

Cell Death Modalities in Therapy of Melanoma


Abstract
1. Introduction
2. Pathogenesis
3. Classification
4. Clinical Features
5. The Cell Death Mechanisms of Current Systematic Therapy
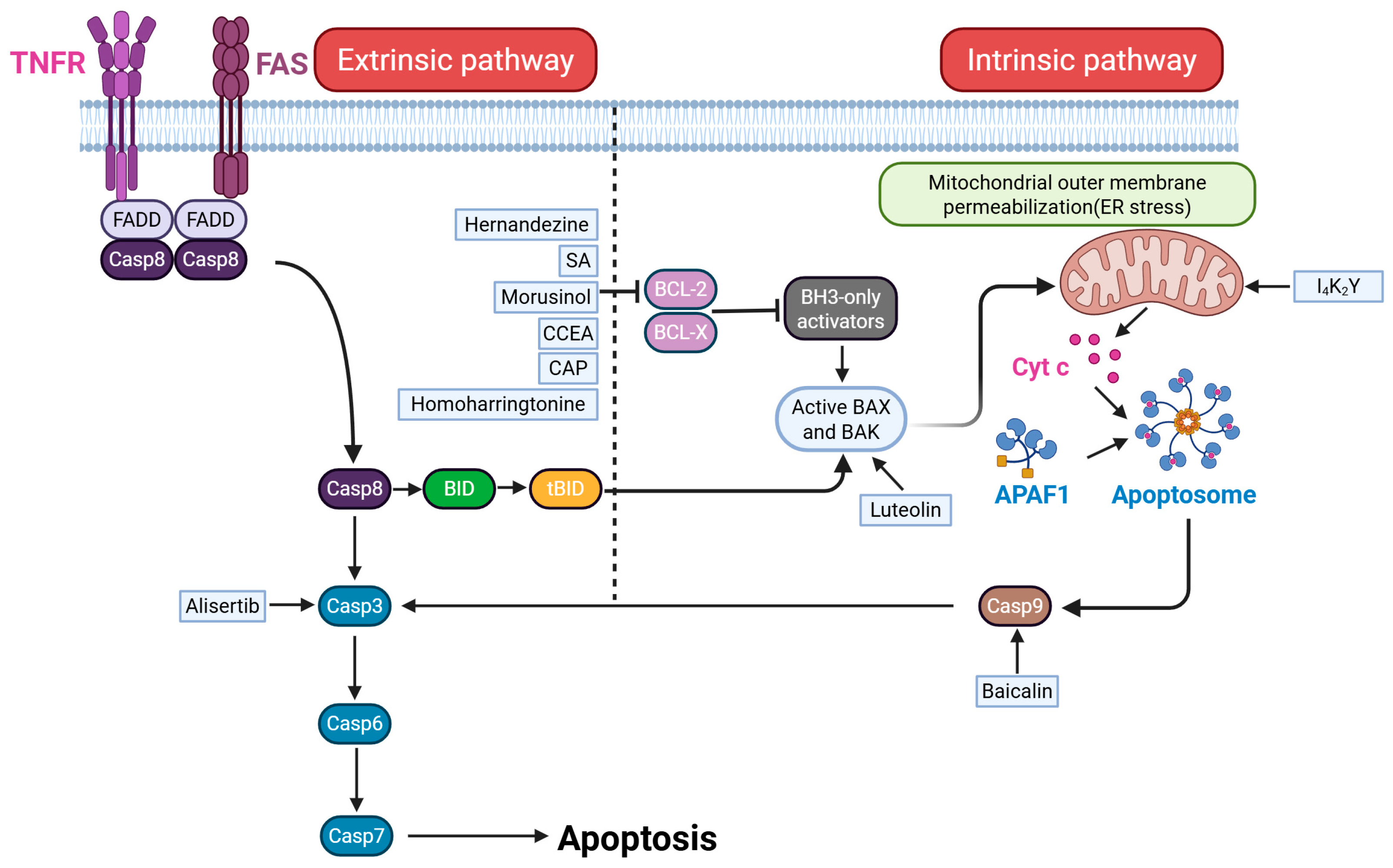
5.1. Apoptosis
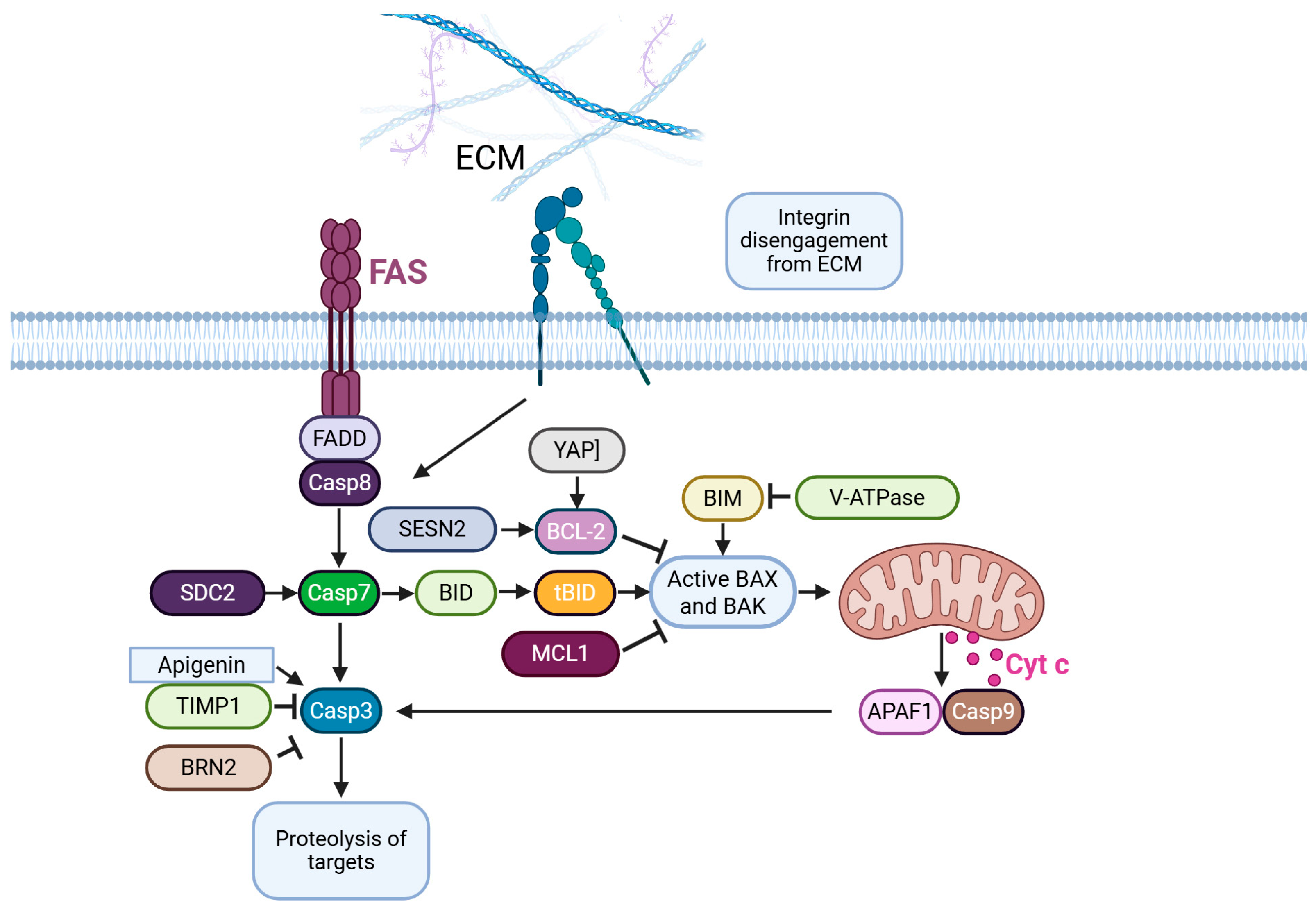
5.2. Anoikis
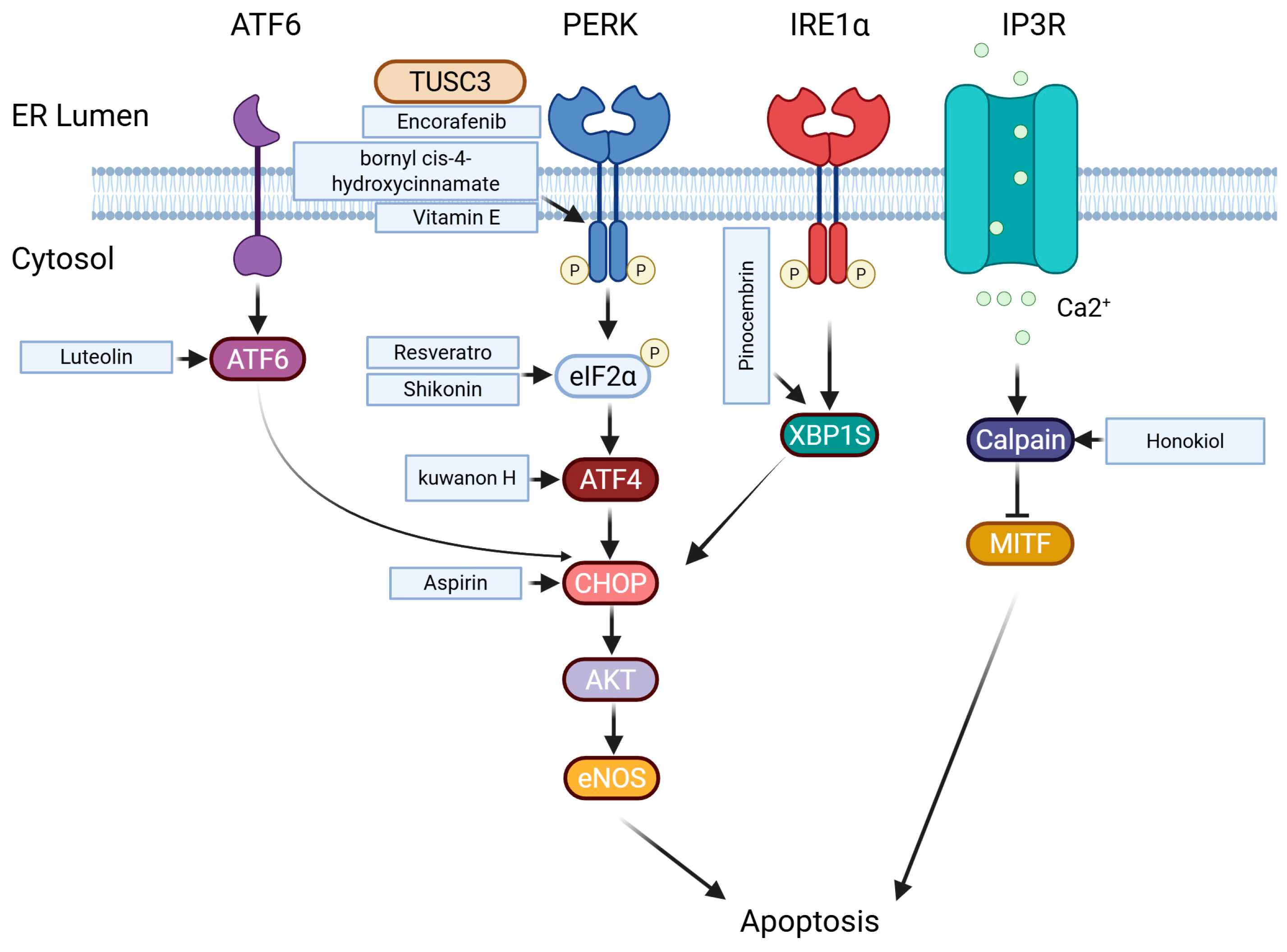
5.3. Endoplasmic Reticulum Stress
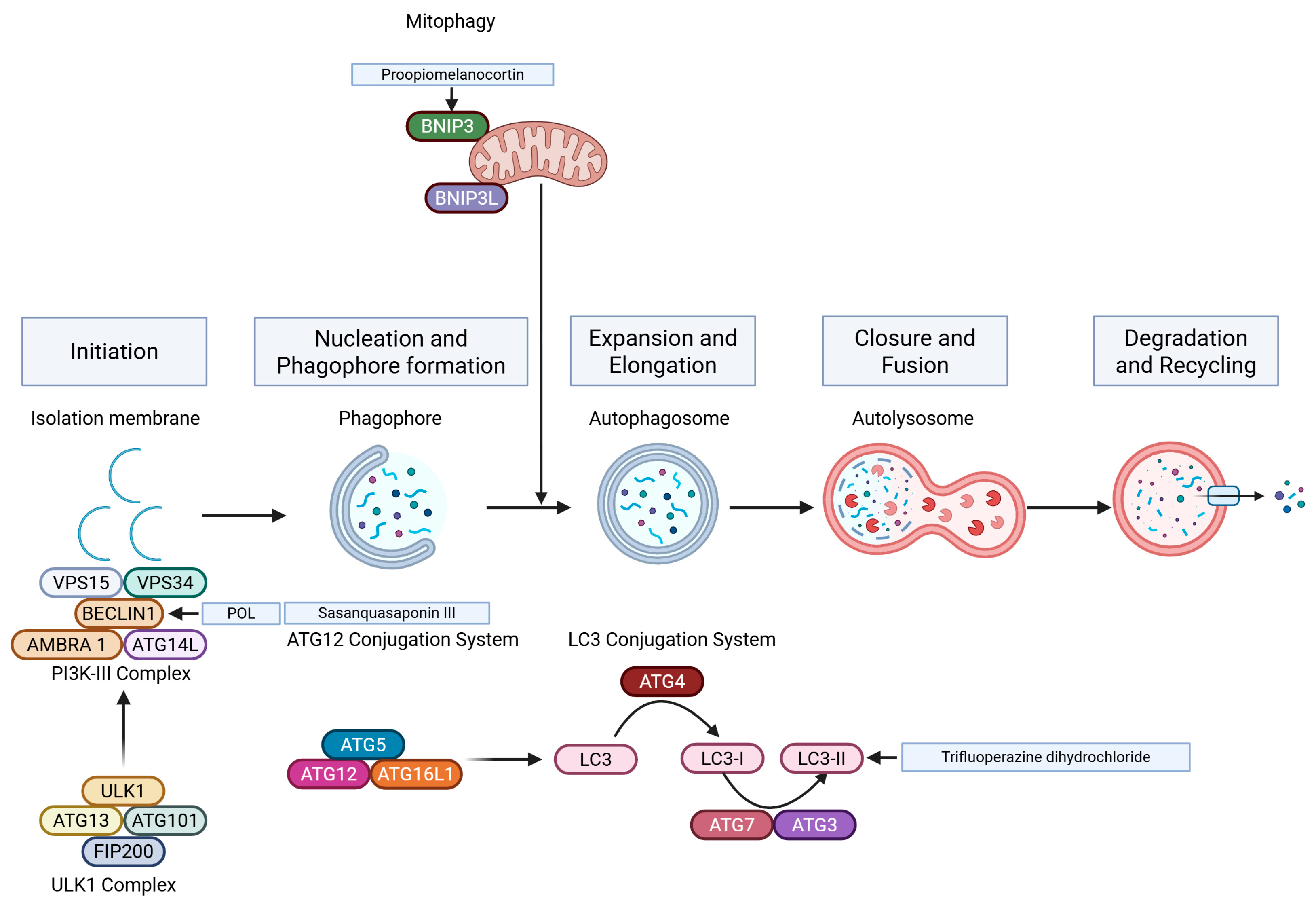
5.4. Autophagy
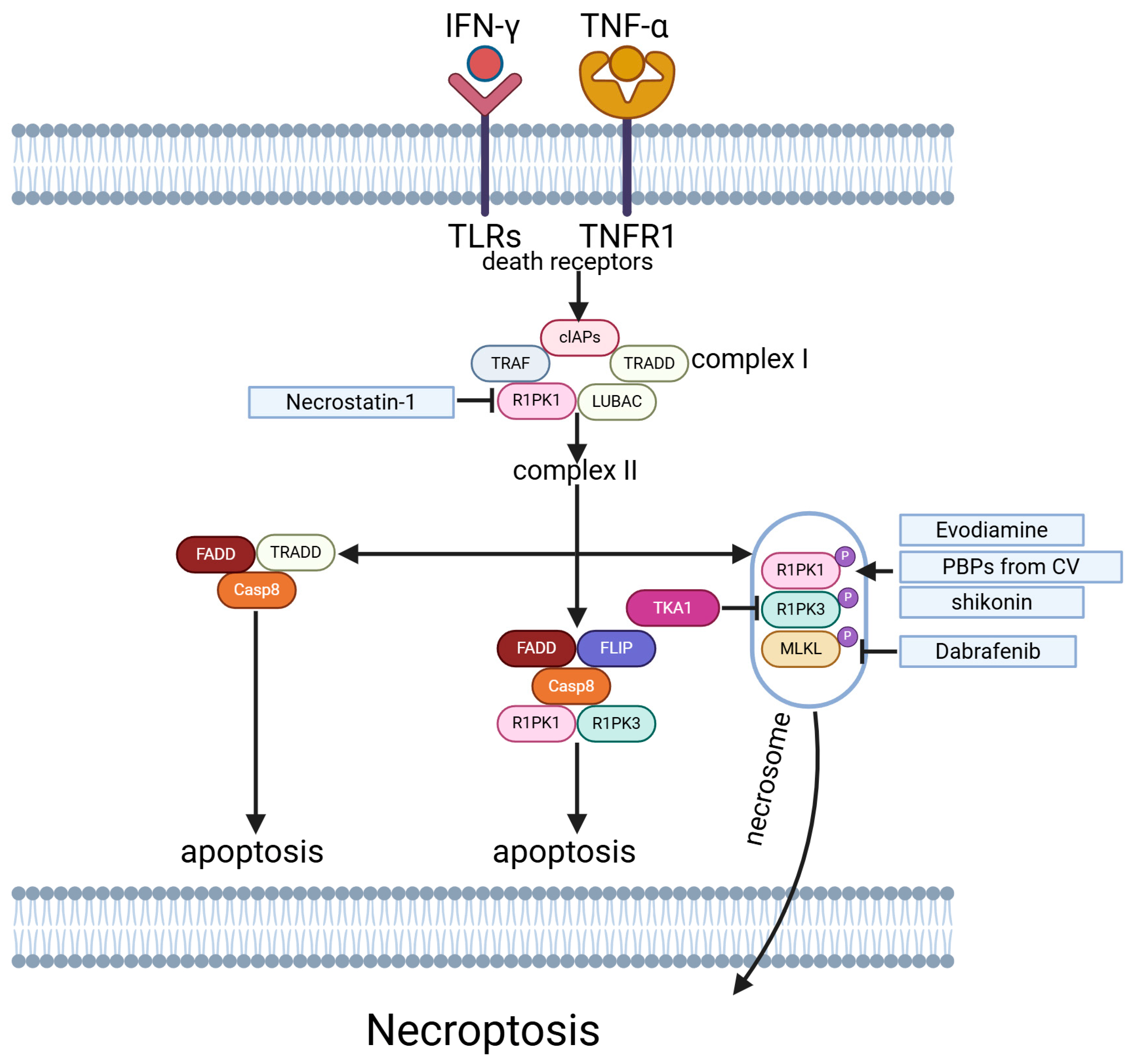
5.5. Necroptosis
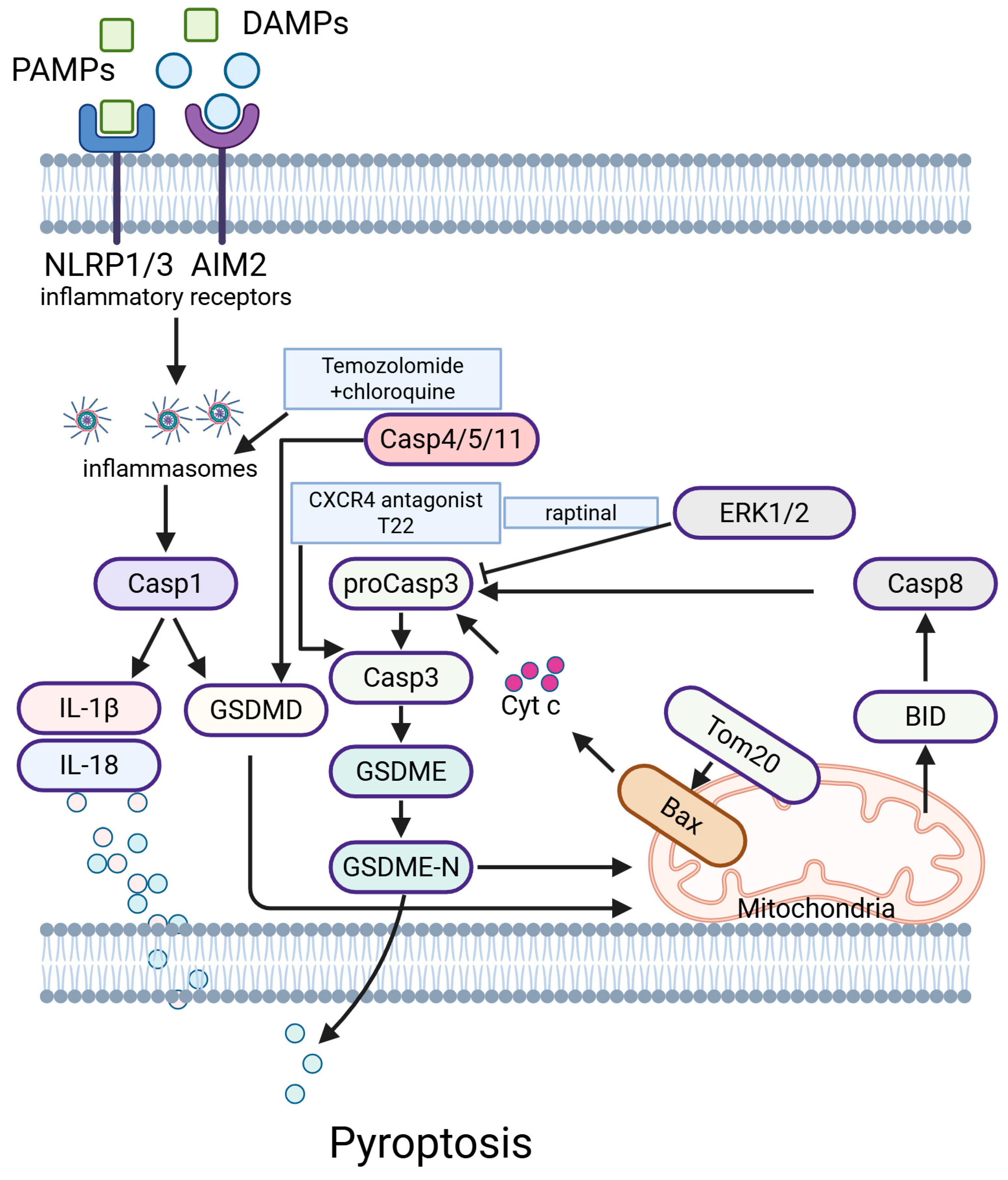
5.6. Pyroptosis
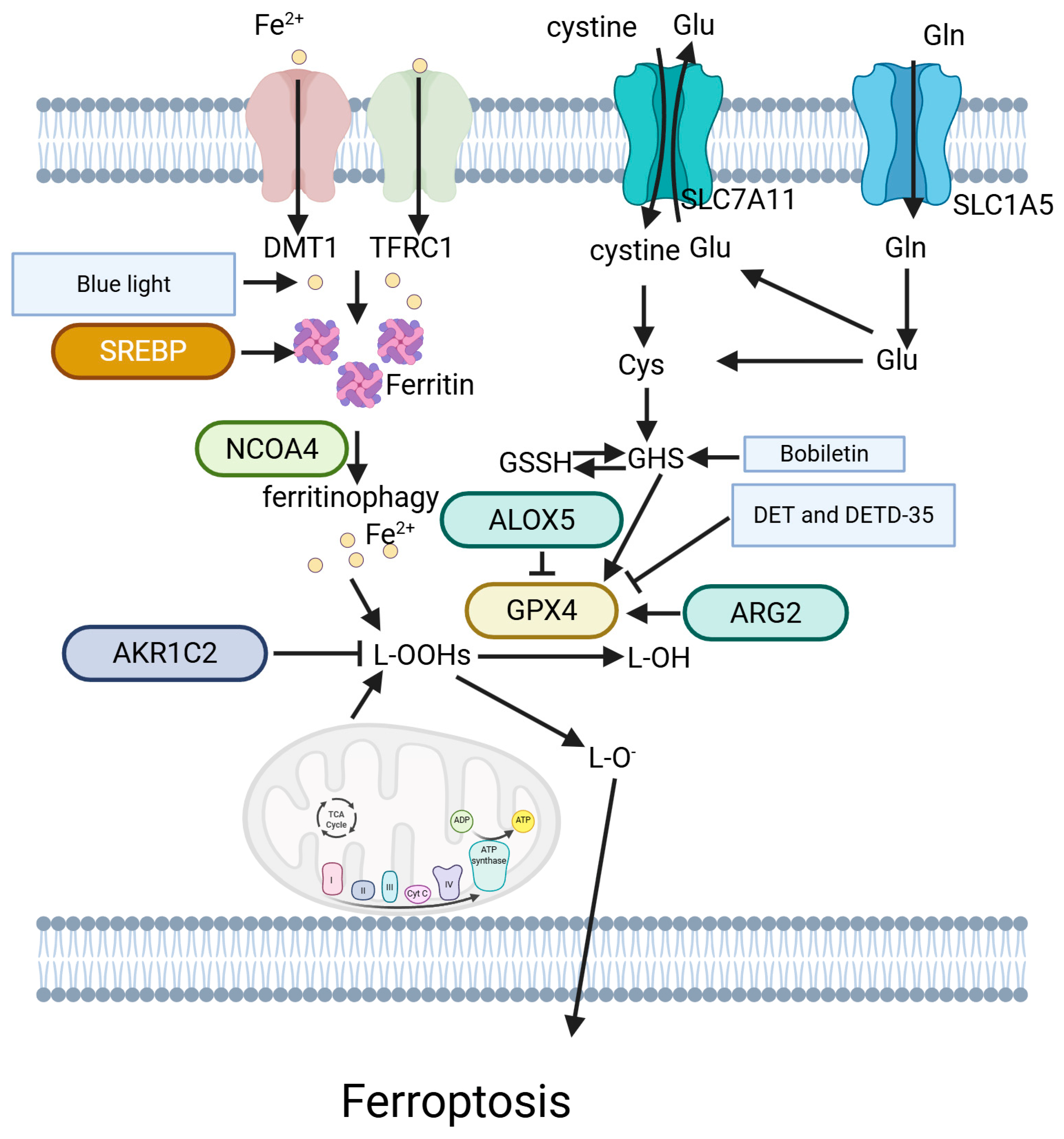
5.7. Ferroptosis
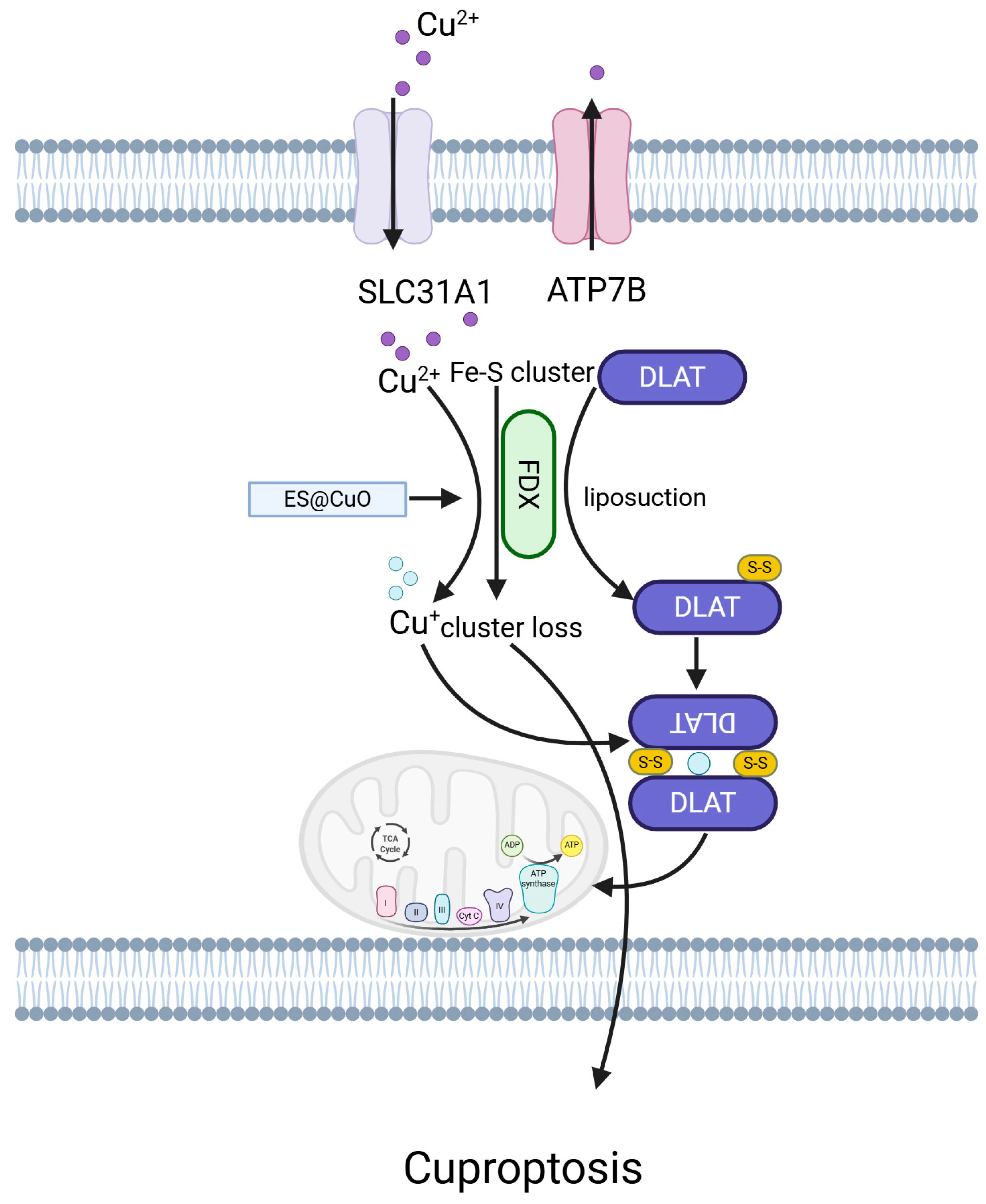
5.8. Cuproptosis
6. Current Clinical Treatment for Melanoma
7. Discussion
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lo, J.A.; Fisher, D.E. The melanoma revolution: From UV carcinogenesis to a new era in therapeutics. Science 2014, 346, 945–949. [Google Scholar] [PubMed]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [PubMed]
- Arozarena, I.; Wellbrock, C. Targeting invasive properties of melanoma cells. FEBS J. 2017, 284, 2148–2162. [Google Scholar] [PubMed]
- Sanz-Moreno, V.; Marshall, C.J. Rho-GTPase signaling drives melanoma cell plasticity. Cell Cycle 2009, 8, 1484–1487. [Google Scholar]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar]
- Couetil, J.; Liu, Z.; Huang, K.; Zhang, J.; Alomari, A.K. Predicting melanoma survival and metastasis with interpretable histopathological features and machine learning models. Front. Med. 2022, 9, 1029227. [Google Scholar]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar]
- Dimitriou, F.; Long, G.V.; Menzies, A.M. Novel adjuvant options for cutaneous melanoma. Ann. Oncol. 2021, 32, 854–865. [Google Scholar]
- Lazaroff, J.; Bolotin, D. Targeted Therapy and Immunotherapy in Melanoma. Dermatol. Clin. 2023, 41, 65–77. [Google Scholar] [PubMed]
- Atkins, M.B.; Lee, S.J.; Chmielowski, B.; Tarhini, A.A.; Cohen, G.I.; Truong, T.G.; Moon, H.H.; Davar, D.; O’Rourke, M.; Stephenson, J.J.; et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients With Advanced BRAF-Mutant Melanoma: The DREAMseq Trial-ECOG-ACRIN EA6134. J. Clin. Oncol. 2023, 41, 186–197. [Google Scholar]
- Liu, N.; Zhang, J.; Yin, M.; Liu, H.; Zhang, X.; Li, J.; Yan, B.; Guo, Y.; Zhou, J.; Tao, J.; et al. Inhibition of xCT suppresses the efficacy of anti-PD-1/L1 melanoma treatment through exosomal PD-L1-induced macrophage M2 polarization. Mol. Ther. 2021, 29, 2321–2334. [Google Scholar] [PubMed]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar]
- Delyon, J.; Lebbe, C.; Dumaz, N. Targeted therapies in melanoma beyond BRAF: Targeting NRAS-mutated and KIT-mutated melanoma. Curr. Opin. Oncol. 2020, 32, 79–84. [Google Scholar] [PubMed]
- Grimaldi, A.M.; Simeone, E.; Festino, L.; Vanella, V.; Strudel, M.; Ascierto, P.A. MEK Inhibitors in the Treatment of Metastatic Melanoma and Solid Tumors. Am. J. Clin. Dermatol. 2017, 18, 745–754. [Google Scholar]
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar]
- Herzberg, B.; Fisher, D.E. Metastatic melanoma and immunotherapy. Clin. Immunol. 2016, 172, 105–110. [Google Scholar]
- Ziogas, D.C.; Konstantinou, F.; Bouros, S.; Theochari, M.; Gogas, H. Combining BRAF/MEK Inhibitors with Immunotherapy in the Treatment of Metastatic Melanoma. Am. J. Clin. Dermatol. 2021, 22, 301–314. [Google Scholar]
- Buchbinder, E.I.; Giobbie-Hurder, A.; Ott, P.A. A phase I/II study of MCS110 with BRAF/MEK inhibition in patients with melanoma after progression on BRAF/MEK inhibition. Investig. New Drugs 2023, 41, 365–370. [Google Scholar]
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114–122. [Google Scholar] [PubMed]
- Robles-Espinoza, C.D.; Roberts, N.D.; Chen, S.; Leacy, F.P.; Alexandrov, L.B.; Pornputtapong, N.; Halaban, R.; Krauthammer, M.; Cui, R.; Timothy Bishop, D.; et al. Germline MC1R status influences somatic mutation burden in melanoma. Nat. Commun. 2016, 7, 12064. [Google Scholar]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar]
- Saldanha, G.; Potter, L.; Daforno, P.; Pringle, J.H. Cutaneous melanoma subtypes show different BRAF and NRAS mutation frequencies. Clin. Cancer Res. 2006, 12, 4499–4505. [Google Scholar] [PubMed]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar]
- Monsel, G.; Ortonne, N.; Bagot, M.; Bensussan, A.; Dumaz, N. c-Kit mutants require hypoxia-inducible factor 1alpha to transform melanocytes. Oncogene 2010, 29, 227–236. [Google Scholar] [PubMed]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., 3rd; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar]
- Berwick, M.; Orlow, I.; Hummer, A.J.; Armstrong, B.K.; Kricker, A.; Marrett, L.D.; Millikan, R.C.; Gruber, S.B.; Anton-Culver, H.; Zanetti, R.; et al. The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: An international population-based study. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1520–1525. [Google Scholar]
- Conde-Perez, A.; Larue, L. PTEN and melanomagenesis. Future Oncol. 2012, 8, 1109–1120. [Google Scholar]
- Steelman, L.S.; Stadelman, K.M.; Chappell, W.H.; Horn, S.; Basecke, J.; Cervello, M.; Nicoletti, F.; Libra, M.; Stivala, F.; Martelli, A.M.; et al. Akt as a therapeutic target in cancer. Expert Opin. Ther. Targets 2008, 12, 1139–1165. [Google Scholar] [PubMed]
- Karst, A.M.; Dai, D.L.; Cheng, J.Q.; Li, G. Role of p53 up-regulated modulator of apoptosis and phosphorylated Akt in melanoma cell growth, apoptosis, and patient survival. Cancer Res. 2006, 66, 9221–9226. [Google Scholar] [PubMed]
- Mehnert, J.M.; Kluger, H.M. Driver mutations in melanoma: Lessons learned from bench-to-bedside studies. Curr. Oncol. Rep. 2012, 14, 449–457. [Google Scholar] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar]
- Slominski, A.; Tobin, D.J.; Shibahara, S.; Wortsman, J. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol. Rev. 2004, 84, 1155–1228. [Google Scholar]
- Slominski, A.T.; Slominski, R.M.; Raman, C.; Chen, J.Y.; Athar, M.; Elmets, C. Neuroendocrine signaling in the skin with a special focus on the epidermal neuropeptides. Am. J. Physiol.-Cell Physiol. 2022, 323, C1757–C1776. [Google Scholar]
- Slominski, R.M.; Sarna, T.; Plonka, P.M.; Raman, C.; Brozyna, A.A.; Slominski, A.T. Melanoma, Melanin, and Melanogenesis: The Yin and Yang Relationship. Front. Oncol. 2022, 12, 842496. [Google Scholar]
- Omholt, K.; Platz, A.; Kanter, L.; Ringborg, U.; Hansson, J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin. Cancer Res. 2003, 9, 6483–6488. [Google Scholar]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar]
- Abecunas, C.; Whitehead, C.E.; Ziemke, E.K.; Baumann, D.G.; Frankowski-McGregor, C.L.; Sebolt-Leopold, J.S.; Fallahi-Sichani, M. Loss of NF1 in Melanoma Confers Sensitivity to SYK Kinase Inhibition. Cancer Res. 2023, 83, 316–331. [Google Scholar]
- Ward, K.A.; Lazovich, D.; Hordinsky, M.K. Germline melanoma susceptibility and prognostic genes: A review of the literature. J. Am. Acad. Dermatol. 2012, 67, 1055–1067. [Google Scholar]
- Kreuger, I.Z.M.; Slieker, R.C.; van Groningen, T.; van Doorn, R. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Investig. Dermatol. 2023, 143, 18–25.e1. [Google Scholar]
- Garutti, M.; Targato, G.; Buriolla, S.; Palmero, L.; Minisini, A.M.; Puglisi, F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells 2021, 10, 1334. [Google Scholar] [CrossRef]
- Serman, N.; Vranic, S.; Glibo, M.; Serman, L.; Bukvic Mokos, Z. Genetic risk factors in melanoma etiopathogenesis and the role of genetic counseling: A concise review. Bosn. J. Basic Med. Sci. 2022, 22, 673–682. [Google Scholar]
- Rice, C.; Shastrula, P.K.; Kossenkov, A.V.; Hills, R.; Baird, D.M.; Showe, L.C.; Doukov, T.; Janicki, S.; Skordalakes, E. Structural and functional analysis of the human POT1-TPP1 telomeric complex. Nat. Commun. 2017, 8, 14928. [Google Scholar]
- Teixido, C.; Castillo, P.; Martinez-Vila, C.; Arance, A.; Alos, L. Molecular Markers and Targets in Melanoma. Cells 2021, 10, 2320. [Google Scholar] [CrossRef]
- Ribeiro Moura Brasil Arnaut, J.; Dos Santos Guimaraes, I.; Evangelista Dos Santos, A.C.; de Moraes Lino da Silva, F.; Machado, J.R.; de Melo, A.C. Molecular landscape of Hereditary Melanoma. Crit. Rev. Oncol. Hematol. 2021, 164, 103425. [Google Scholar]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar]
- Guida, S.; Guida, G.; Goding, C.R. MC1R Functions, Expression, and Implications for Targeted Therapy. J. Investig. Dermatol. 2022, 142, 293–302.e1. [Google Scholar]
- Marzuka-Alcala, A.; Gabree, M.J.; Tsao, H. Melanoma susceptibility genes and risk assessment. Methods Mol. Biol. 2014, 1102, 381–393. [Google Scholar]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dreno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [PubMed]
- Bobos, M. Histopathologic classification and prognostic factors of melanoma: A 2021 update. Ital. J. Dermatol. Venereol. 2021, 156, 300–321. [Google Scholar]
- Elder, D.E. Skin cancer. Melanoma and other specific nonmelanoma skin cancers. Cancer 1995, 75 (Suppl. S1), 245–256. [Google Scholar]
- Burbidge, T.E.; Bastian, B.C.; Guo, D.; Li, H.; Morris, D.G.; Monzon, J.G.; Leung, G.; Yang, H.; Cheng, T. Association of Indoor Tanning Exposure with Age at Melanoma Diagnosis and BRAF V600E Mutations. J. Natl. Cancer Inst. 2019, 111, 1228–1231. [Google Scholar] [PubMed]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Abeni, D.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur. J. Cancer 2005, 41, 28–44. [Google Scholar]
- Viros, A.; Fridlyand, J.; Bauer, J.; Lasithiotakis, K.; Garbe, C.; Pinkel, D.; Bastian, B.C. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med. 2008, 5, e120. [Google Scholar]
- Broekaert, S.M.; Roy, R.; Okamoto, I.; van den Oord, J.; Bauer, J.; Garbe, C.; Barnhill, R.L.; Busam, K.J.; Cochran, A.J.; Cook, M.G.; et al. Genetic and morphologic features for melanoma classification. Pigment Cell Melanoma Res. 2010, 23, 763–770. [Google Scholar]
- Lin, M.J.; Mar, V.; McLean, C.; Wolfe, R.; Kelly, J.W. Diagnostic accuracy of malignant melanoma according to subtype. Australas. J. Dermatol. 2014, 55, 35–42. [Google Scholar]
- Grob, J.J.; Bonerandi, J.J. The ‘ugly duckling’ sign: Identification of the common characteristics of nevi in an individual as a basis for melanoma screening. Arch. Dermatol. 1998, 134, 103–104. [Google Scholar]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [PubMed]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar]
- Hussein, M.R.; Haemel, A.K.; Wood, G.S. Apoptosis and melanoma: Molecular mechanisms. J. Pathol. 2003, 199, 275–288. [Google Scholar]
- Shang, Y.Y.; Yao, M.; Zhou, Z.W.; Jian, C.; Li, X.; Hu, R.Y.; Yu, Y.Y.; Qiong, G.; Biao, Y.; Liu, Y.X.; et al. Alisertib promotes apoptosis and autophagy in melanoma through p38 MAPK-mediated aurora a signaling. Oncotarget 2017, 8, 107076–107088. [Google Scholar]
- Wang, X.; Li, X.; Xia, Y.; Wang, D.; Li, X.; Liu, L.; Zheng, Q.; Li, D.; Jiang, Q. Hernandezine Regulates Proliferation and Autophagy-Induced Apoptosis in Melanoma Cells. J. Nat. Prod. 2022, 85, 1351–1362. [Google Scholar]
- Wang, D.; Li, Q.; Wang, Y.; Jiang, Y. (S)-(-)-N-[2-(3-Hydroxy-2-oxo-2,3-dihydro-1H-indol-3-yl)-ethyl]-acetamide inhibits melanoma cell growth through inducing apoptosis and autophagy. Cutan. Ocul. Toxicol. 2021, 40, 293–299. [Google Scholar]
- Guo, L.; Dong, Z.; Zhang, X.; Yang, Y.; Hu, X.; Ji, Y.; Li, C.; Wan, S.; Xu, J.; Liu, C.; et al. Morusinol extracted from Morus alba induces cell cycle arrest and apoptosis via inhibition of DNA damage response in melanoma by CHK1 degradation through the ubiquitin-proteasome pathway. Phytomedicine 2023, 114, 154765. [Google Scholar]
- Li, Y.; Hou, H.; Liu, Z.; Tang, W.; Wang, J.; Lu, L.; Fu, J.; Gao, D.; Zhao, F.; Gao, X.; et al. CD44 targeting nanodrug based on chondroitin sulfate for melanoma therapy by inducing mitochondrial apoptosis pathways. Carbohydr. Polym. 2023, 320, 121255. [Google Scholar]
- Peng, X.; Hao, J.; Tao, W.; Guo, D.; Liang, T.; Hu, X.; Xu, H.; Fan, X.; Chen, C. Amyloid-like aggregates of short self-assembly peptide selectively induce melanoma cell apoptosis. J. Colloid Interface Sci. 2023, 640, 498–509. [Google Scholar] [PubMed]
- Xiao, J.R.; Do, C.W.; To, C.H. Potential therapeutic effects of baicalein, baicalin, and wogonin in ocular disorders. J. Ocul. Pharmacol. Ther. 2014, 30, 605–614. [Google Scholar]
- Huang, L.; Peng, B.; Nayak, Y.; Wang, C.; Si, F.; Liu, X.; Dou, J.; Xu, H.; Peng, G. Baicalein and Baicalin Promote Melanoma Apoptosis and Senescence via Metabolic Inhibition. Front. Cell Dev. Biol. 2020, 8, 836. [Google Scholar]
- Golpour, M.; Alimohammadi, M.; Sohbatzadeh, F.; Fattahi, S.; Bekeschus, S.; Rafiei, A. Cold atmospheric pressure plasma treatment combined with starvation increases autophagy and apoptosis in melanoma in vitro and in vivo. Exp. Dermatol. 2022, 31, 1016–1028. [Google Scholar]
- Tang, J.F.; Li, G.L.; Zhang, T.; Du, Y.M.; Huang, S.Y.; Ran, J.H.; Li, J.; Chen, D.L. Homoharringtonine inhibits melanoma cells proliferation in vitro and vivo by inducing DNA damage, apoptosis, and G2/M cell cycle arrest. Arch. Biochem. Biophys. 2021, 700, 108774. [Google Scholar]
- Yao, X.; Jiang, W.; Yu, D.; Yan, Z. Luteolin inhibits proliferation and induces apoptosis of human melanoma cells in vivo and in vitro by suppressing MMP-2 and MMP-9 through the PI3K/AKT pathway. Food Funct. 2019, 10, 703–712. [Google Scholar] [PubMed]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F.; et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [PubMed]
- Broussard, L.; Howland, A.; Ryu, S.; Song, K.; Norris, D.; Armstrong, C.A.; Song, P.I. Melanoma Cell Death Mechanisms. Chonnam Med. J. 2018, 54, 135–142. [Google Scholar]
- Buchheit, C.L.; Weigel, K.J.; Schafer, Z.T. Cancer cell survival during detachment from the ECM: Multiple barriers to tumour progression. Nat. Rev. Cancer 2014, 14, 632–641. [Google Scholar]
- Neuendorf, H.M.; Simmons, J.L.; Boyle, G.M. Therapeutic targeting of anoikis resistance in cutaneous melanoma metastasis. Front. Cell Dev. Biol. 2023, 11, 1183328. [Google Scholar]
- He, Y.W.; Fan, Q.P.; Hua, A.L.; Liu, Q. Identification of hub anoikis-associated genes and risk signature in cutaneous melanoma. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 5662–5676. [Google Scholar] [PubMed]
- Tseng, T.; Uen, W.; Tseng, J.; Lee, S. Enhanced chemosensitization of anoikis-resistant melanoma cells through syndecan-2 upregulation upon anchorage independency. Oncotarget 2017, 8, 61528–61537. [Google Scholar]
- Hasnat, M.A.; Pervin, M.; Lim, J.H.; Lim, B.O. Apigenin Attenuates Melanoma Cell Migration by Inducing Anoikis through Integrin and Focal Adhesion Kinase Inhibition. Molecules 2015, 20, 21157–21166. [Google Scholar] [CrossRef] [PubMed]
- Toricelli, M.; Melo, F.H.M.; Hunger, A.; Zanatta, D.; Strauss, B.E.; Jasiulionis, M.G. Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma. Cancers 2017, 9, 37. [Google Scholar] [CrossRef]
- Adeshakin, F.O.; Adeshakin, A.O.; Liu, Z.; Lu, X.; Cheng, J.; Zhang, P.; Yan, D.; Zhang, G.; Wan, X. Upregulation of V-ATPase by STAT3 Activation Promotes Anoikis Resistance and Tumor Metastasis. J. Cancer 2021, 12, 4819–4829. [Google Scholar] [PubMed]
- Weems, A.D.; Welf, E.S.; Driscoll, M.K.; Zhou, F.Y.; Mazloom-Farsibaf, H.; Chang, B.J.; Murali, V.S.; Gihana, G.M.; Weiss, B.G.; Chi, J.; et al. Blebs promote cell survival by assembling oncogenic signalling hubs. Nature 2023, 615, 517–525. [Google Scholar]
- Zhao, B.; Xie, J.; Zhou, X.; Zhang, L.; Cheng, X.; Liang, C. YAP activation in melanoma contributes to anoikis resistance and metastasis. Exp. Biol. Med. 2021, 246, 888–896. [Google Scholar]
- Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Andreucci, E.; Nediani, C.; Laurenzana, A.; Margheri, F.; Fibbi, G.; Calorini, L. Anoikis Resistance as a Further Trait of Acidic-Adapted Melanoma Cells. J. Oncol. 2019, 2019, 8340926. [Google Scholar]
- Pierce, C.J.; Simmons, J.L.; Broit, N.; Karunarathne, D.; Ng, M.F.; Boyle, G.M. BRN2 expression increases anoikis resistance in melanoma. Oncogenesis 2020, 9, 64. [Google Scholar]
- Zhu, G.; Xu, P.; Guo, S.; Yi, X.; Wang, H.; Yang, Y.; Liu, L.; Shi, Q.; Gao, T.; Li, C. Metastatic Melanoma Cells Rely on Sestrin2 to Acquire Anoikis Resistance via Detoxifying Intracellular ROS. J. Investig. Dermatol. 2020, 140, 666–675.e2. [Google Scholar]
- Boisvert-Adamo, K.; Longmate, W.; Abel, E.V.; Aplin, A.E. Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 2009, 7, 549–556. [Google Scholar] [PubMed]
- Scutti, J.A.; Matsuo, A.L.; Pereira, F.V.; Massaoka, M.H.; Figueiredo, C.R.; Moreira, D.F.; Belizario, J.E.; Travassos, L.R. Role of SOCS-1 Gene on Melanoma Cell Growth and Tumor Development. Transl. Oncol. 2011, 4, 101–109. [Google Scholar]
- Marciniak, S.J.; Chambers, J.E.; Ron, D. Pharmacological targeting of endoplasmic reticulum stress in disease. Nat. Rev. Drug Discov. 2022, 21, 115–140. [Google Scholar] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar]
- Hill, D.S.; Lovat, P.E.; Haass, N.K. Induction of endoplasmic reticulum stress as a strategy for melanoma therapy: Is there a future? Melanoma Manag. 2014, 1, 127–137. [Google Scholar] [PubMed]
- Hu, X.; Pan, G.; Luo, J.; Gao, X.; Mu, Y.; Wang, Z.; Hu, X.; Li, C.; Abbas, M.N.; Zhang, K.; et al. Kuwanon H Inhibits Melanoma Growth through Cytotoxic Endoplasmic Reticulum Stress and Impaired Autophagy Flux. J. Agric. Food Chem. 2023, 71, 13768–13782. [Google Scholar] [PubMed]
- Ausina, P.; Branco, J.R.; Demaria, T.M.; Esteves, A.M.; Leandro, J.G.B.; Ochioni, A.C.; Mendonca, A.P.M.; Palhano, F.L.; Oliveira, M.F.; Abou-Kheir, W.; et al. Acetylsalicylic acid and salicylic acid present anticancer properties against melanoma by promoting nitric oxide-dependent endoplasmic reticulum stress and apoptosis. Sci. Rep. 2020, 10, 19617. [Google Scholar]
- Heo, J.R.; Kim, S.M.; Hwang, K.A.; Kang, J.H.; Choi, K.C. Resveratrol induced reactive oxygen species and endoplasmic reticulum stress-mediated apoptosis, and cell cycle arrest in the A375SM malignant melanoma cell line. Int. J. Mol. Med. 2018, 42, 1427–1435. [Google Scholar]
- Liu, Y.; Kang, X.; Niu, G.; He, S.; Zhang, T.; Bai, Y.; Li, Y.; Hao, H.; Chen, C.; Shou, Z.; et al. Shikonin induces apoptosis and prosurvival autophagy in human melanoma A375 cells via ROS-mediated ER stress and p38 pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 626–635. [Google Scholar]
- Grzywa, T.M.; Klicka, K.; Paskal, W.; Dudkiewicz, J.; Wejman, J.; Pyzlak, M.; Wlodarski, P.K. miR-410-3p is induced by vemurafenib via ER stress and contributes to resistance to BRAF inhibitor in melanoma. PLoS ONE 2020, 15, e0234707. [Google Scholar]
- Wang, S.; Zhu, W. Tumour suppressor candidate 3 inhibits biological function and increases endoplasmic reticulum stress of melanoma cells WM451 by regulating AKT/GSK3-beta/beta-catenin pathway. Cell Biochem. Funct. 2020, 38, 604–612. [Google Scholar] [PubMed]
- Chiu, C.S.; Tsai, C.H.; Hsieh, M.S.; Tsai, S.C.; Jan, Y.J.; Lin, W.Y.; Lai, D.W.; Wu, S.M.; Hsing, H.Y.; Arbiser, J.L.; et al. Exploiting Honokiol-induced ER stress CHOP activation inhibits the growth and metastasis of melanoma by suppressing the MITF and beta-catenin pathways. Cancer Lett. 2019, 442, 113–125. [Google Scholar] [PubMed]
- Zheng, Y.; Wang, K.; Wu, Y.; Chen, Y.; Chen, X.; Hu, C.W.; Hu, F. Pinocembrin induces ER stress mediated apoptosis and suppresses autophagy in melanoma cells. Cancer Lett. 2018, 431, 31–42. [Google Scholar]
- Kim, J.K.; Kang, K.A.; Ryu, Y.S.; Piao, M.J.; Han, X.; Oh, M.C.; Boo, S.J.; Jeong, S.U.; Jeong, Y.J.; Chae, S.; et al. Induction of Endoplasmic Reticulum Stress via Reactive Oxygen Species Mediated by Luteolin in Melanoma Cells. Anticancer Res. 2016, 36, 2281–2289. [Google Scholar]
- Niessner, H.; Sinnberg, T.; Kosnopfel, C.; Smalley, K.S.M.; Beck, D.; Praetorius, C.; Mai, M.; Beissert, S.; Kulms, D.; Schaller, M.; et al. BRAF Inhibitors Amplify the Proapoptotic Activity of MEK Inhibitors by Inducing ER Stress in NRAS-Mutant Melanoma. Clin. Cancer Res. 2017, 23, 6203–6214. [Google Scholar] [PubMed]
- Yang, T.Y.; Wu, Y.J.; Chang, C.I.; Chiu, C.C.; Wu, M.L. The Effect of Bornyl cis-4-Hydroxycinnamate on Melanoma Cell Apoptosis is Associated with Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2018, 19, 1370. [Google Scholar] [CrossRef]
- Riaz Ahmed, K.B.; Kanduluru, A.K.; Feng, L.; Fuchs, P.L.; Huang, P. Antitumor agent 25-epi Ritterostatin GN1N induces endoplasmic reticulum stress and autophagy mediated cell death in melanoma cells. Int. J. Oncol. 2017, 50, 1482–1490. [Google Scholar]
- Montagnani Marelli, M.; Marzagalli, M.; Moretti, R.M.; Beretta, G.; Casati, L.; Comitato, R.; Gravina, G.L.; Festuccia, C.; Limonta, P. Vitamin E delta-tocotrienol triggers endoplasmic reticulum stress-mediated apoptosis in human melanoma cells. Sci. Rep. 2016, 6, 30502. [Google Scholar]
- Onwuha-Ekpete, L.; Tack, L.; Knapinska, A.; Smith, L.; Kaushik, G.; Lavoi, T.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. Novel pyrrolidine diketopiperazines selectively inhibit melanoma cells via induction of late-onset apoptosis. J. Med. Chem. 2014, 57, 1599–1608. [Google Scholar]
- Palrasu, M.; Knapinska, A.M.; Diez, J.; Smith, L.; LaVoi, T.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. A Novel Probe for Spliceosomal Proteins that Induces Autophagy and Death of Melanoma Cells Reveals New Targets for Melanoma Drug Discovery. Cell. Physiol. Biochem. 2019, 53, 656–686. [Google Scholar]
- Velayutham, S.; Seerattan, R.; Sultan, M.; Seal, T.; Danthurthy, S.; Chinnappan, B.; Landi, J.; Pearl, K.; Singh, A.; Smalley, K.S.M.; et al. Novel Anti-Melanoma Compounds Are Efficacious in A375 Cell Line Xenograft Melanoma Model in Nude Mice. Biomolecules 2023, 13, 1276. [Google Scholar] [CrossRef] [PubMed]
- Pangilinan, C.; Klionsky, D.J.; Liang, C. Emerging dimensions of autophagy in melanoma. Autophagy 2024, 20, 1700–1711. [Google Scholar] [PubMed]
- Liu, H.; He, Z.; von Rutte, T.; Yousefi, S.; Hunger, R.E.; Simon, H.U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci. Transl. Med. 2013, 5, 202ra123. [Google Scholar]
- Wang, W.J.; Wang, Y.; Chen, H.Z.; Xing, Y.Z.; Li, F.W.; Zhang, Q.; Zhou, B.; Zhang, H.K.; Zhang, J.; Bian, X.L.; et al. Orphan nuclear receptor TR3 acts in autophagic cell death via mitochondrial signaling pathway. Nat. Chem. Biol. 2014, 10, 133–140. [Google Scholar]
- Luan, W.; Qian, Y.; Ni, X.; Chanda, T.K.; Xia, Y.; Wang, J.; Yan, Y.; Xu, B. Polygonatum odoratum lectin promotes BECN1 expression and induces autophagy in malignant melanoma by regulation of miR1290. OncoTargets Ther. 2017, 10, 4569–4577. [Google Scholar]
- Xiao, Y.; Diao, Q.; Liang, Y.; Peng, Y.; Zeng, K. MicroRNA-24-1-5p promotes malignant melanoma cell autophagy and apoptosis via regulating ubiquitin D. Mol. Med. Rep. 2017, 16, 8448–8454. [Google Scholar]
- Wu, J.C.; Tsai, H.E.; Liu, G.S.; Wu, C.S.; Tai, M.H. Autophagic cell death participates in POMC-induced melanoma suppression. Cell Death Discov. 2018, 4, 11. [Google Scholar]
- Liu, Y.; Wang, X.; Zhu, W.; Sui, Z.; Wei, X.; Zhang, Y.; Qi, J.; Xing, Y.; Wang, W. TRPML1-induced autophagy inhibition triggers mitochondrial mediated apoptosis. Cancer Lett. 2022, 541, 215752. [Google Scholar]
- Rok, J.; Rzepka, Z.; Kowalska, J.; Banach, K.; Beberok, A.; Wrzesniok, D. The Anticancer Potential of Doxycycline and Minocycline—A Comparative Study on Amelanotic Melanoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 831. [Google Scholar] [CrossRef]
- Xia, Y.; Xu, F.; Xiong, M.; Yang, H.; Lin, W.; Xie, Y.; Xi, H.; Xue, Q.; Ye, T.; Yu, L. Repurposing of antipsychotic trifluoperazine for treating brain metastasis, lung metastasis and bone metastasis of melanoma by disrupting autophagy flux. Pharmacol. Res. 2021, 163, 105295. [Google Scholar]
- Liang, Q.P.; Xu, T.Q.; Liu, B.L.; Lei, X.P.; Hambrook, J.R.; Zhang, D.M.; Zhou, G.X. Sasanquasaponin IotaIotaIota from Schima crenata Korth induces autophagy through Akt/mTOR/p70S6K pathway and promotes apoptosis in human melanoma A375 cells. Phytomedicine 2019, 58, 152769. [Google Scholar] [PubMed]
- Yin, S.; Xia, C.; Wang, Y.; Wan, D.; Rao, J.; Tang, X.; Wei, J.; Wang, X.; Li, M.; Zhang, Z.; et al. Dual receptor recognizing liposomes containing paclitaxel and hydroxychloroquine for primary and metastatic melanoma treatment via autophagy-dependent and independent pathways. J. Control. Release 2018, 288, 148–160. [Google Scholar] [PubMed]
- Song, B.; Wu, P.; Liang, Z.; Wang, J.; Zheng, Y.; Wang, Y.; Chi, H.; Li, Z.; Song, Y.; Yin, X.; et al. A Novel Necroptosis-Related Gene Signature in Skin Cutaneous Melanoma Prognosis and Tumor Microenvironment. Front. Genet. 2022, 13, 917007. [Google Scholar]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [PubMed]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015, 6, e1884. [Google Scholar]
- Basit, F.; van Oppen, L.M.; Schockel, L.; Bossenbroek, H.M.; van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; Hgm Willems, P.; et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar]
- Kong, Q.; Lv, J.; Yan, S.; Chang, K.J.; Wang, G. A Novel Naphthyridine Derivative, 3u, Induces Necroptosis at Low Concentrations and Apoptosis at High Concentrations in Human Melanoma A375 Cells. Int. J. Mol. Sci. 2018, 19, 2975. [Google Scholar] [CrossRef]
- Yang, L.; Joseph, S.; Sun, T.; Hoffmann, J.; Thevissen, S.; Offermanns, S.; Strilic, B. TAK1 regulates endothelial cell necroptosis and tumor metastasis. Cell Death Differ. 2019, 26, 1987–1997. [Google Scholar]
- Liu, J.; Luo, B.; Zhang, P.; Jiang, K.; Hou, Z.; Cao, X.; Tang, J. Necroptosis-related LncRNAs in skin cutaneous melanoma: Evaluating prognosis, predicting immunity, and guiding therapy. BMC Cancer 2023, 23, 752. [Google Scholar]
- Liu, N.; Li, Y.; Chen, G.; Ge, K. Evodiamine induces reactive oxygen species-dependent apoptosis and necroptosis in human melanoma A-375 cells. Oncol. Lett. 2020, 20, 121. [Google Scholar] [PubMed]
- Zhang, K.; Song, W.; Wei, M.; Sun, Y.; Wang, N.; Ma, L.; Yu, X.; Gao, R.; Wang, R.; Zhang, Y.; et al. A Novel Anticancer Stem Cell Compound Derived from Pleuromutilin Induced Necroptosis of Melanoma Cells. J. Med. Chem. 2021, 64, 15825–15845. [Google Scholar] [PubMed]
- Pawlikowska, M.; Jedrzejewski, T.; Brozyna, A.A.; Wrotek, S. Protein-Bound Polysaccharides from Coriolus Versicolor Induce RIPK1/RIPK3/MLKL-Mediated Necroptosis in ER-Positive Breast Cancer and Amelanotic Melanoma Cells. Cell. Physiol. Biochem. 2020, 54, 591–604. [Google Scholar]
- Ahmad, H.; Crotts, M.S.; Jacobs, J.C.; Baer, R.W.; Cox, J.L. Shikonin Causes Non-apoptotic Cell Death in B16F10 Melanoma. Anti-Cancer Agents Med. Chem. 2023, 23, 1880–1887. [Google Scholar]
- Yang, H.; Liu, C.; Zhang, Y.Q.; Ge, L.T.; Chen, J.; Jia, X.Q.; Gu, R.X.; Sun, Y.; Sun, W.D. Ilexgenin A induces B16-F10 melanoma cell G1/S arrest in vitro and reduces tumor growth in vivo. Int. Immunopharmacol. 2015, 24, 423–431. [Google Scholar]
- Zaffaroni, N.; Beretta, G.L. The Therapeutic Potential of Pyroptosis in Melanoma. Int. J. Mol. Sci. 2023, 24, 1285. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, J.Y.; Liu, X.S.; Chen, H.Z.; Ai, Y.L.; Cheng, K.; Sun, R.Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar]
- Zhao, Z.; Huang, Y.; Wang, J.; Lin, H.; Cao, F.; Li, S.; Li, Y.; Li, Z.; Liu, X. A self-assembling CXCR4-targeted pyroptosis nanotoxin for melanoma therapy. Biomater. Sci. 2023, 11, 2200–2210. [Google Scholar] [PubMed]
- Vernon, M.; Wilski, N.A.; Kotas, D.; Cai, W.; Pomante, D.; Tiago, M.; Alnemri, E.S.; Aplin, A.E. Raptinal Induces Gasdermin E-Dependent Pyroptosis in Naive and Therapy-Resistant Melanoma. Mol. Cancer Res. 2022, 20, 1811–1821. [Google Scholar]
- Ahmed, F.; Tseng, H.Y.; Ahn, A.; Gunatilake, D.; Alavi, S.; Eccles, M.; Rizos, H.; Gallagher, S.J.; Tiffen, J.C.; Hersey, P.; et al. Repurposing Melanoma Chemotherapy to Activate Inflammasomes in the Treatment of BRAF/MAPK Inhibitor Resistant Melanoma. J. Investig. Dermatol. 2022, 142, 1444–1455.e10. [Google Scholar]
- Zeng, B.; Chen, Y.; Chen, H.; Zhao, Q.; Sun, Z.; Liu, D.; Li, X.; Zhang, Y.; Wang, J.; Xing, H.R. Exosomal miR-211-5p regulates glucose metabolism, pyroptosis, and immune microenvironment of melanoma through GNA15. Pharmacol. Res. 2023, 188, 106660. [Google Scholar] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar]
- Wang, Q.; Wang, Y.; Ding, J.; Wang, C.; Zhou, X.; Gao, W.; Huang, H.; Shao, F.; Liu, Z. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 2020, 579, 421–426. [Google Scholar]
- Talty, R.; Bosenberg, M. The role of ferroptosis in melanoma. Pigment Cell Melanoma Res. 2022, 35, 18–25. [Google Scholar]
- Meng, Y.; Sun, H.Y.; He, Y.; Zhou, Q.; Liu, Y.H.; Su, H.; Yin, M.Z.; Zeng, F.R.; Chen, X.; Deng, G.T. BET inhibitors potentiate melanoma ferroptosis and immunotherapy through AKR1C2 inhibition. Mil. Med. Res. 2023, 10, 61. [Google Scholar]
- Guo, W.; Wu, Z.; Chen, J.; Guo, S.; You, W.; Wang, S.; Ma, J.; Wang, H.; Wang, X.; Wang, H.; et al. Nanoparticle delivery of miR-21-3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J. Immunother. Ther. Cancer 2022, 10, e004381. [Google Scholar]
- Zhang, K.; Wu, L.; Zhang, P.; Luo, M.; Du, J.; Gao, T.; O’Connell, D.; Wang, G.; Wang, H.; Yang, Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol. Carcinog. 2018, 57, 1566–1576. [Google Scholar]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [PubMed]
- Yu, Y.; Ren, Y.; Wang, C.; Li, Z.; Niu, F.; Li, Z.; Ye, Q.; Wang, J.; Yan, Y.; Liu, P.; et al. Arginase 2 negatively regulates sorafenib-induced cell death by mediating ferroptosis in melanoma. Acta Biochim. Biophys. Sin. 2022, 54, 1658–1670. [Google Scholar]
- Wang, M.; Cheng, H.; Wu, H.; Liu, C.; Li, S.; Li, B.; Su, J.; Luo, S.; Li, Q. Gambogenic acid antagonizes the expression and effects of long non-coding RNA NEAT1 and triggers autophagy and ferroptosis in melanoma. Biomed. Pharmacother. 2022, 154, 113636. [Google Scholar]
- Wang, M.; Zeng, G.; Xiong, B.; Zhu, X.; Guo, J.; Chen, D.; Zhang, S.; Luo, M.; Guo, L.; Cai, L. ALOX5 promotes autophagy-dependent ferroptosis by activating the AMPK/mTOR pathway in melanoma. Biochem. Pharmacol. 2023, 212, 115554. [Google Scholar]
- Li, G.; Zhou, C.; Wang, L.; Zheng, Y.; Zhou, B.; Li, G.; Ma, Z.; Sun, P.; Deng, Y.; Su, L.; et al. MitoCur-1 induces ferroptosis to reverse vemurafenib resistance in melanoma through inhibition of USP14. Pigment Cell Melanoma Res. 2024, 37, 316–328. [Google Scholar]
- Chang, M.T.; Tsai, L.C.; Nakagawa-Goto, K.; Lee, K.H.; Shyur, L.F. Phyto-sesquiterpene lactones DET and DETD-35 induce ferroptosis in vemurafenib sensitive and resistant melanoma via GPX4 inhibition and metabolic reprogramming. Pharmacol. Res. 2022, 178, 106148. [Google Scholar] [PubMed]
- Li, M.; Zheng, J.; Wu, T.; He, Y.; Guo, J.; Xu, J.; Gao, C.; Qu, S.; Zhang, Q.; Zhao, J.; et al. Activation of TRPV4 Induces Exocytosis and Ferroptosis in Human Melanoma Cells. Int. J. Mol. Sci. 2022, 23, 4146. [Google Scholar] [CrossRef]
- Yang, R.; Deng, F.; Yang, Y.; Tian, Q.; Huangfu, S.; Yang, L.; Hou, J.; Yang, G.; Pang, W.; Lu, J.; et al. Blue light promotes vitamin C-mediated ferroptosis of melanoma through specifically upregulating transporter SVCT2 and generating Fe(2). Biomaterials 2023, 299, 122186. [Google Scholar]
- Khorsandi, K.; Kianmehr, Z.; Hosseinmardi, Z.; Hosseinzadeh, R. Anti-cancer effect of gallic acid in presence of low level laser irradiation: ROS production and induction of apoptosis and ferroptosis. Cancer Cell Int. 2020, 20, 18. [Google Scholar] [PubMed]
- Vadarevu, H.; Juneja, R.; Lyles, Z.; Vivero-Escoto, J.L. Light-Activated Protoporphyrin IX-Based Polysilsesquioxane Nanoparticles Induce Ferroptosis in Melanoma Cells. Nanomaterials 2021, 11, 2324. [Google Scholar] [CrossRef]
- Feng, S.; Zhou, Y.; Huang, H.; Lin, Y.; Zeng, Y.; Han, S.; Huang, K.; Liu, Q.; Zhu, W.; Yuan, Z.; et al. Nobiletin Induces Ferroptosis in Human Skin Melanoma Cells Through the GSK3beta-Mediated Keap1/Nrf2/HO-1 Signalling Pathway. Front. Genet. 2022, 13, 865073. [Google Scholar]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar]
- Chen, L.; Min, J.; Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar]
- Liu, J.Y.; Liu, L.P.; Li, Z.; Luo, Y.W.; Liang, F. The role of cuproptosis-related gene in the classification and prognosis of melanoma. Front. Immunol. 2022, 13, 986214. [Google Scholar]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAF(V600E)-Driven Melanomagenesis and Counters Resistance to BRAF(V600E) and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [PubMed]
- Lu, X.; Chen, X.; Lin, C.; Yi, Y.; Zhao, S.; Zhu, B.; Deng, W.; Wang, X.; Xie, Z.; Rao, S.; et al. Elesclomol Loaded Copper Oxide Nanoplatform Triggers Cuproptosis to Enhance Antitumor Immunotherapy. Adv. Sci. 2024, 11, e2309984. [Google Scholar]
- Sun, Q.; Hong, Z.; Zhang, C.; Wang, L.; Han, Z.; Ma, D. Immune checkpoint therapy for solid tumours: Clinical dilemmas and future trends. Signal Transduct. Target. Ther. 2023, 8, 320. [Google Scholar] [PubMed]
- Ernst, M.; Giubellino, A. The Current State of Treatment and Future Directions in Cutaneous Malignant Melanoma. Biomedicines 2022, 10, 822. [Google Scholar] [CrossRef]
- Steininger, J.; Gellrich, F.F.; Schulz, A.; Westphal, D.; Beissert, S.; Meier, F. Systemic Therapy of Metastatic Melanoma: On the Road to Cure. Cancers 2021, 13, 1430. [Google Scholar] [CrossRef]
- McCulloch, J.A.; Davar, D.; Rodrigues, R.R.; Badger, J.H.; Fang, J.R.; Cole, A.M.; Balaji, A.K.; Vetizou, M.; Prescott, S.M.; Fernandes, M.R.; et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med. 2022, 28, 545–556. [Google Scholar]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; Dick-Necula, D.; Raskin, S.; Bloch, N.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar]
- Heinzerling, L.; Eigentler, T.K.; Fluck, M.; Hassel, J.C.; Heller-Schenck, D.; Leipe, J.; Pauschinger, M.; Vogel, A.; Zimmer, L.; Gutzmer, R. Tolerability of BRAF/MEK inhibitor combinations: Adverse event evaluation and management. ESMO Open 2019, 4, e000491. [Google Scholar]
- Broman, K.K.; Dossett, L.A.; Sun, J.; Eroglu, Z.; Zager, J.S. Update on BRAF and MEK inhibition for treatment of melanoma in metastatic, unresectable, and adjuvant settings. Expert Opin. Drug Saf. 2019, 18, 381–392. [Google Scholar] [PubMed]
- Menzies, A.M.; Long, G.V. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar]
- Anonymous. New Medical Devices. P T 2015, 40, 792–806. [Google Scholar]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. elife 2017, 6, e18970. [Google Scholar] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar]
- Postow, M.A.; Carvajal, R.D. Therapeutic implications of KIT in melanoma. Cancer J. 2012, 18, 137–141. [Google Scholar]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar]
- Aleksandrova, K.; Leise, J.; Priesner, C.; Aktas, M.; Apel, M.; Assenmacher, M.; Burger, I.; Richter, A.; Altefrohne, P.; Schubert, C.; et al. Automated manufacturing and characterization of clinical grade autologous CD20 CAR T cells for the treatment of patients with stage III/IV melanoma. Front. Immunol. 2024, 15, 1328368. [Google Scholar]
- Anagnostou, V.; Bruhm, D.C.; Niknafs, N.; White, J.R.; Shao, X.M.; Sidhom, J.W.; Stein, J.; Tsai, H.L.; Wang, H.; Belcaid, Z.; et al. Integrative Tumor and Immune Cell Multi-omic Analyses Predict Response to Immune Checkpoint Blockade in Melanoma. Cell Rep. Med. 2020, 1, 100139. [Google Scholar]
- Pollack, L.A.; Li, J.; Berkowitz, Z.; Weir, H.K.; Wu, X.C.; Ajani, U.A.; Ekwueme, D.U.; Li, C.; Pollack, B.P. Melanoma survival in the United States, 1992 to 2005. J. Am. Acad. Dermatol. 2011, 65 (Suppl. S1), S78.E1–S78.E10. [Google Scholar] [CrossRef] [PubMed]
- Niebling, M.G.; Haydu, L.E.; Lo, S.N.; Rawson, R.V.; Lamboo, L.G.E.; Stollman, J.T.; Karim, R.Z.; Thompson, J.F.; Scolyer, R.A. The prognostic significance of microsatellites in cutaneous melanoma. Mod. Pathol. 2020, 33, 1369–1379. [Google Scholar]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar]
- Najjar, Y.G.; Kirkwood, J.M. Neoadjuvant treatment for melanoma: Current challenges and future perspectives. Melanoma Manag. 2016, 3, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Yan, H.; Han, X.; Weng, L.; Wei, Q.; Sun, X.; Lu, W.; Wei, Q.; Ye, J.; Cai, X.; et al. Ginseng-derived nanoparticles alter macrophage polarization to inhibit melanoma growth. J. ImmunoTher. Cancer 2019, 7, 326. [Google Scholar] [CrossRef]
- Kato, M.; Liu, W.; Yi, H.; Asai, N.; Hayakawa, A.; Kozaki, K.; Takahashi, M.; Nakashima, I. The herbal medicine Sho-saiko-to inhibits growth and metastasis of malignant melanoma primarily developed in ret-transgenic mice. J. Investig. Dermatol. 1998, 111, 640–644. [Google Scholar] [PubMed]
- Liu, Y.X.; Bai, J.X.; Li, T.; Fu, X.Q.; Chen, Y.J.; Zhu, P.L.; Chou, J.Y.; Yin, C.L.; Li, J.K.; Wang, Y.P.; et al. MiR-let-7a/f-CCR7 signaling is involved in the anti-metastatic effects of an herbal formula comprising Sophorae Flos and Lonicerae Japonicae Flos in melanoma. Phytomedicine 2019, 64, 153084. [Google Scholar]
- Albuquerque, K.R.S.; Pacheco, N.M.; Del Rosario Loyo Casao, T.; de Melo, F.; Novaes, R.D.; Goncalves, R.V. Applicability of Plant Extracts in Preclinical Studies of Melanoma: A Systematic Review. Mediat. Inflamm. 2018, 2018, 6797924. [Google Scholar]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef]
- Davids, M.S.; Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 2012, 30, 3127–3135. [Google Scholar] [CrossRef]
- Cerezo, M.; Tichet, M.; Abbe, P.; Ohanna, M.; Lehraiki, A.; Rouaud, F.; Allegra, M.; Giacchero, D.; Bahadoran, P.; Bertolotto, C.; et al. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol. Cancer Ther. 2013, 12, 1605–1615. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Jiang, C.C.; Kiejda, K.A.; Gillespie, S.; Zhang, X.D.; Hersey, P. Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin. Cancer Res. 2007, 13, 4934–4942. [Google Scholar]
- Al Subeh, Z.Y.; Poschel, D.B.; Redd, P.S.; Klement, J.D.; Merting, A.D.; Yang, D.; Mehta, M.; Shi, H.; Colson, Y.L.; Oberlies, N.H.; et al. Lipid Nanoparticle Delivery of Fas Plasmid Restores Fas Expression to Suppress Melanoma Growth In Vivo. ACS Nano 2022, 16, 12695–12710. [Google Scholar]
- Slominski, R.M.; Chen, J.Y.; Raman, C.; Slominski, A.T. Photo-neuro-immuno-endocrinology: How the ultraviolet radiation regulates the body, brain, and immune system. Proc. Natl. Acad. Sci. USA 2024, 121, e2308374121. [Google Scholar]
- Slominski, A.T.; Zmijewski, M.A.; Plonka, P.M.; Szaflarski, J.P.; Paus, R. How UV Light Touches the Brain and Endocrine System Through Skin, and Why. Endocrinology 2018, 159, 1992–2007. [Google Scholar]
- Slominski, R.M.; Kim, T.K.; Janjetovic, Z.; Brozyna, A.A.; Podgorska, E.; Dixon, K.M.; Mason, R.S.; Tuckey, R.C.; Sharma, R.; Crossman, D.K.; et al. Malignant Melanoma: An Overview, New Perspectives, and Vitamin D Signaling. Cancers 2024, 16, 2262. [Google Scholar] [CrossRef] [PubMed]
- Slominski, R.M.; Raman, C.; Chen, J.Y.; Slominski, A.T. How cancer hijacks the body’s homeostasis through the neuroen-docrine system. Trends Neurosci. 2023, 46, 263–275. [Google Scholar] [PubMed]
- Verykiou, S.; Alexander, M.; Edwards, N.; Plummer, R.; Chaudhry, B.; Lovat, P.E.; Hill, D.S. Harnessing autophagy to overcome mitogen-activated protein kinase kinase inhibitor-induced resistance in metastatic melanoma. Br. J. Dermatol. 2019, 180, 346–356. [Google Scholar] [PubMed]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [PubMed]
- Degan, S.; May, B.L.; Jin, Y.J.; Hammoda, M.B.; Sun, H.; Zhang, G.; Wang, Y.; Erdmann, D.; Warren, W.; Zhang, J.Y. Co-Treatment of Chloroquine and Trametinib Inhibits Melanoma Cell Proliferation and Decreases Immune Cell Infiltration. Front. Oncol. 2022, 12, 782877. [Google Scholar]
- Du, J.; Dong, Z.; Tan, L.; Tan, M.; Zhang, F.; Zhang, K.; Pan, G.; Li, C.; Shi, S.; Zhang, Y.; et al. Tubeimoside I Inhibits Cell Proliferation and Induces a Partly Disrupted and Cytoprotective Autophagy Through Rapidly Hyperactivation of MEK1/2-ERK1/2 Cascade via Promoting PTP1B in Melanoma. Front. Cell Dev. Biol. 2020, 8, 607757. [Google Scholar]
- Goodall, M.L.; Wang, T.; Martin, K.R.; Kortus, M.G.; Kauffman, A.L.; Trent, J.M.; Gately, S.; MacKeigan, J.P. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 2014, 10, 1120–1136. [Google Scholar]
- Blank, C.U.; Lucas, M.W.; Scolyer, R.A.; van de Wiel, B.A.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.P.M.; Lijnsvelt, J.M.; Maher, N.G.; et al. Neoadjuvant Nivolumab and Ipilimumab in Resectable Stage III Melanoma. N. Engl. J. Med. 2024, 391, 1696–1708. [Google Scholar]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, P.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. Final, 10-Year Outcomes with Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2025, 392, 11–22. [Google Scholar]
- Meyer, N.; Henkel, L.; Linder, B.; Zielke, S.; Tascher, G.; Trautmann, S.; Geisslinger, G.; Munch, C.; Fulda, S.; Tegeder, I.; et al. Autophagy activation, lipotoxicity and lysosomal membrane permeabilization synergize to promote pimozide- and loperamide-induced glioma cell death. Autophagy 2021, 17, 3424–3443. [Google Scholar]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar]
- Balkwill, F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar]
- Qian, X.; Bi, Q.Y.; Wang, Z.N.; Han, F.; Liu, L.M.; Song, L.B.; Li, C.Y.; Zhang, A.Q.; Ji, X.M. Qingyihuaji Formula promotes apoptosis and autophagy through inhibition of MAPK/ERK and PI3K/Akt/mTOR signaling pathway on pancreatic cancer in vivo and in vitro. J. Ethnopharmacol. 2023, 307, 116198. [Google Scholar]
- Karpel-Massler, G.; Ishida, C.T.; Bianchetti, E.; Shu, C.; Perez-Lorenzo, R.; Horst, B.; Banu, M.; Roth, K.A.; Bruce, J.N.; Canoll, P.; et al. Inhibition of Mitochondrial Matrix Chaperones and Antiapoptotic Bcl-2 Family Proteins Empower Antitumor Therapeutic Responses. Cancer Res. 2017, 77, 3513–3526. [Google Scholar]
- Wang, J.; Zhang, Y.; Cao, J.; Wang, Y.; Anwar, N.; Zhang, Z.; Zhang, D.; Ma, Y.; Xiao, Y.; Xiao, L.; et al. The role of autophagy in bone metabolism and clinical significance. Autophagy 2023, 19, 2409–2427. [Google Scholar]
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Yang, Y.; Wu, L.; Wu, X.; Zhao, Y.; Ren, J. RIPK3-MLKL necroptotic signalling amplifies STING pathway and exacerbates lethal sepsis. Clin. Transl. Med. 2023, 13, e1334. [Google Scholar]
- Carter, B.Z.; Mak, P.Y.; Muftuoglu, M.; Tao, W.; Ke, B.; Pei, J.; Bedoy, A.D.; Ostermann, L.B.; Nishida, Y.; Isgandarova, S.; et al. Epichaperome inhibition targets TP53-mutant AML and AML stem/progenitor cells. Blood 2023, 142, 1056–1070. [Google Scholar]
- Bai, F.L.; Yu, Y.H.; Tian, H.; Ren, G.P.; Wang, H.; Zhou, B.; Han, X.H.; Yu, Q.Z.; Li, D.S. Genetically engineered Newcastle disease virus expressing interleukin-2 and TNF-related apoptosis-inducing ligand for cancer therapy. Cancer Biol. Ther. 2014, 15, 1226–1238. [Google Scholar] [PubMed]
- Mohapatra, A.; Gupta, P.; Ratra, D. Accelerated hydroxychloroquine toxic retinopathy. Doc. Ophthalmol. 2024, 148, 37–45. [Google Scholar]
- Luckemann, L.; Unteroberdorster, M.; Martinez Gomez, E.; Schedlowski, M.; Hadamitzky, M. Behavioral conditioning of anti-proliferative and immunosuppressive properties of the mTOR inhibitor rapamycin. Brain Behav. Immun. 2019, 79, 326–331. [Google Scholar] [PubMed]
- Boshchenko, A.A.; Maslov, L.N.; Mukhomedzyanov, A.V.; Zhuravleva, O.A.; Slidnevskaya, A.S.; Naryzhnaya, N.V.; Zinovieva, A.S.; Ilinykh, P.A. Peptides Are Cardioprotective Drugs of the Future: The Receptor and Signaling Mechanisms of the Cardioprotective Effect of Glucagon-like Peptide-1 Receptor Agonists. Int. J. Mol. Sci. 2024, 25, 4900. [Google Scholar] [CrossRef]
- Messaoud-Nacer, Y.; Culerier, E.; Rose, S.; Maillet, I.; Rouxel, N.; Briault, S.; Ryffel, B.; Quesniaux, V.F.J.; Togbe, D. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. 2022, 13, 269. [Google Scholar] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar]
- Schadendorf, D.; Dummer, R.; Flaherty, K.T.; Robert, C.; Arance, A.; de Groot, J.W.B.; Garbe, C.; Gogas, H.J.; Gutzmer, R.; Krajsova, I.; et al. COLUMBUS 7-year update: A randomized, open-label, phase III trial of encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF V600E/K-mutant melanoma. Eur. J. Cancer 2024, 204, 114073. [Google Scholar]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar]
- Tang, J.Q.; Hou, X.Y.; Yang, C.S.; Li, Y.X.; Xin, Y.; Guo, W.W.; Wei, Z.P.; Liu, Y.Q.; Jiang, G. Recent developments in nanomedicine for melanoma treatment. Int. J. Cancer 2017, 141, 646–653. [Google Scholar]
- Gharpure, K.M.; Wu, S.Y.; Li, C.; Lopez-Berestein, G.; Sood, A.K. Nanotechnology: Future of Oncotherapy. Clin. Cancer Res. 2015, 21, 3121–3130. [Google Scholar]
- Ragelle, H.; Riva, R.; Vandermeulen, G.; Naeye, B.; Pourcelle, V.; Le Duff, C.S.; D’Haese, C.; Nysten, B.; Braeckmans, K.; De Smedt, S.C.; et al. Chitosan nanoparticles for siRNA delivery: Optimizing formulation to increase stability and efficiency. J. Control. Release 2014, 176, 54–63. [Google Scholar]
- Wang, Y.; Xu, Z.; Guo, S.; Zhang, L.; Sharma, A.; Robertson, G.P.; Huang, L. Intravenous delivery of siRNA targeting CD47 effectively inhibits melanoma tumor growth and lung metastasis. Mol. Ther. 2013, 21, 1919–1929. [Google Scholar] [PubMed]
- Alshamsan, A.; Hamdy, S.; Samuel, J.; El-Kadi, A.O.; Lavasanifar, A.; Uludag, H. The induction of tumor apoptosis in B16 melanoma following STAT3 siRNA delivery with a lipid-substituted polyethylenimine. Biomaterials 2010, 31, 1420–1428. [Google Scholar] [PubMed]
- Yang, J.; Pan, Z.; Zhao, F.; Feng, X.; Liu, Q.; Li, Y.; Lyu, J. A nomogram for predicting survival in patients with nodular melanoma: A population-based study. Medicine 2019, 98, e16059. [Google Scholar] [CrossRef] [PubMed]
- Rauwerdink, D.J.W.; van Doorn, R.; van der Hage, J.; Van den Eertwegh, A.J.M.; Haanen, J.; Aarts, M.; Berkmortel, F.; Blank, C.U.; Boers-Sonderen, M.J.; De Groot, J.W.B.; et al. Systemic Therapy in Advanced Nodular Melanoma versus Superficial Spreading Melanoma: A Nation-Wide Study of the Dutch Melanoma Treatment Registry. Cancers 2022, 14, 5694. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Mao, L.; Qi, Z.; Zhang, L.; Guo, J.; Si, L. Immunotherapy in Acral and Mucosal Melanoma: Current Status and Future Directions. Front. Immunol. 2021, 12, 680407. [Google Scholar] [CrossRef]
- Eroglu, Z.; Zaretsky, J.M.; Hu-Lieskovan, S.; Kim, D.W.; Algazi, A.; Johnson, D.B.; Liniker, E.; Ben, K.; Munhoz, R.; Rapisuwon, S.; et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018, 553, 347–350. [Google Scholar]
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF Heterogeneity in Melanoma. Curr. Treat. Options Oncol. 2021, 22, 20. [Google Scholar]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Mutations | Mechanism |
|---|---|
| NARS/BRAF | Activates the RAS/RAF/MAPK/ERK signaling pathway [25,38] |
| KIT | Leads to constitutive activation of c-KIT tyrosine kinase activity and subsequent induction of both MAPK and PI3K/AKT pathways [27,39] |
| NF1 | Negative feedback on RAS, thereby causing hyperactivation of the MAPK and PI3K/mTOR signaling pathways [40] |
| CDKN2A | Impairs two of the most important tumor suppressor pathways, Rb and p53 [41,42] |
| CDK4 | Leads to the amino acid substitution and prevents the binding of p16 to the catalytic subunit in the Rb pathway, triggering constitutive activation of the CDK4 kinase [43] |
| POT1 | Impairs the function of shelterin complex [44] and confers a telomere instability phenotype [45] |
| TERT | Activates the MAPK pathway [46] and increases telomerase activity resulting in chromosomal instability [47] |
| MITF | Regulates multiple biological processes in melanoma cells such as differentiation, proliferation, migration, and senescence by activating several pathways such as the BRAFV600E/ERK1/2 and Wnt/β-catenin pathways [48] |
| MC1R | Impairs the cAMP pathway mediated by ligand–receptor interaction and increases risk for melanoma secondary to intensified UV-mediated DNA damage in the setting of absent photoprotective eumelanin [49,50] |
| PTEN | Activates the PI3K/AKT/PTEN pathway [51] |
| Manner of Death | Regulated Pathways of Cell Death | Corresponding Treatment |
|---|---|---|
| Apoptosis | Inhibition of the p38/MAPK pathway | Alisertib (ALS) [66] |
| Inhibition of the PI3K/AKT and activation of MAPK pathway | (S)-(-)-N-[2-(3-Hydroxy-2-oxo-2,3-dihydro-1H-indol-3-yl)-ethyl]-acetamide (SA) [68] | |
| Activation of the AMPK–mTOR pathway | Hernandezine [67] | |
| Inhibiting DNA damage response through CHK1 degradation via the ubiquitin–proteasome pathway | Morusinol [69] | |
| Upregulation of Bax and downregulation of Bcl-2 | CCEA (composed of chondroitin, antiangiogenic peptide, and cisplatin) [70] | |
| Blocking the exchange of biomolecules between the nucleus and cytoplasm | Ac-IIIIKKDopa-NH2 [71] | |
| Anoikis | Inhibition of the FAK/ERK1/2 pathways | Apigenin [83] |
| Inhibition of the Timp1-mediated PI3K/PDK1/PKC pathway | shTimp1/AKT-siRNA [84] | |
| Inhibition of YAP activity | CA3 [87] | |
| Inhibition of c-MET by targeting BRN2 | Foretinib or capmatinib [89] | |
| Increasing the cellular level of ROS | shSesn2 [90] | |
| Inhibition of the Mcl-1-mediated BRAF/MEK pathway | BH3 mimetic targeting Mcl-1 [91] | |
| Endoplasmic reticulum stress | Regulation of the ATF4-DDIT3-TRIB3-AKT-mTOR axis | Kuwanon H (KuH) [96] |
| Upregulation of C/EBP homologous protein (CHOP) | Acetylsalicylic acid (ASA) and salicylic acid (SA) [97] | |
| Upregulation of eIF2α, CHOP, and caspase-3 | Resveratrol [98], Shikonin [99] | |
| Inhibition of the AKT/GSK3-β/β-catenin pathway | Tumor suppressor candidate 3 (TUSC3) [101] | |
| Inhibition of MITF and β-catenin pathways | Honokiol [102] | |
| Activation of the IRE1α/Xbp1 pathway | Pinocembrin [103] | |
| Upregulation of ROS generation | Luteolin [104] | |
| Inhibition of the BRAF and RAF/MEK/ERK (MAPK) pathways | Vemurafenib with binimetinib [105] | |
| Upregulation of eIF2α | Bornyl cis-4-hydroxy cinnamate [106] | |
| Inhibition of GRP78 and upregulation of CHOP | 25-epi Ritterostatin GN1N [107] | |
| Activation of the PERK/p-eIF2α/ATF4/CHOP pathway | Vitamin E δ-tocotrienol (δ-TT) [108] | |
| Binding of hnRNP H1 and H2 | Novel Pyrrolidine Diketopiperazines 2155–14 and 2155–18 [109,110,111] | |
| Autophagy | Upregulation of BECN1 by miR1290 inhibition | Polygonatum odoratum lectin (POL) [115] |
| Activation of the JNK pathway | miR-24-1-5p [116] | |
| Activation of HIF-1α/BNIP3/BNIP3L pathway | Proopiomelanocortin (POMC) [117] | |
| Accumulation of damaged mitochondria via blocking the mitophagic flux to lysosomes | siTRPML1 [118] | |
| Downregulation of LC3, ERK1/2 and MITF | Combination of doxycycline and minocycline [119] | |
| Autophagosome accumulation | Sasanquasaponin ΙΙΙ [121] | |
| Regulation of the CXCR4/CXCL12 axis | Liposomes containing paclitaxel and hydroxychloroquine [122] | |
| Necroptosis | Inhibition of mitochondrial complex I-mediated cellular ROS level increase | BAY 87-2243 [127] |
| Upregulation of death receptors and scaffold protein by activating caspase-8 | 10-Methoxy-1,2,3,4-tetrahydrobenzo(g) (1,3) diazepino(1,2-a)-(1,8) naphthyridin-6-yl) (phenyl) methanone (3u) [128] | |
| Activation of caspase-3, caspase-9, and poly (ADP-ribose) polymerase 1 | Evodiamine (EVO) [131] | |
| Activation of the RIPK1/RIPK3/MLKL pathway | Protein-bound polysaccharides (PBPs) [133] | |
| Increasing levels of CHOP and RIP1 | Shikonin [134] | |
| Increasing the cellular level of ROS | Pleuromutilin derivative compound 38 [132] | |
| Pyroptosis | Activation of the Tom20-Bax-caspase-3-GSDME pathway | CCCP and several chemotherapeutic drugs, such as SSZ [137] |
| Targeting CXCR4 | A self-assembling nanotoxin (T22 peptide) [138] | |
| Activation of caspase 3 | Raptinal [139] | |
| T-cell accumulation/activation, GSDM E cleavage, and release of HMGB1 | PLX4720/PD0325901 [142] | |
| Activation of caspase 3 and GSDM E/D cleavage | Temozolomide/chloroquine [140] | |
| Activation of gasdermin | Phe-BF3 [143] | |
| Ferroptosis | GPX4 inhibition | BET inhibitor [145], |
| IFN-γ release and driving action | nanoparticle of miR-21-3p [146] | |
| Upregulation of GOT1 | anti-miR-9 [147] | |
| SLC1A5-mediated glutamine uptake and MDA accumulation | anti-miR-137 [148] | |
| Inhibition of the AKT/GPX4 pathway | ARG2 KO [149] | |
| Inhibition of NEAT1 | Gambogenic acid (GNA) [150] | |
| Activation of the AMPK/mTOR pathway | Arachidonate 5-lipoxygenase (ALOX5) [151] | |
| Inhibition of USP14 | MitoCur-1 [152] | |
| GPX4 inhibition-mediated metabolic reprogramming | DET and DETD-35 [153] | |
| Upregulation of SVCT2 | Blue light [155] | |
| Increasing lipid peroxidation | Gallic acid (GA) with low-level laser [156],protoporphyrin IX-based polysilsesquioxane nanoparticles. (PpIX-PSilQ NPs) [157] | |
| Inhibition of the GSK3β-mediated Keap1/Nrf2/HO-1 pathway | Nobiletin [158] | |
| Cuproptosis | Inhibiting the MAPK signaling pathway | Chelating copper [162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Zhao, J.-H.; Tang, M.-X.; Li, M.; Zhao, H.; Li, Z.-Y.; Liu, A.-D. Cell Death Modalities in Therapy of Melanoma. Int. J. Mol. Sci. 2025, 26, 3475. https://doi.org/10.3390/ijms26083475
Wang M, Zhao J-H, Tang M-X, Li M, Zhao H, Li Z-Y, Liu A-D. Cell Death Modalities in Therapy of Melanoma. International Journal of Molecular Sciences. 2025; 26(8):3475. https://doi.org/10.3390/ijms26083475
Chicago/Turabian StyleWang, Meng, Jia-Hui Zhao, Ming-Xuan Tang, Meng Li, Hu Zhao, Zhong-Yu Li, and An-Dong Liu. 2025. "Cell Death Modalities in Therapy of Melanoma" International Journal of Molecular Sciences 26, no. 8: 3475. https://doi.org/10.3390/ijms26083475
APA StyleWang, M., Zhao, J.-H., Tang, M.-X., Li, M., Zhao, H., Li, Z.-Y., & Liu, A.-D. (2025). Cell Death Modalities in Therapy of Melanoma. International Journal of Molecular Sciences, 26(8), 3475. https://doi.org/10.3390/ijms26083475