
MiRNA Omics Reveal the Mechanisms of the Dual Effects of Selenium Supplementation on the Development of the Silkworm (Bombyx mori)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
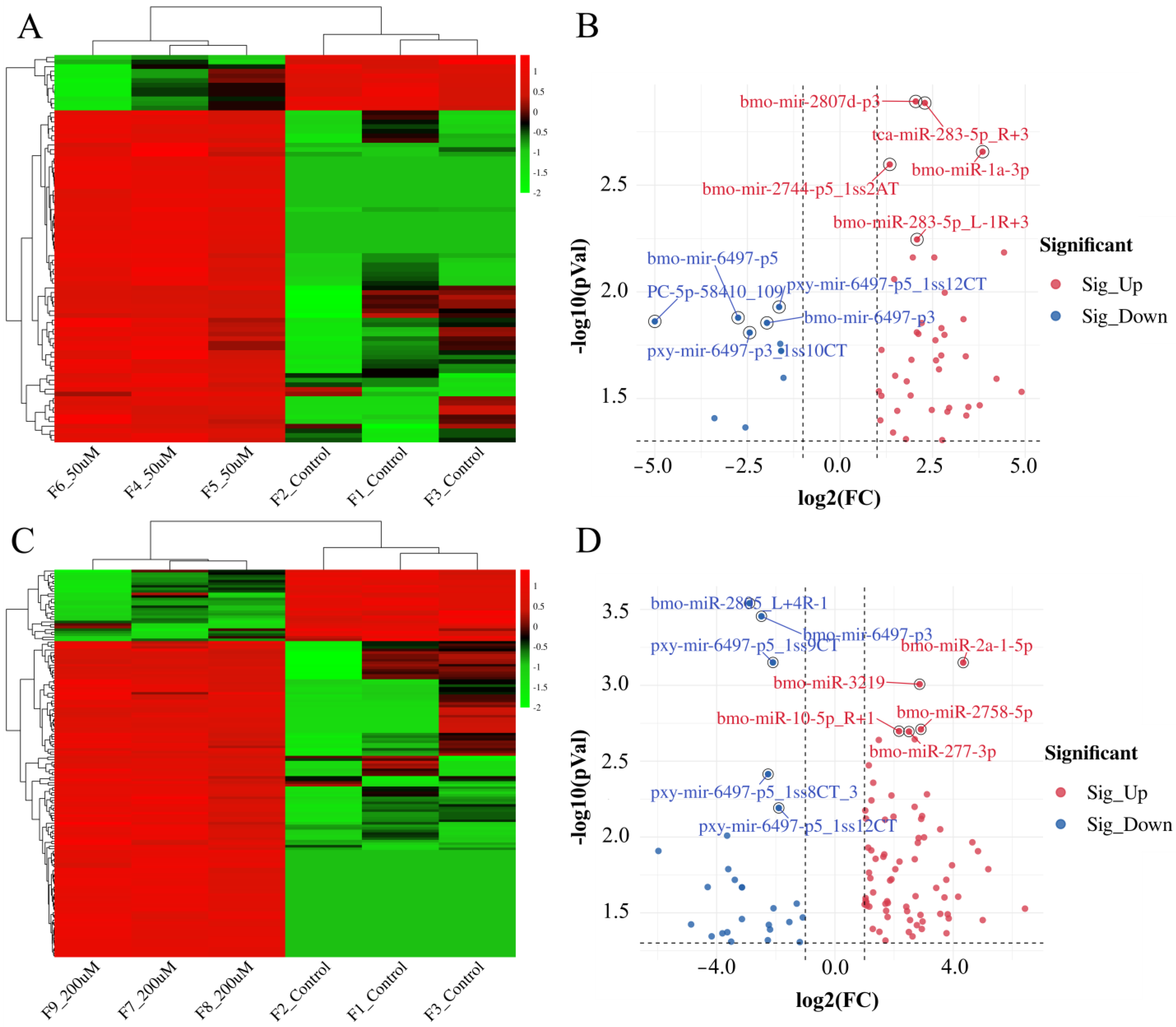
2.1. Analysis of Differentially Expressed miRNAs
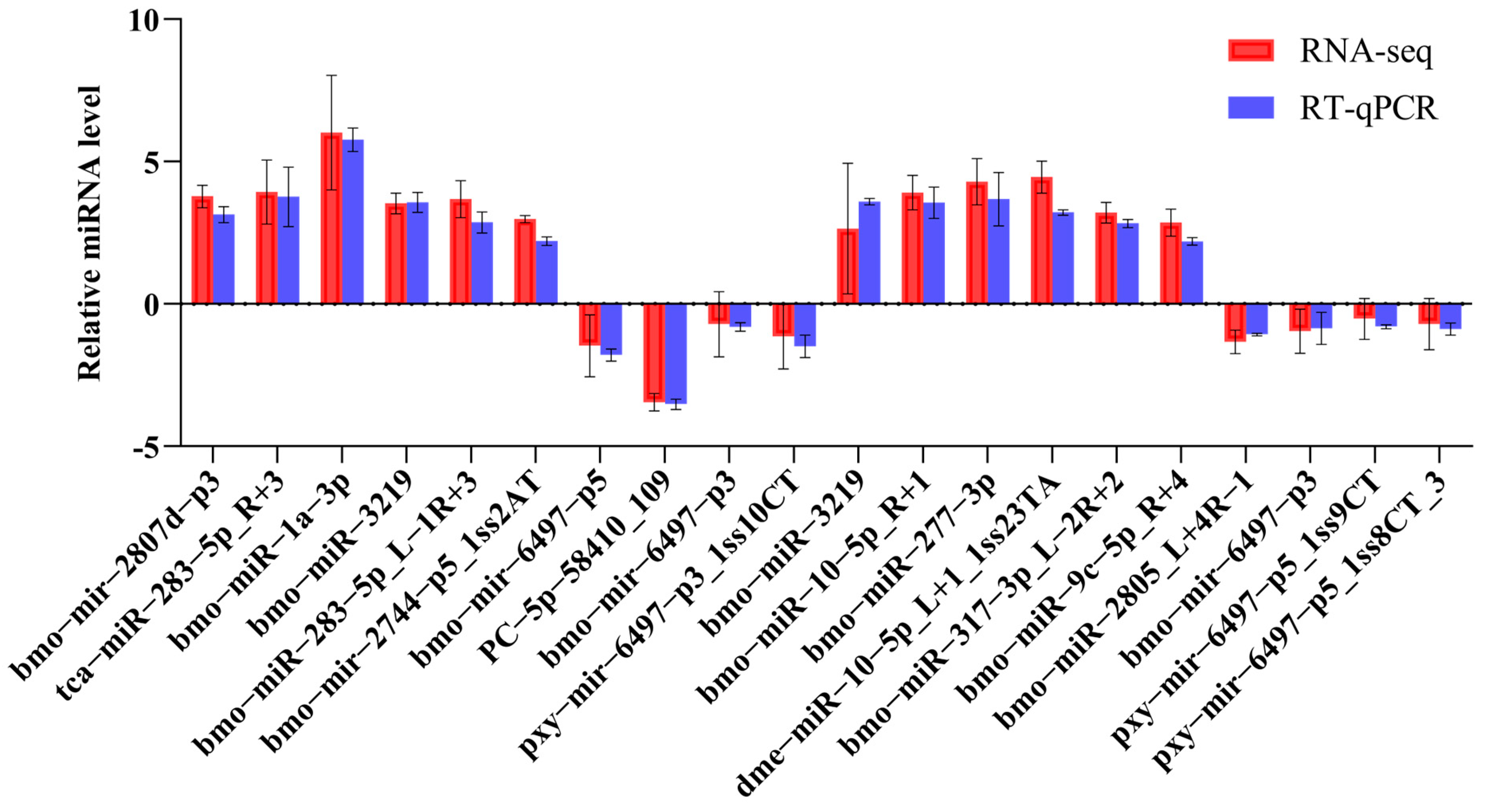
2.2. Validation of DEMs by RT-qPCR
2.3. Prediction of Targets of Differentially Expressed miRNAs
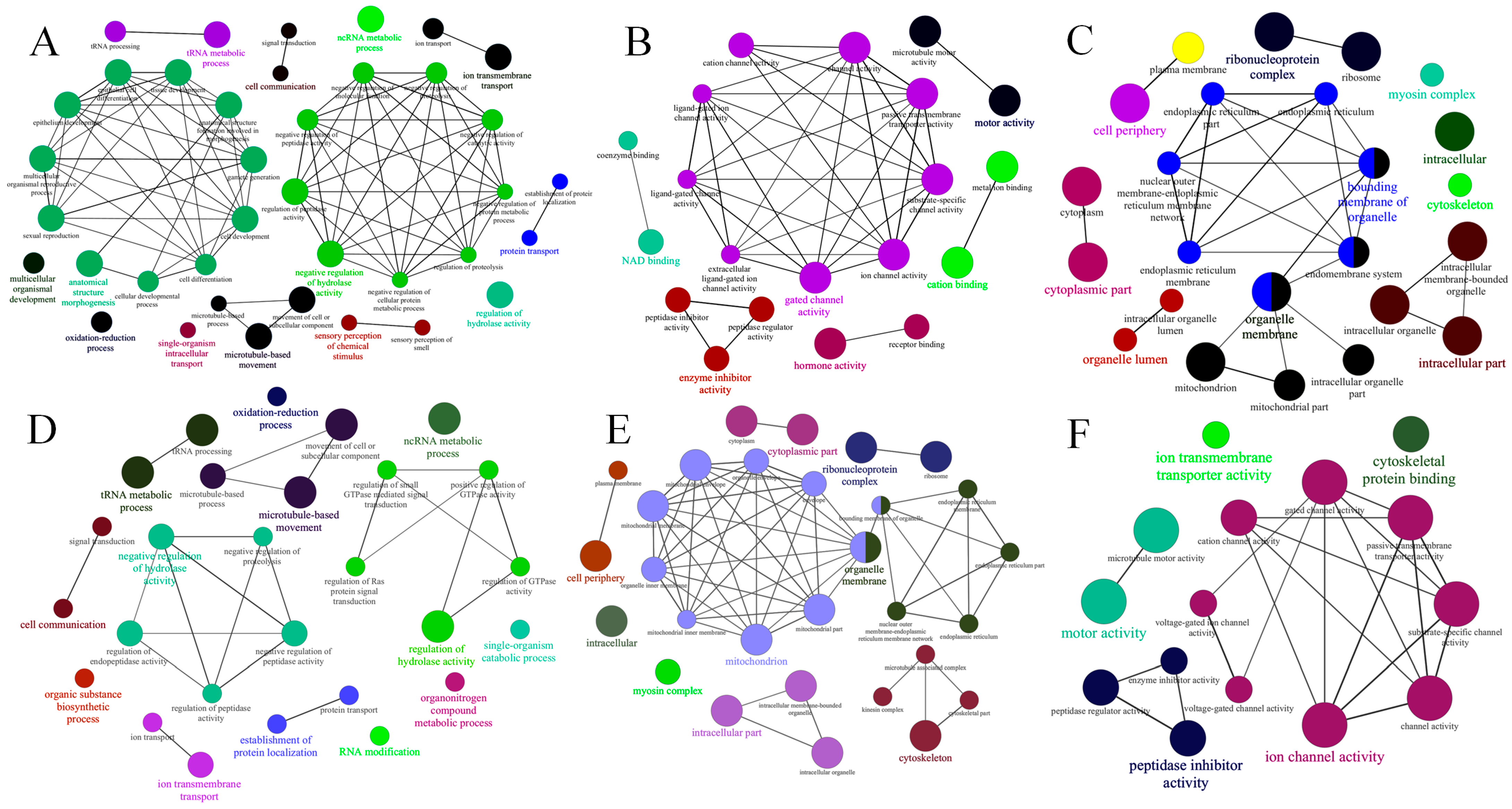
2.4. GO Enrichment Analysis of Target Genes and GO Networks
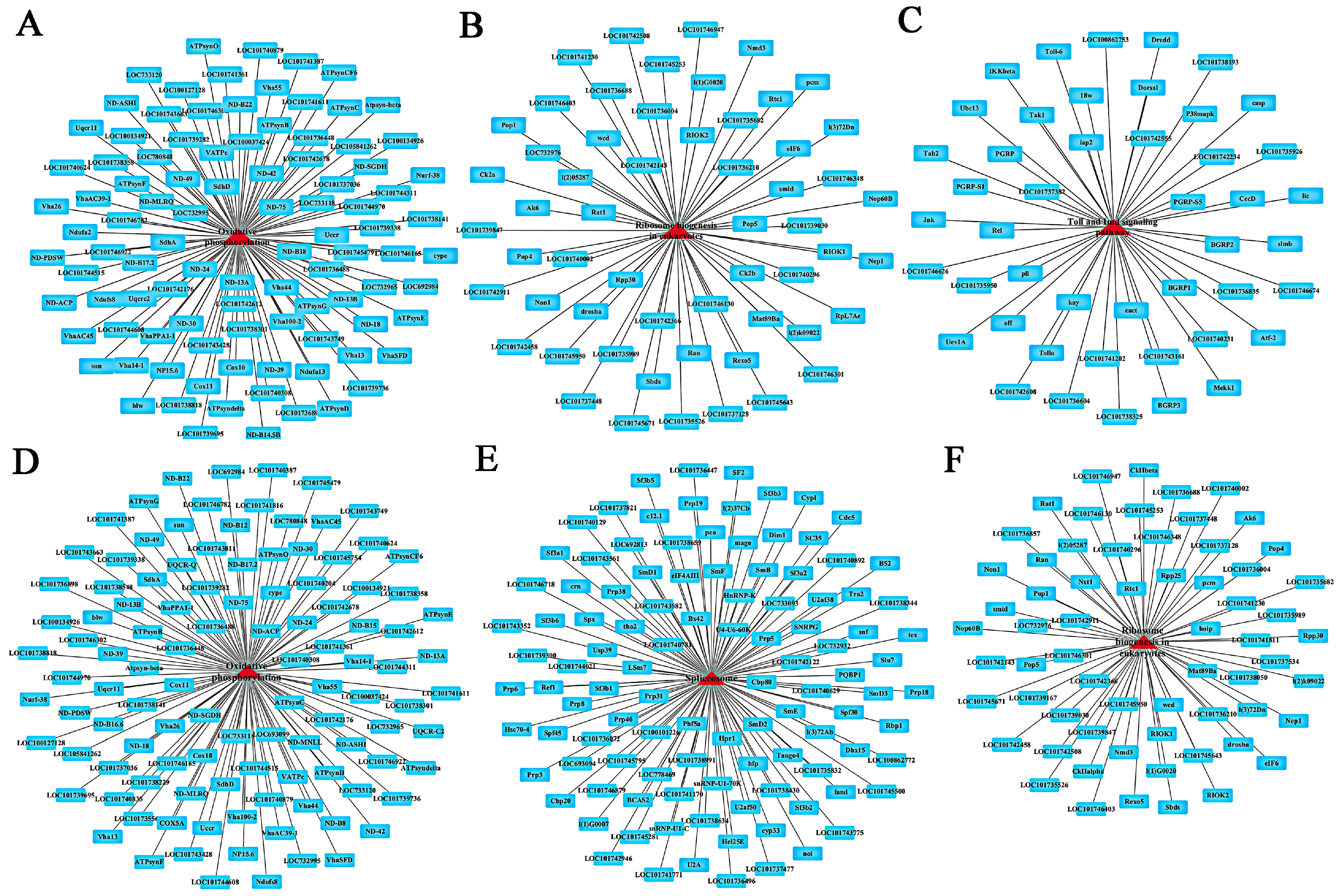
2.5. KEGG Pathway Enrichment Analysis
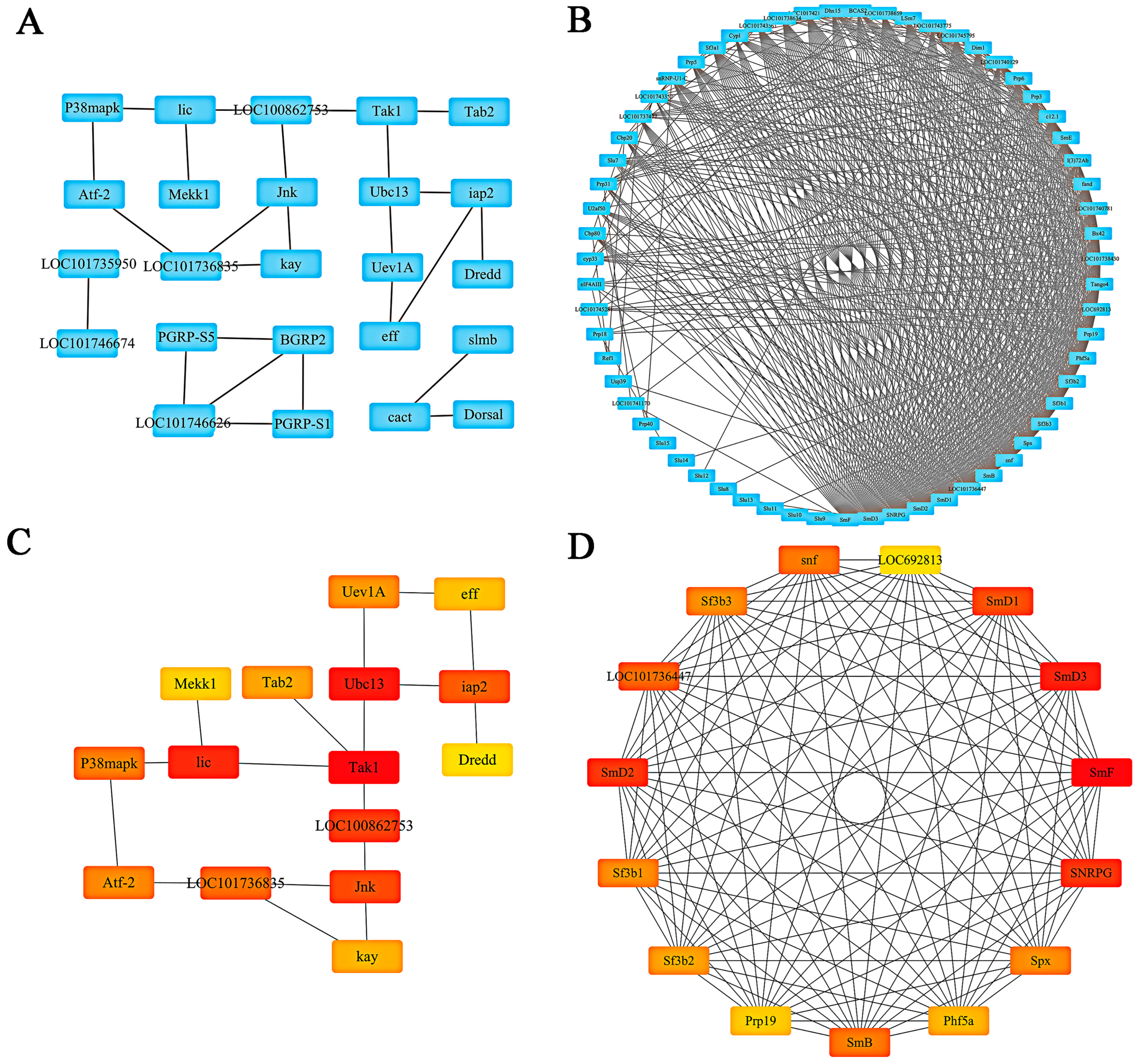
2.6. PPI Network Establishment and Hub Gene Analysis of Target Genes
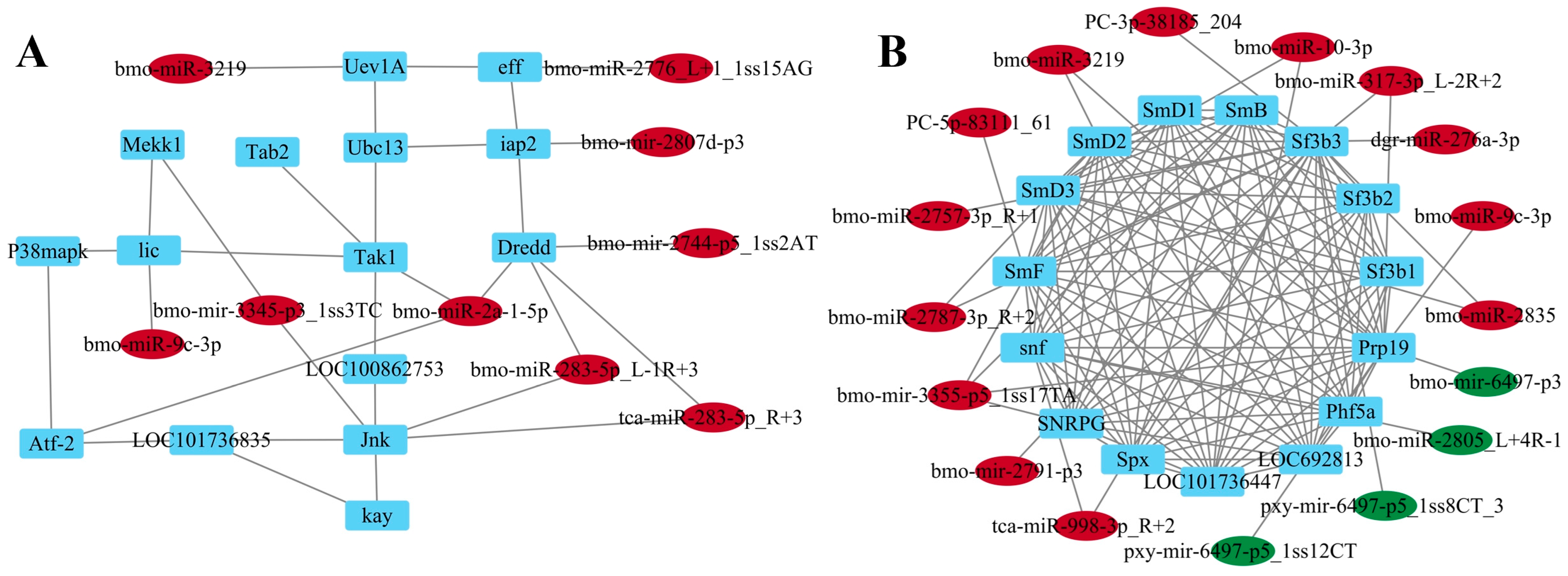
2.7. Construction of the miRNA-Target Regulatory Network
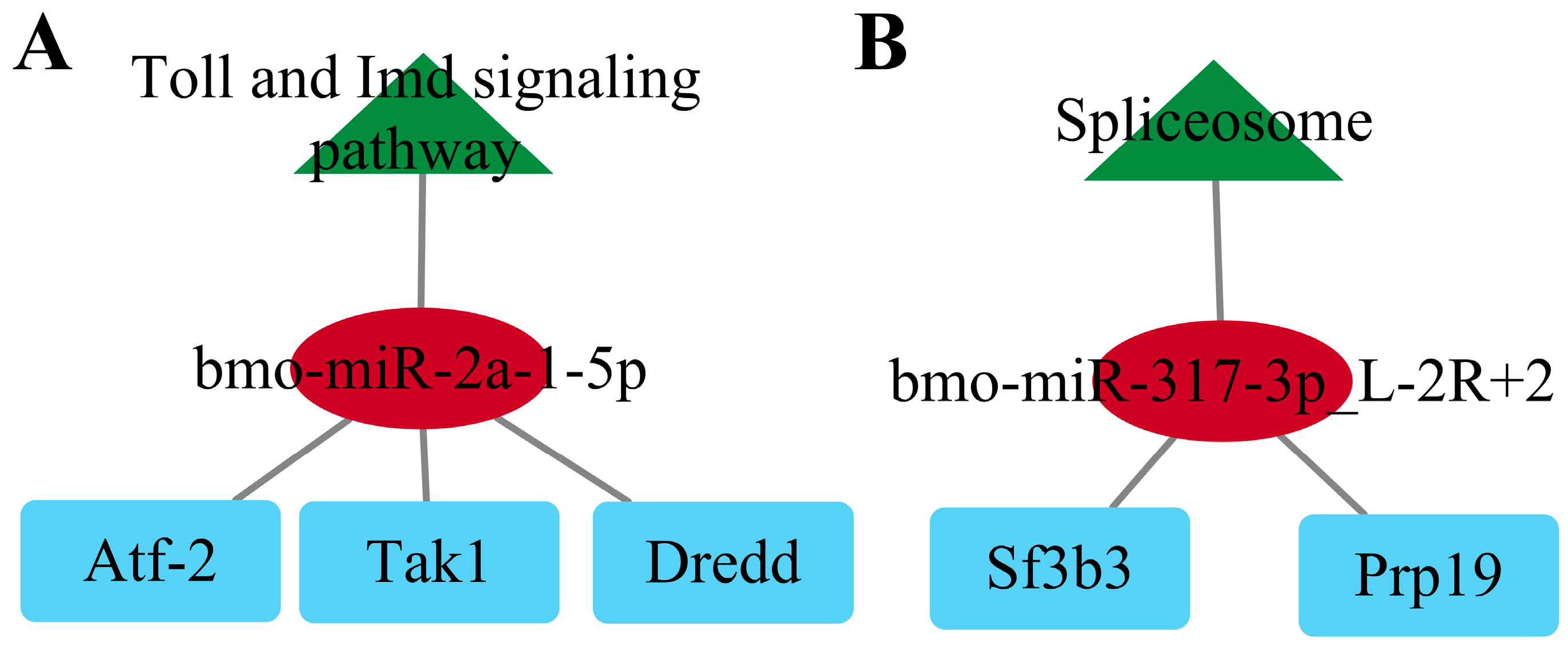
2.8. Construction of the KEGG-miRNA-Targeting Network
3. Discussion
4. Materials and Methods
4.1. Experimental Material
4.2. miRNA Sequencing
4.3. Validation of miRNA by RT-qPCR Analysis
4.4. Target Gene Prediction
4.5. Differential miRNA-Target Gene Enrichment Analysis
4.6. Protein Interaction Network Construction and Hub Gene Screening
4.7. Construction of the KEGG-miRNA-Target Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Genchi, G.; Lauria, G.; Catalano, A.; Sinicropi, M.S.; Carocci, A. Biological activity of selenium and its impact on human health. Int. J. Mol. Sci. 2023, 24, 2633. [Google Scholar] [CrossRef] [PubMed]
- Kuršvietienė, L.; Mongirdienė, A.; Bernatonienė, J.; Šulinskienė, J.; Stanevičienė, I. Selenium anticancer properties and impact on cellular redox status. Antioxidants 2020, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Constans, J.; Sire, S.; Sergeant, C.; Simonoff, M.; Ragnaud, J.M. Dilated cardiomyopathy and selenium deficiency in AIDS. Apropos of a case. Rev. Med. Interne 1997, 18, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Trippe, R.C., III; Pilon-Smits, E.A.H. Selenium transport and metabolism in plants: Phytoremediation and biofortification implications. J. Hazard. Mater. 2021, 404, 12. [Google Scholar] [CrossRef]
- Zafar, S.; Hasnain, Z.; Danish, S.; Battaglia, M.L.; Fahad, S.; Ansari, M.J.; Alharbi, S.A. Modulations of wheat growth by selenium nanoparticles under salinity stress. BMC Plant Biol. 2024, 24, 14. [Google Scholar] [CrossRef]
- Ferrari, L.; Cattaneo, D.; Abbate, R.; Manoni, M.; Ottoboni, M.; Luciano, A.; von Holst, C.; Pinotti, L. Advances in selenium supplementation: From selenium-enriched yeast to potential selenium-enriched insects, and selenium nanoparticles. Anim. Nutr. 2023, 14, 193–203. [Google Scholar] [CrossRef]
- So, J.; Choe, D.H.; Rust, M.K.; Trumble, J.T.; Lee, C.Y. The impact of selenium on insects. J. Econ. Entomol. 2023, 116, 1041–1062. [Google Scholar] [CrossRef]
- Feng, J.; Li, X.; Xiao, Y.; Zhang, F.R.; Liu, Z.Q.; Zhang, H.F.; Yang, X.H. Effects of Se-enriched Chrysanthemum morifolium on lifespan and antioxidant defense-related gene expression of Drosophila melanogaster model. J. Food Biochem. 2022, 46, 11. [Google Scholar] [CrossRef]
- Dong, Z.; Lin, Y.Y.; Wu, H.; Zhang, M.M. Selenium accumulation in protein fractions of Tenebrio molitor larvae and the antioxidant and immunoregulatory activity of protein hydrolysates. Food Chem. 2021, 334, 127475. [Google Scholar] [CrossRef]
- Liu, J.; Kong, T.Y.; Xiong, H.M. Mulberry-leaves-derived red-emissive carbon dots for feeding silkworms to produce brightly fluorescent silk. Adv. Mater. 2022, 34, e2200152. [Google Scholar] [CrossRef]
- Pongworn, S.; Wongsa, J.; Vatanyoopaisarn, S.; Thumthanaruk, B.; Uttapap, D.; Lamsal, B.; Rungsardthong, V. Influence of dietary mulberry leaf on the nutritional composition of mulberry silkworm, Bombyx mori L. at different developmental stages. J. Insects Food Feed. 2024, 10, 1933–1946. [Google Scholar]
- Hong, T.T.; Hu, F.; Ge, W.J.; Zhang, R.; Du, J.; Thakur, K.; Tang, S.M.; Wei, Z.J. Selenium treatment alleviates the inhibition caused by Nep-L gene knockdown in silkworm (Bombyx mori). Biol. Trace Elem. Res. 2025, 203, 1656–1666. [Google Scholar] [PubMed]
- Hong, T.T.; Hu, S.; Hu, F.; Ge, W.J.; Thakur, K.; Tang, S.M.; Wei, Z.J. Selenium treatment ameliorates the adverse effects caused by Dynamin gene knockdown in Bombyx mori. Arch. Insect Biochem. Physiol. 2024, 117, e22148. [Google Scholar] [CrossRef]
- Jiang, L.; Peng, L.L.; Cao, Y.Y.; Thakur, K.; Hu, F.; Tang, S.M.; Wei, Z.J. Effect of dietary selenium supplementation on growth and reproduction of silkworm Bombyx mori L. Biol. Trace Elem. Res. 2020, 193, 271–281. [Google Scholar]
- Jiang, L.; Peng, L.L.; Cao, Y.Y.; Thakur, K.; Hu, F.; Tang, S.M.; Wei, Z.J. Transcriptome analysis reveals gene expression changes of the fat body of silkworm (Bombyx mori L.) in response to selenium treatment. Chemosphere 2020, 245, 125660. [Google Scholar]
- Dissanayake, E.; Inoue, Y. MicroRNAs in Allergic Disease. Curr. Allergy Asthma Rep. 2016, 16, 67. [Google Scholar]
- Singh, C.P. In vitro treatment of seroin proteins to BmNPV budded virions suppresses viral proliferation in Bombyx mori larvae and ectopic overexpression of host-miRNAs downregulates the expression of Seroin2 mRNA in BmN cells. Int. J. Trop. Insect Sci. 2021, 41, 1485–1491. [Google Scholar]
- Cao, H.H.; Kong, W.W.; Ling, B.; Wang, Z.Y.; Zhang, Y.; Guo, Z.X.; Liu, S.H.; Xu, J.P. Bmo-miR-3351 modulates glutathione content and inhibits BmNPV proliferation by targeting BmGSTe6 in Bombyx mori. Insect Sci. 2024, 31, 1378–1396. [Google Scholar] [CrossRef]
- Zhang, R.; Peng, L.L.; Zhu, H.T.; Jiang, L.; Thakur, K.; Tang, S.M.; Hu, F.; Wei, Z.J. Metabonomic analysis of the hemolymph reveals the dual effects of dietary selenium supplementations on growth of silkworm Bombyx mori. J. Insects Food Feed. 2023, 9, 475–490. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Y.X.; Zhang, X.L.; Liu, J.H.; Gong, P.; Su, Z.Q.; Fan, L.H.; Li, G.L. Emerging nanoparticles in food: Sources, application, and safety. J. Agric. Food Chem. 2023, 71, 3564–3582. [Google Scholar]
- Kong, W.A.; Tian, Q.Q.; Yang, Q.L.; Liu, Y.; Wang, G.T.; Cao, Y.J.; Wang, L.P.; Xia, S.Z.; Sun, Y.M.; Zhao, C.; et al. Sodium Selenite Enhances antibiotics sensitivity of Pseudomonas aeruginosa and deceases its pathogenicity by inducing oxidative stress and inhibiting quorum sensing system. Antioxidants 2021, 10, 1873. [Google Scholar] [CrossRef] [PubMed]
- El-Ashram, S.; Kheirallah, D.A.M.; El-Samad, L.M.; Toto, N.A. Relative expression of microRNAs, apoptosis, and ultrastructure anomalies induced by gold nanoparticles in Trachyderma hispida (Coleoptera: Tenebrionidae). PLoS ONE 2020, 15, e0241837. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, M.M.; Dowidar, M.F.; Ali, H.A.; Ghonimi, W.A.M.; Al-Farga, A.; Ahmed, A.I. Effect of selenium nanoparticles and/or bee venom against stz-induced diabetic cardiomyopathy and nephropathy. Metabolites 2023, 13, 400. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.L.; Xia, D.G.; Qiu, Z.Y.; Gao, P.; Tang, S.M.; Shen, X.J.; Zhu, F.; Zhao, Q.L. Expression of a vitelline membrane protein, BmVMP23, is repressed by bmo-miR-1a-3p in silkworm, Bombyx mori. FEBS Lett. 2013, 587, 970–975. [Google Scholar] [CrossRef]
- Cai, Y.; Yu, X.; Zhou, Q.; Yu, C.; Hu, H.; Liu, J.; Lin, H.; Yang, J.; Zhang, B.; Cui, P.; et al. Novel microRNAs in silkworm (Bombyx mori). Funct. Integr. Genom. 2010, 10, 405–415. [Google Scholar] [CrossRef]
- Luo, Q.; Zhou, Q.; Yu, X.; Lin, H.; Hu, S.; Yu, J. Genome-wide mapping of conserved microRNAs and their host transcripts in Tribolium castaneum. J. Genet. Genom. 2008, 35, 349–355. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, P.; Gao, Q.; Feng, X.; Han, L.; Zhang, F.; Bai, Y.; Han, M.; Hu, H.; Dai, F.; et al. Comparative transcriptome analysis reveals bmo-mir-6497-3p regulate circadian clock genes during the embryonic diapause induction process in bivoltine silkworm. Insects 2021, 12, 739. [Google Scholar] [CrossRef]
- Liang, P.; Feng, B.; Zhou, X.; Gao, X. Identification and developmental profiling of microRNAs in diamondback moth, Plutellaxylostella (L.). PLoS ONE 2013, 8, e78787. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, A.; Zhang, Y.; Xue, C.; Zhao, M.; Zhang, J. Activating pathway of three metabolic detoxification phases via down-regulated endogenous microRNAs, modulates triflumezopyrim tolerance in the small brown planthopper, Laodelphax striatellus (Fallén). Int. J. Biol. Macromol. 2022, 222, 2439–2451. [Google Scholar] [CrossRef]
- Wang, X.; Tang, S.; Song, F.; Chen, C.; Guo, X.; Shen, X. Bmo-miR-2758 targets BmFMBP-1 (Lepidoptera: Bombycidae) and suppresses its expression in BmN Cells. J. Insect Sci. 2016, 16, 28. [Google Scholar] [CrossRef]
- Fan, Y.X.; Andoh, V.; Chen, L. Multi-omics study and ncRNA regulation of anti-BmNPV in silkworms, Bombyx mori: An update. Front. Microbiol. 2023, 14, 1123448. [Google Scholar]
- Wu, P.; Jiang, X.X.; Guo, X.J.; Li, L.; Chen, T. Genome-Wide Analysis of Differentially Expressed microRNA in Bombyx mori Infected with Nucleopolyhedrosis Virus. PLoS ONE 2016, 11, e0165865. [Google Scholar]
- He, P.-A.; Nie, Z.; Chen, J.; Chen, J.; Lv, Z.; Sheng, Q.; Zhou, S.; Gao, X.; Kong, L.; Wu, X.; et al. Identification and characteristics of microRNAs from Bombyx mori. BMC Genom. 2008, 9, 248. [Google Scholar]
- Chen, Y.; Jiang, T.; Tan, Z.; Xue, P.; Xu, J.; Tang, S.; Yi, Y.; Shen, X. Bom-miR-2805 upregulates the expression of Bombyx mori fibroin light chain gene in vivo. J. Cell. Biochem. 2019, 120, 14326–14335. [Google Scholar]
- Zafar, J.; Huang, J.; Xu, X.; Jin, F. Analysis of long non-coding rna-mediated regulatory networks of plutella xylostella in response to metarhizium anisopliae infection. Insects 2022, 13, 916. [Google Scholar] [CrossRef]
- Myllymaki, H.; Valanne, S.; Ramet, M. The Drosophila Imd Signaling Pathway. J. Immunol. 2014, 192, 3455–3462. [Google Scholar]
- Wu, H.; Tan, M.; Li, Y.; Zheng, L.; Xu, J.; Jiang, D. The immunotoxicity of Cd exposure to gypsy moth larvae: An integrated analysis of cellular immunity and humoral immunity. Ecotoxicol. Environ. Saf. 2022, 235, 113434. [Google Scholar]
- Tanaka, H.; Ishibashi, J.; Fujita, K.; Nakajima, Y.; Sagisaka, A.; Tomimoto, K.; Suzuki, N.; Yoshiyama, M.; Kaneko, Y.; Iwasaki, T.; et al. A genome-wide analysis of genes and gene families involved in innate immunity of Bombyx mori. Insect Biochem. Mol. Biol. 2008, 38, 1087–1110. [Google Scholar]
- Gottar, M.; Gobert, V.; Michel, T.; Belvin, M.; Duyk, G.; Hoffmann, J.A.; Ferrandon, D.; Royet, J. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 2002, 416, 640–644. [Google Scholar]
- Choe, K.-M.; Werner, T.; Stöven, S.; Hultmark, D.; Anderson, K.V. Requirement for a peptidoglycan recognition protein (PGRP) in relish activation and antibacterial immune responses in Drosophila. Science 2002, 296, 359–362. [Google Scholar]
- Fogaça, A.C.; Sousa, G.; Pavanelo, D.B.; Esteves, E.; Martins, L.A.; Urbanová, V.; Kopáček, P.; Daffre, S. Tick immune system: What is known, the interconnections, the gaps, and the challenges. Front. Immunol. 2021, 12, 628054. [Google Scholar]
- Zumaya-Estrada, F.A.; Martínez-Barnetche, J.; Lavore, A.; Rivera-Pomar, R.; Rodríguez, M.H. Comparative genomics analysis of triatomines reveals common first line and inducible immunity-related genes and the absence of Imd canonical components among hemimetabolous arthropods. Parasites Vectors 2018, 11, 48. [Google Scholar] [PubMed]
- Takahasi, K.; Ochiai, M.; Horiuchi, M.; Kumeta, H.; Ogura, K.; Ashida, M.; Inagaki, F. Solution structure of the silkworm βGRP/GNBP3 N-terminal domain reveals the mechanism for β-1,3-glucan-specific recognition. Proc. Natl. Acad. Sci. USA 2009, 106, 11679–11684. [Google Scholar] [CrossRef]
- Lo, P.C.; Mount, S.M. Drosophila melanogaster genes for U1 snRNA variants and their expression during development. Nucleic Acids Res. 1990, 18, 6971–6979. [Google Scholar]
- Sierra-Montes, J.M.; Pereira-Simon, S.; Smail, S.S.; Herrera, R.J. The silk moth Bombyx mori U1 and U2 snRNA variants are differentially expressed. Gene 2005, 352, 127–136. [Google Scholar]
- Saluz, H.; Dudler, R.; Schmidt, T.; Kubli, E. The localization and estimated copy number of Drosophila melanogaster U1, U4, U5 and U6 snRNA genes. Nucleic Acids Res. 1988, 16, 3582. [Google Scholar]
- Hardin, S.B.; Ortler, C.J.; McNamara-Schroeder, K.J.; Stumph, W.E. Similarities and differences in the conformation of protein-DNA complexes at the U1 and U6 snRNA gene promoters. Nucleic Acids Res. 2000, 28, 2771–2778. [Google Scholar]
- Chen, J.; Liang, Z.; Liang, Y.; Pang, R.; Zhang, W. Conserved microRNAs miR-8-5p and miR-2a-3p modulate chitin biosynthesis in response to 20-hydroxyecdysone signaling in the brown planthopper, Nilaparvata lugens. Insect Biochem. Mol. Biol. 2013, 43, 839–848. [Google Scholar]
- Agwunobi, D.O.; Pei, T.; Bai, R.; Wang, Z.; Shi, X.; Zhang, M.; Yu, Z.; Liu, J. miR-2a and miR-279 are functionally associated with cold tolerance in Dermacentor silvarum (Acari: Ixodidae). Comp. Biochem. Physiol. Part D Genom. Proteom. 2022, 41, 100946. [Google Scholar]
- Stronach, B.; Lennox, A.L.; Garlena, R.A. Domain specificity of MAP3K family members, MLK and Tak1, for JNK signaling in Drosophila. Genetics 2014, 197, 497–513. [Google Scholar]
- Li, R.; Huang, Y.; Zhang, Q.; Zhou, H.; Jin, P.; Ma, F. The miR-317 functions as a negative regulator of Toll immune response and influences Drosophila survival. Dev. Comp. Immunol. 2019, 95, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Dou, W.; Song, Z.H.; Jin, T.J.; Yuan, G.R.; De Schutter, K.; Smagghe, G.; Wang, J.J. Identification and profiling of Bactrocera dorsalis microRNAs and their potential roles in regulating the developmental transitions of egg hatching, molting, pupation and adult eclosion. Insect Biochem. Mol. Biol. 2020, 127, 103475. [Google Scholar] [CrossRef] [PubMed]
- Tsapras, P.; Petridi, S.; Chan, S.; Geborys, M.; Jacomin, A.C.; Sagona, A.P.; Meier, P.; Nezis, I.P. Selective autophagy controls innate immune response through a TAK1/TAB2/SH3PX1 axis. Cell Rep. 2022, 38, 110286. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.H.; Sun, L.; Kong, L.; Hu, J.H.; Yu, M.C.; Reinach, P.; Zang, J.W.; Ge, B.X. Drosophila TAB2 is required for the immune activation of JNK and NF-kappaB. Cell. Signal. 2006, 18, 964–970. [Google Scholar] [CrossRef]
- Kohyama-Koganeya, A.; Nabetani, T.; Miura, M.; Hirabayashi, Y. Glucosylceramide synthase in the fat body controls energy metabolism in Drosophila. J. Lipid Res. 2011, 52, 1392–1399. [Google Scholar] [CrossRef]
- Kleino, A.; Silverman, N. The Drosophila IMD pathway in the activation of the humoral immune response. Dev. Comp. Immunol. 2014, 42, 25–35. [Google Scholar] [CrossRef]
- Pan, W.; Yao, X.; Lin, L.; Liu, X.; Jin, P.; Ma, F. The Relish/miR-275/Dredd mediated negative feedback loop is crucial to restoring immune homeostasis of Drosophila Imd pathway. Insect Biochem. Mol. Biol. 2023, 162, 104013. [Google Scholar] [CrossRef]
- Karousis, E.D.; Mühlemann, O. Nonsense-mediated mRNA decay begins where translation ends. Cold Spring Harb. Perspect. Biol. 2019, 11, a032862. [Google Scholar] [CrossRef]
- Yi, Y.H.; Xu, H.; Li, M.; Wu, G.Q. RNA-seq profiles of putative genes involved in specific immune priming in Bombyx mori haemocytes. Infect. Genet. Evol. 2019, 74, 103921. [Google Scholar] [CrossRef]
- Mardis, E.; McCombie, W.R. Library Quantification Using SYBR Green-Quantitative Polymerase Chain Reaction (qPCR). Cold Spring Harb. Protoc. 2017, 2017, pdb-prot094714. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Y.Y.; Ma, R.H.; Thakur, K.; Han, J.; Hu, F.; Zhang, J.G.; Wei, Z.J. Multi-omics reveals the anticancer mechanism of asparagus saponin-asparanin A on endometrial cancer Ishikawa cells. Food Funct. 2021, 12, 614–632. [Google Scholar] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar]
- Tang, Y.; Li, M.; Wang, J.X.; Pan, Y.; Wu, F.X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015, 127, 67–72. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, W.-J.; Hu, F.; Hong, T.-T.; Thakur, K.; Tang, S.-M.; Zhang, J.-G.; Wei, Z.-J. MiRNA Omics Reveal the Mechanisms of the Dual Effects of Selenium Supplementation on the Development of the Silkworm (Bombyx mori). Int. J. Mol. Sci. 2025, 26, 3394. https://doi.org/10.3390/ijms26073394
Ge W-J, Hu F, Hong T-T, Thakur K, Tang S-M, Zhang J-G, Wei Z-J. MiRNA Omics Reveal the Mechanisms of the Dual Effects of Selenium Supplementation on the Development of the Silkworm (Bombyx mori). International Journal of Molecular Sciences. 2025; 26(7):3394. https://doi.org/10.3390/ijms26073394
Chicago/Turabian StyleGe, Wen-Jie, Fei Hu, Ting-Ting Hong, Kiran Thakur, Shun-Ming Tang, Jian-Guo Zhang, and Zhao-Jun Wei. 2025. "MiRNA Omics Reveal the Mechanisms of the Dual Effects of Selenium Supplementation on the Development of the Silkworm (Bombyx mori)" International Journal of Molecular Sciences 26, no. 7: 3394. https://doi.org/10.3390/ijms26073394
APA StyleGe, W.-J., Hu, F., Hong, T.-T., Thakur, K., Tang, S.-M., Zhang, J.-G., & Wei, Z.-J. (2025). MiRNA Omics Reveal the Mechanisms of the Dual Effects of Selenium Supplementation on the Development of the Silkworm (Bombyx mori). International Journal of Molecular Sciences, 26(7), 3394. https://doi.org/10.3390/ijms26073394