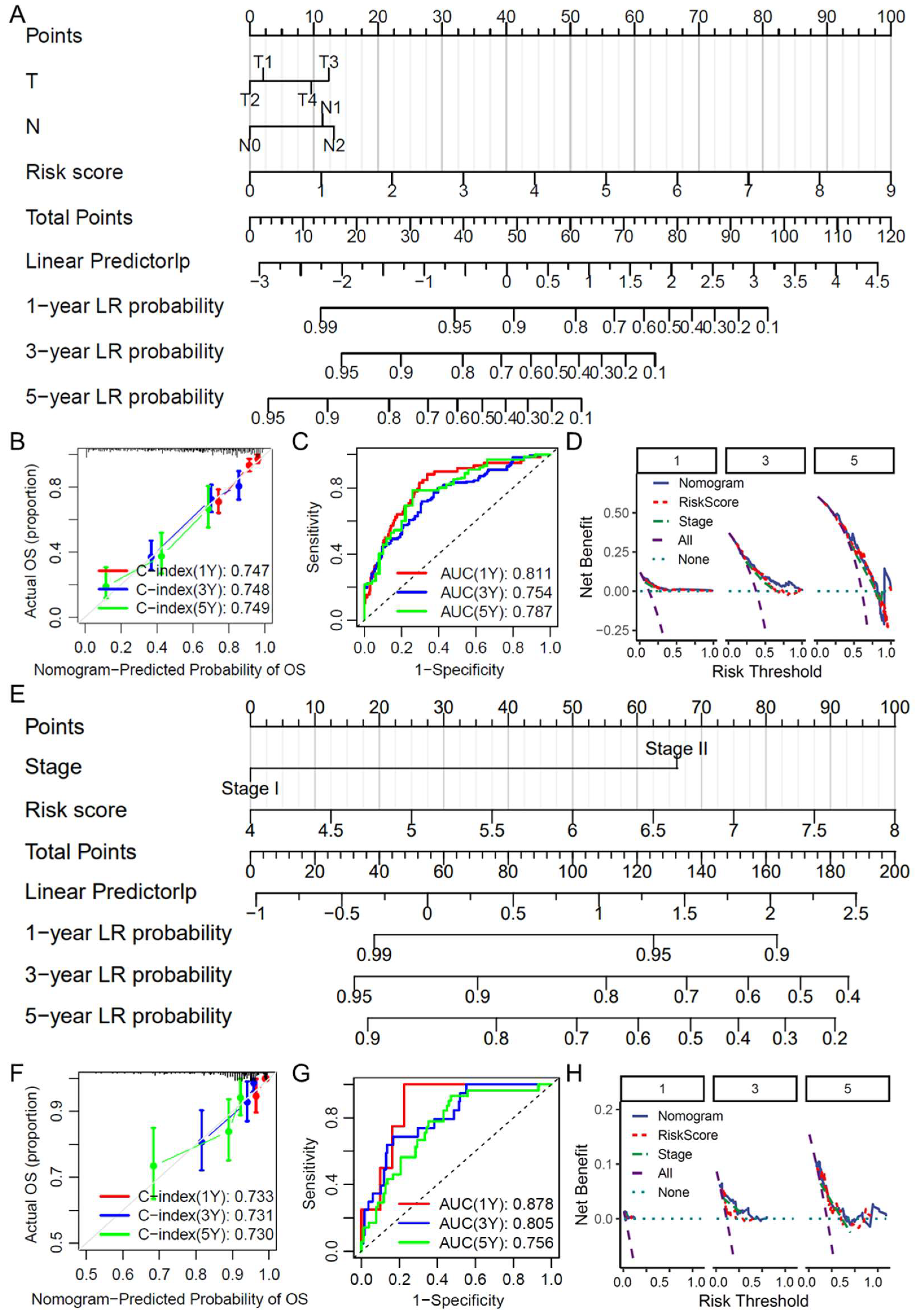
2.5. Construction and Evaluation of a Clinical Prediction Nomogram
2.6. Construction of a Nomogram-Based Clinical Prediction Tool
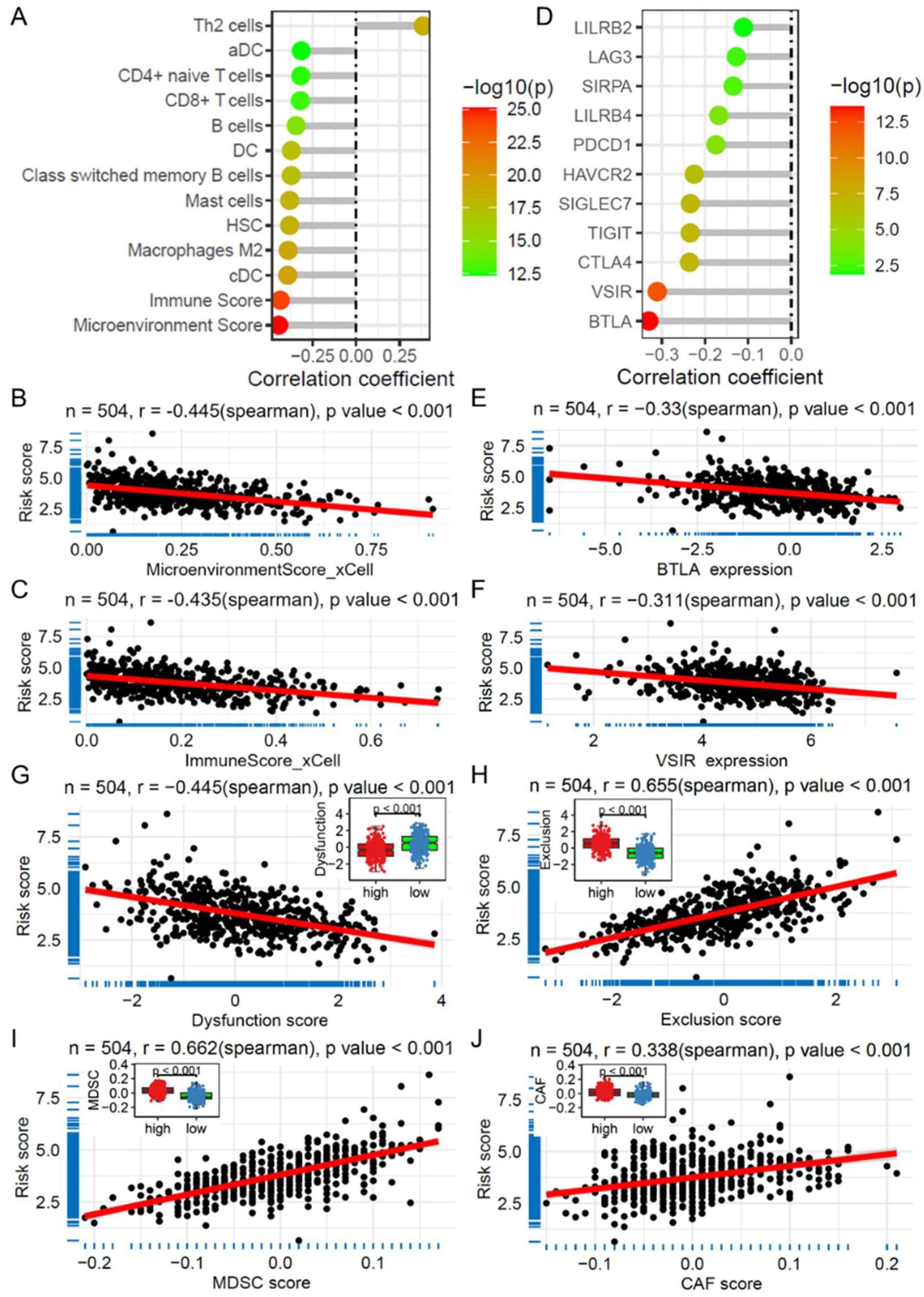
2.7. Risk Score and Its Association with Immune Cell Infiltration and Immunotherapy
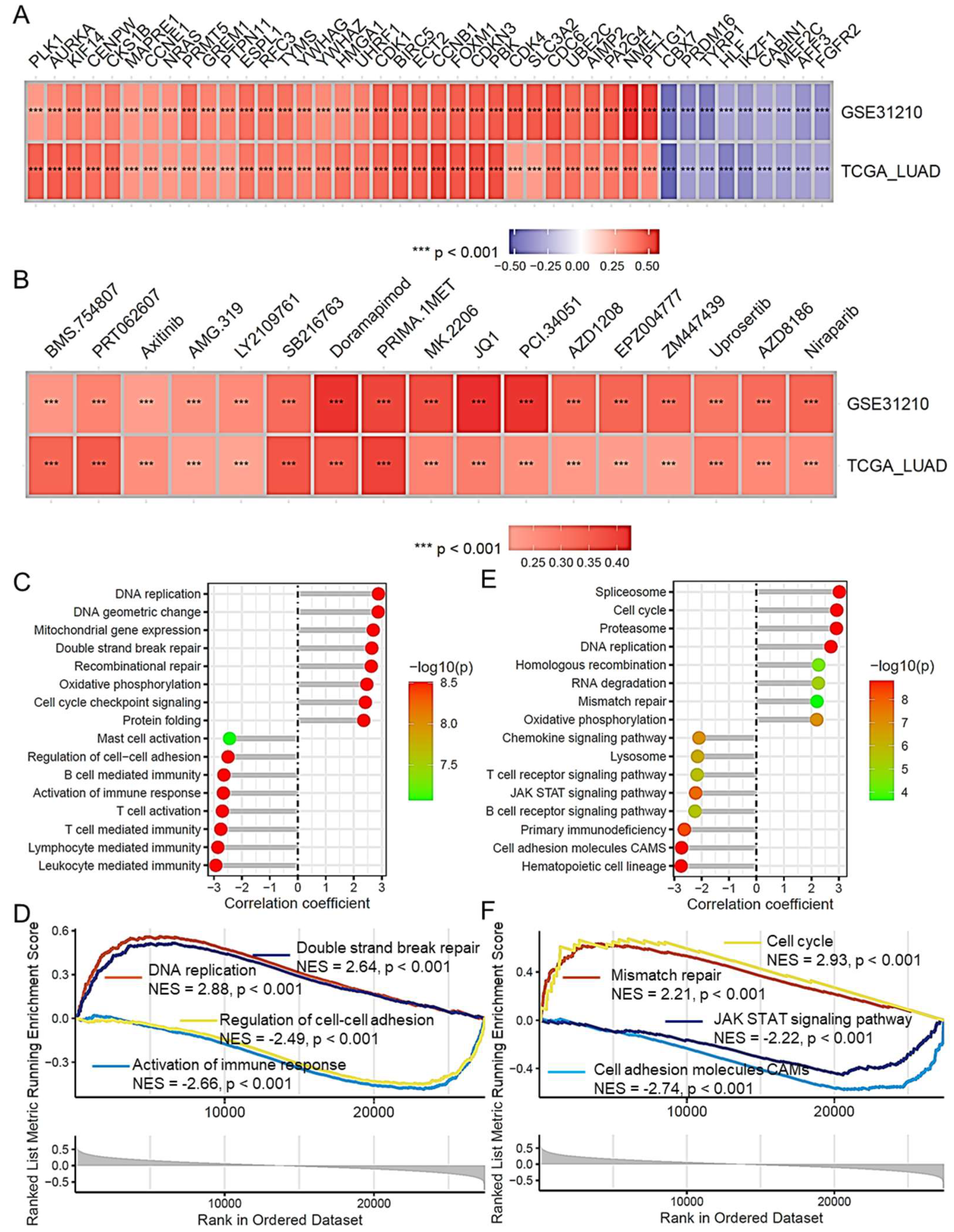
2.8. Risk Score and Its Association with Lung Adenocarcinoma Progression
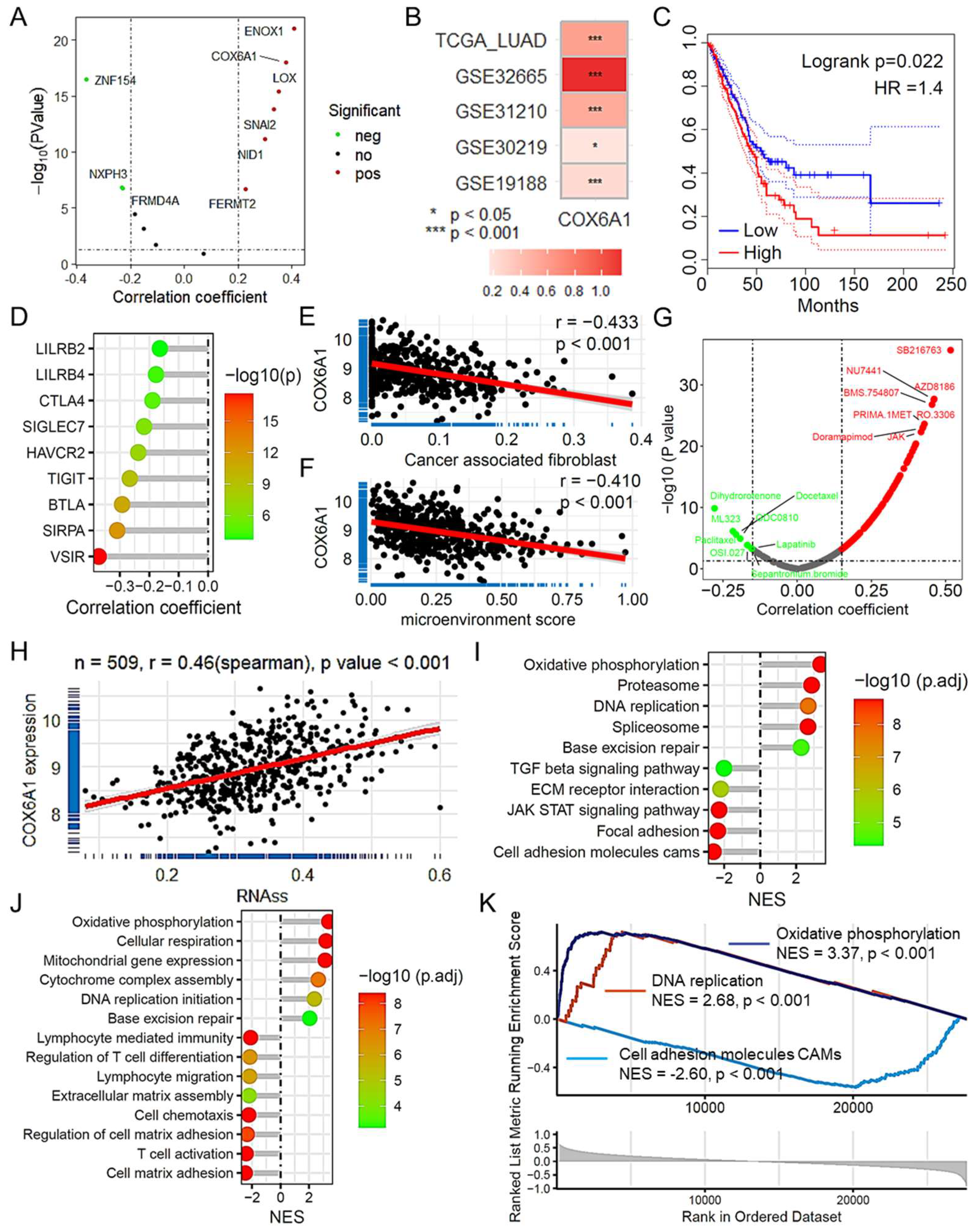
2.9. COX6A1 as a Key Gene Promoting Tumor Progression in LUAD
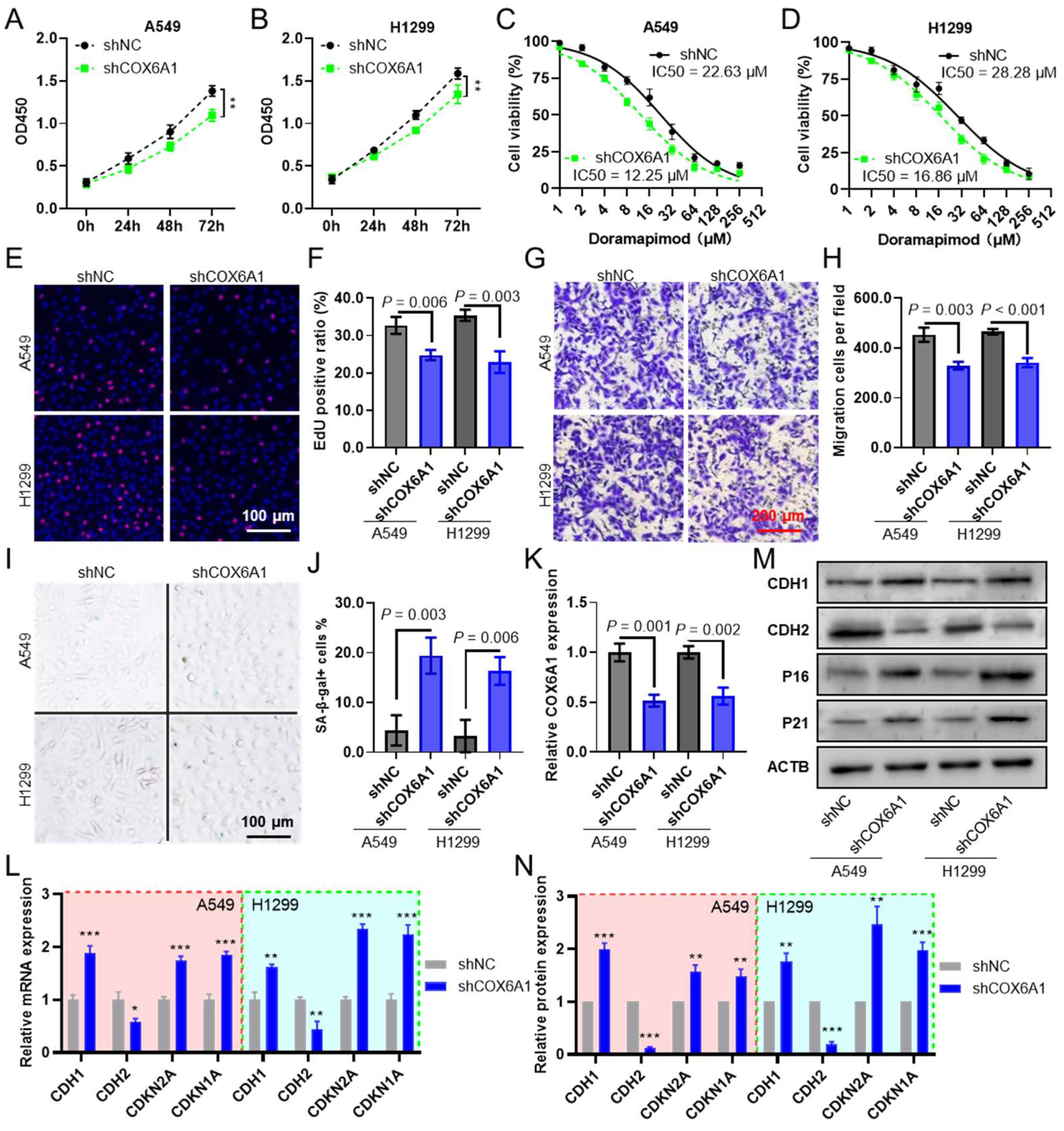
2.10. COX6A1 Is a Gene That Promotes Tumor Progression in the Model
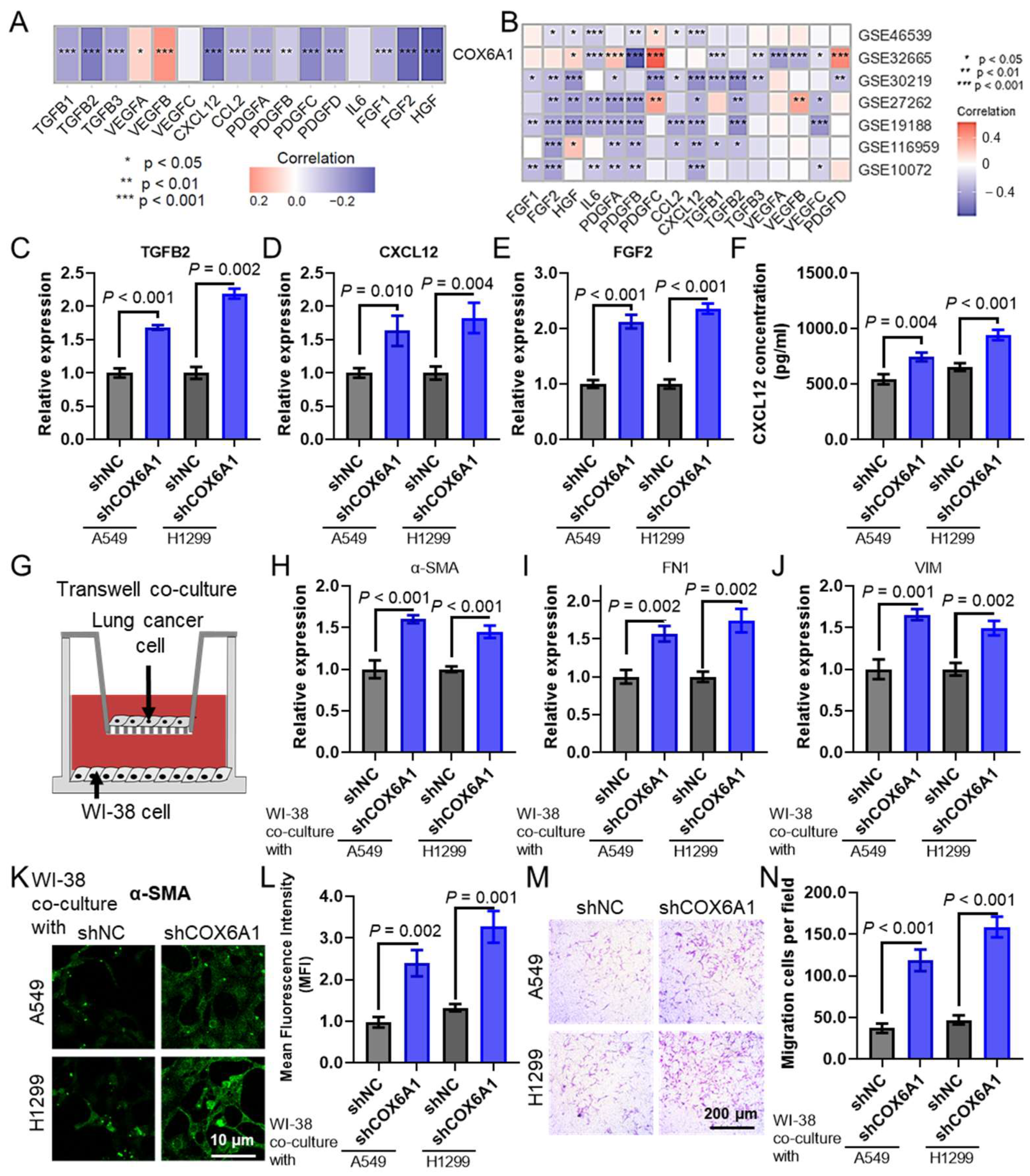
2.11. COX6A1 Knockdown in Lung Cancer Cells Promotes CAF Infiltration
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Immune Infiltration Analysis
4.3. Prognostic Model Construction and Validation
4.4. Nomogram Construction and Evaluation
4.5. Clinical Prediction Tool Development
4.6. Drug Sensitivity Analysis
4.7. GSEA Enrichment Analysis
4.8. Cell Culture
4.9. shRNA Construction and Transfection
4.10. CCK-8 Assay
4.11. Doramapimod Dose–Response Curve
4.12. Transwell Cell Migration Assay
4.13. EdU Cell Proliferation Assay
4.14. β-Galactosidase Assay
4.15. Quantitative PCR (qPCR)
4.16. Western Blotting
4.17. ELISA (Enzyme-Linked Immunosorbent Assay)
4.18. Co-Culture System
4.19. Immunofluorescence Analysis
4.20. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AUC | Area Under the Curve |
| CAF | Cancer-Associated Fibroblast |
| CAFRG | Cancer-Associated Fibroblast-Related Genes |
| COX | Cox regression analysis |
| DCA | Decision Curve Analysis |
| GEO | Gene Expression Omnibus |
| GSEA | Gene Set Enrichment Analysis |
| HR | Hazard Ratio |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| LUAD | Lung Adenocarcinoma |
| NES | Normalized Enrichment Score |
| OXPHOS | Oxidative Phosphorylation |
| qPCR | Quantitative Polymerase Chain Reaction |
| ROC | Receiver Operating Characteristic |
| TCGA | The Cancer Genome Atlas |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| TIMER | Tumor Immune Estimation Resource |
References
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Chen, P.; Liu, Y.; Wen, Y.; Zhou, C. Non-small cell lung cancer in China. Cancer Commun. 2022, 42, 937–970. [Google Scholar] [CrossRef]
- Li, Y.; Yan, B.; He, S. Advances and challenges in the treatment of lung cancer. Biomed. Pharmacother. 2023, 169, 115891. [Google Scholar] [CrossRef]
- Sen, T.; Takahashi, N.; Chakraborty, S.; Takebe, N.; Nassar, A.H.; Karim, N.A.; Puri, S.; Naqash, A.R. Emerging advances in defining the molecular and therapeutic landscape of small-cell lung cancer. Nat. Rev. Clin. Oncol. 2024, 21, 610–627. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: From basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, H.; Zhu, L.; Li, J.; Ma, S. Cancer-associated fibroblasts in non-small cell lung cancer: Recent advances and future perspectives. Cancer Lett. 2021, 514, 38–47. [Google Scholar] [CrossRef]
- Cords, L.; Engler, S.; Haberecker, M.; Rüschoff, J.H.; Moch, H.; de Souza, N.; Bodenmiller, B. Cancer-associated fibroblast phenotypes are associated with patient outcome in non-small cell lung cancer. Cancer Cell 2024, 42, 396–412.e5. [Google Scholar] [CrossRef]
- Li, X.; Sun, Z.; Peng, G.; Xiao, Y.; Guo, J.; Wu, B.; Li, X.; Zhou, W.; Li, J.; Li, Z.; et al. Single-cell RNA sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics 2022, 12, 620–638. [Google Scholar] [CrossRef]
- Davidson, G.; Helleux, A.; Vano, Y.A.; Lindner, V.; Fattori, A.; Cerciat, M.; Elaidi, R.T.; Verkarre, V.; Sun, C.M.; Chevreau, C.; et al. Mesenchymal-like Tumor Cells and Myofibroblastic Cancer-Associated Fibroblasts Are Associated with Progression and Immunotherapy Response of Clear Cell Renal Cell Carcinoma. Cancer Res. 2023, 83, 2952–2969. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, J.; Li, J.; Zhu, Z.; Cui, Z.; Liu, R.; Lu, R.; Yao, Z.; Xu, Q. Cancer associated fibroblast-derived CCL5 promotes hepatocellular carcinoma metastasis through activating HIF1α/ZEB1 axis. Cell Death Dis. 2022, 13, 478. [Google Scholar] [CrossRef]
- Fang, T.; Lv, H.; Lv, G.; Li, T.; Wang, C.; Han, Q.; Yu, L.; Su, B.; Guo, L.; Huang, S.; et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018, 9, 191. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Feng, S.; Xu, Y.; Dai, Z.; Yin, H.; Zhang, K.; Shen, Y. Integrative Analysis from Multicenter Studies Identifies a WGCNA-Derived Cancer-Associated Fibroblast Signature for Ovarian Cancer. Front. Immunol. 2022, 13, 951582. [Google Scholar] [CrossRef]
- Herrera, M.; Berral-González, A.; López-Cade, I.; Galindo-Pumariño, C.; Bueno-Fortes, S.; Martín-Merino, M.; Carrato, A.; Ocaña, A.; De La Pinta, C.; López-Alfonso, A.; et al. Cancer-associated fibroblast-derived gene signatures determine prognosis in colon cancer patients. Mol. Cancer 2021, 20, 73. [Google Scholar] [CrossRef]
- Rui, R.; Zhou, L.; He, S. Cancer immunotherapies: Advances and bottlenecks. Front. Immunol. 2023, 14, 1212476. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Chen, P.Y.; Wei, W.F.; Wu, H.Z.; Fan, L.S.; Wang, W. Cancer-Associated Fibroblast Heterogeneity: A Factor That Cannot Be Ignored in Immune Microenvironment Remodeling. Front. Immunol. 2021, 12, 671595. [Google Scholar] [CrossRef]
- Philip, M.; Schietinger, A. CD8(+) T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 2022, 22, 209–223. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef]
- Terlizzi, M.; Colarusso, C.; Pinto, A.; Sorrentino, R. Drug resistance in non-small cell lung Cancer (NSCLC): Impact of genetic and non-genetic alterations on therapeutic regimen and responsiveness. Pharmacol. Ther. 2019, 202, 140–148. [Google Scholar] [CrossRef]
- Wong-Riley, M.; Guo, A.; Bachman, N.J.; Lomax, M.I. Human COX6A1 gene: Promoter analysis, cDNA isolation and expression in the monkey brain. Gene 2000, 247, 63–75. [Google Scholar] [CrossRef]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int. J. Cancer 2020, 146, 10–17. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Uslu, C.; Kapan, E.; Lyakhovich, A. Cancer resistance and metastasis are maintained through oxidative phosphorylation. Cancer Lett. 2024, 587, 216705. [Google Scholar] [CrossRef]
- Wang, L.; Lankhorst, L.; Bernards, R. Exploiting senescence for the treatment of cancer. Nat. Rev. Cancer 2022, 22, 340–355. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- Lunin, S.M.; Novoselova, E.G.; Glushkova, O.V.; Parfenyuk, S.B.; Novoselova, T.V.; Khrenov, M.O. Cell Senescence and Central Regulators of Immune Response. Int. J. Mol. Sci. 2022, 23, 4109. [Google Scholar] [CrossRef]
- Xin, L.; Gao, J.; Zheng, Z.; Chen, Y.; Lv, S.; Zhao, Z.; Yu, C.; Yang, X.; Zhang, R. Fibroblast Activation Protein-α as a Target in the Bench-to-Bedside Diagnosis and Treatment of Tumors: A Narrative Review. Front. Oncol. 2021, 11, 648187. [Google Scholar] [CrossRef]
- Vasiukov, G.; Zou, Y.; Senosain, M.F.; Rahman, J.S.M.; Antic, S.; Young, K.M.; Grogan, E.L.; Kammer, M.N.; Maldonado, F.; Reinhart-King, C.A.; et al. Cancer-associated fibroblasts in early-stage lung adenocarcinoma correlate with tumor aggressiveness. Sci. Rep. 2023, 13, 17604. [Google Scholar] [CrossRef]
- Thapa, P.; Debnath, S.; Bedi, A.; Parashar, M.; Gonzalez, P.; Reus, J.; Hammers, H.; Sun, X. Monovalent and Divalent Designs of Copper Radiotheranostics Targeting Fibroblast Activation Protein in Cancer. Cancers 2024, 16, 4180. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | TCGA LUAD | Chi-Square p Value | |||
|---|---|---|---|---|---|
| Training (n = 353) | Internal Testing (n = 151) | All (n = 504) | |||
| Gender | female | 194 (54.96%) | 76 (50.33%) | 270 (53.57%) | 0.634 |
| male | 159 (45.04%) | 75 (49.67%) | 234 (46.43%) | ||
| Age | ≤60 | 119 (34.59%) | 39 (26.00%) | 158 (31.98%) | 0.170 |
| >60 | 225 (65.41%) | 111 (74.00%) | 336 (68.02%) | ||
| M | M0 | 231 (90.59%) | 100 (95.24%) | 335 (93.06%) | 0.116 |
| M1 | 24 (9.41%) | 5 (4.76%) | 25 (6.94%) | ||
| N | N0 | 225 (65.79%) | 99 (66.89%) | 324 (66.12%) | 0.972 |
| N1/2 | 117 (34.21%) | 49 (33.11%) | 166 (33.88%) | ||
| T | T1/2 | 306 (86.69%) | 132 (87.42%) | 438 (86.90%) | 0.975 |
| T3/4 | 47 (13.31%) | 19 (12.58%) | 66 (13.10%) | ||
| Stage | Stage I/II | 272 (77.05%) | 118 (78.15%) | 390 (77.38%) | 0.965 |
| Stage III/IV | 81 (22.95%) | 33 (21.85%) | 114 (22.62%) | ||
| Smoke history | Nonsmoke | 139 (39.38%) | 61 (40.40%) | 200 (39.68%) | 0.977 |
| Smoke | 214 (60.62%) | 90 (59.60%) | 304 (60.32%) | ||
| Time | ≤2 | 204 (57.79%) | 81 (53.64%) | 285 (56.55%) | 0.691 |
| >2 | 149 (42.21%) | 70 (46.36%) | 219 (43.45%) | ||
| Status | 0 | 220 (62.32%) | 101 (66.89%) | 321 (63.69%) | 0.621 |
| 1 | 133 (37.68%) | 50 (33.11%) | 183 (36.31%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.; Li, B.; Qin, L.; Liang, T.; Hu, W.; Li, J.; Wang, J. Development and Validation of a Prognostic Model for Lung Adenocarcinoma Based on CAF-Related Genes: Unveiling the Role of COX6A1 in Cancer Progression and CAF Infiltration. Int. J. Mol. Sci. 2025, 26, 3478. https://doi.org/10.3390/ijms26083478
Zhu X, Li B, Qin L, Liang T, Hu W, Li J, Wang J. Development and Validation of a Prognostic Model for Lung Adenocarcinoma Based on CAF-Related Genes: Unveiling the Role of COX6A1 in Cancer Progression and CAF Infiltration. International Journal of Molecular Sciences. 2025; 26(8):3478. https://doi.org/10.3390/ijms26083478
Chicago/Turabian StyleZhu, Xinyu, Bo Li, Lexin Qin, Tingting Liang, Wentao Hu, Jianxiang Li, and Jin Wang. 2025. "Development and Validation of a Prognostic Model for Lung Adenocarcinoma Based on CAF-Related Genes: Unveiling the Role of COX6A1 in Cancer Progression and CAF Infiltration" International Journal of Molecular Sciences 26, no. 8: 3478. https://doi.org/10.3390/ijms26083478
APA StyleZhu, X., Li, B., Qin, L., Liang, T., Hu, W., Li, J., & Wang, J. (2025). Development and Validation of a Prognostic Model for Lung Adenocarcinoma Based on CAF-Related Genes: Unveiling the Role of COX6A1 in Cancer Progression and CAF Infiltration. International Journal of Molecular Sciences, 26(8), 3478. https://doi.org/10.3390/ijms26083478