
PTPN2 Regulates Iron Handling Protein Expression in Inflammatory Bowel Disease Patients and Prevents Iron Deficiency in Mice

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
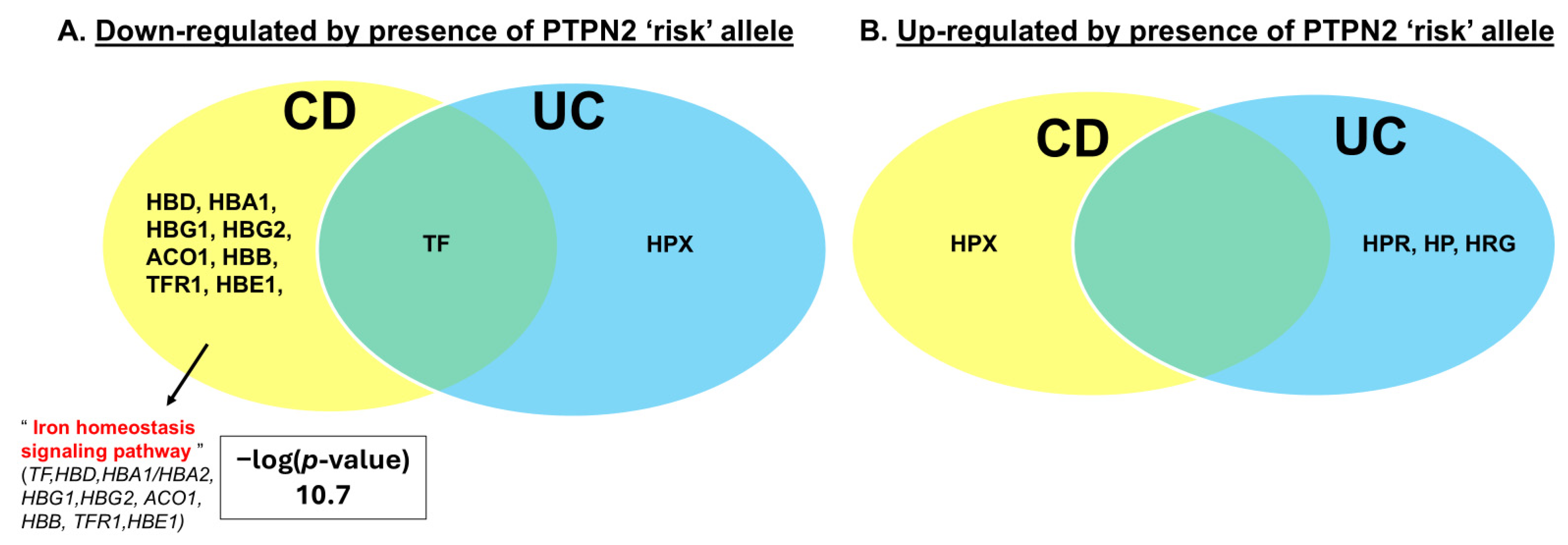
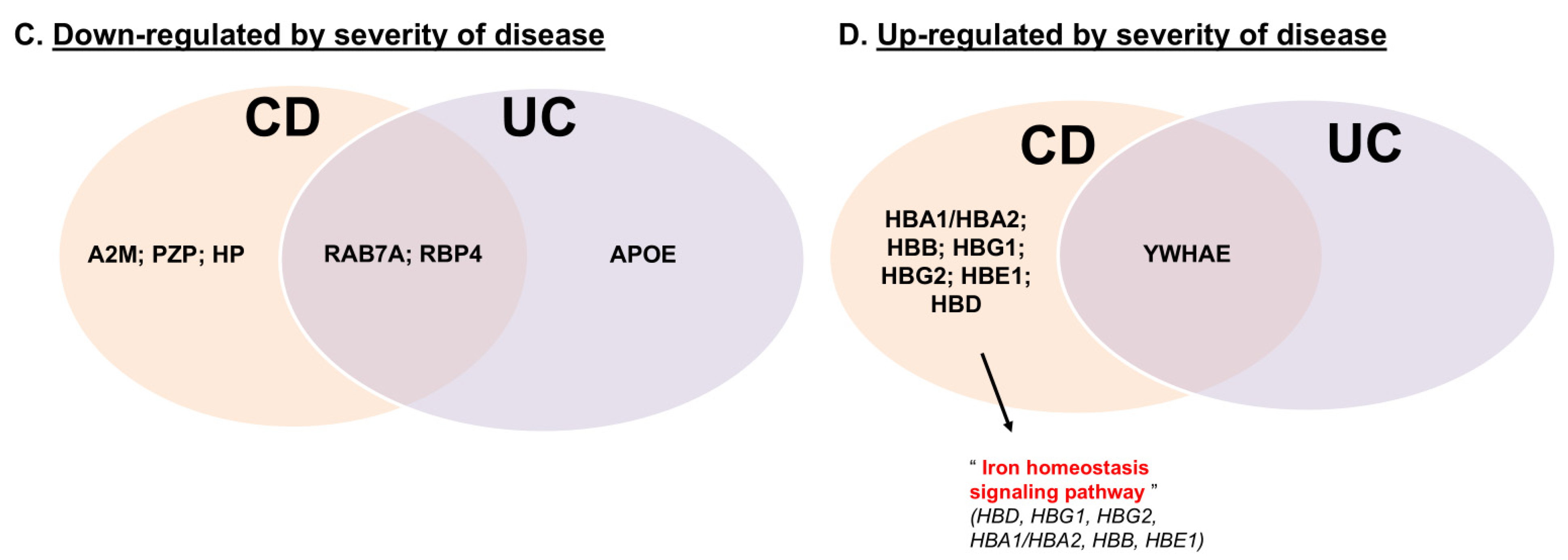
2.1. PTPN2 rs1893217 Risk Allele Alters Serum Iron Handling Proteins in IBD Patients
2.2. Constitutive Loss of Ptpn2 in Mice Causes Features of Anemia Including Iron Deficiency
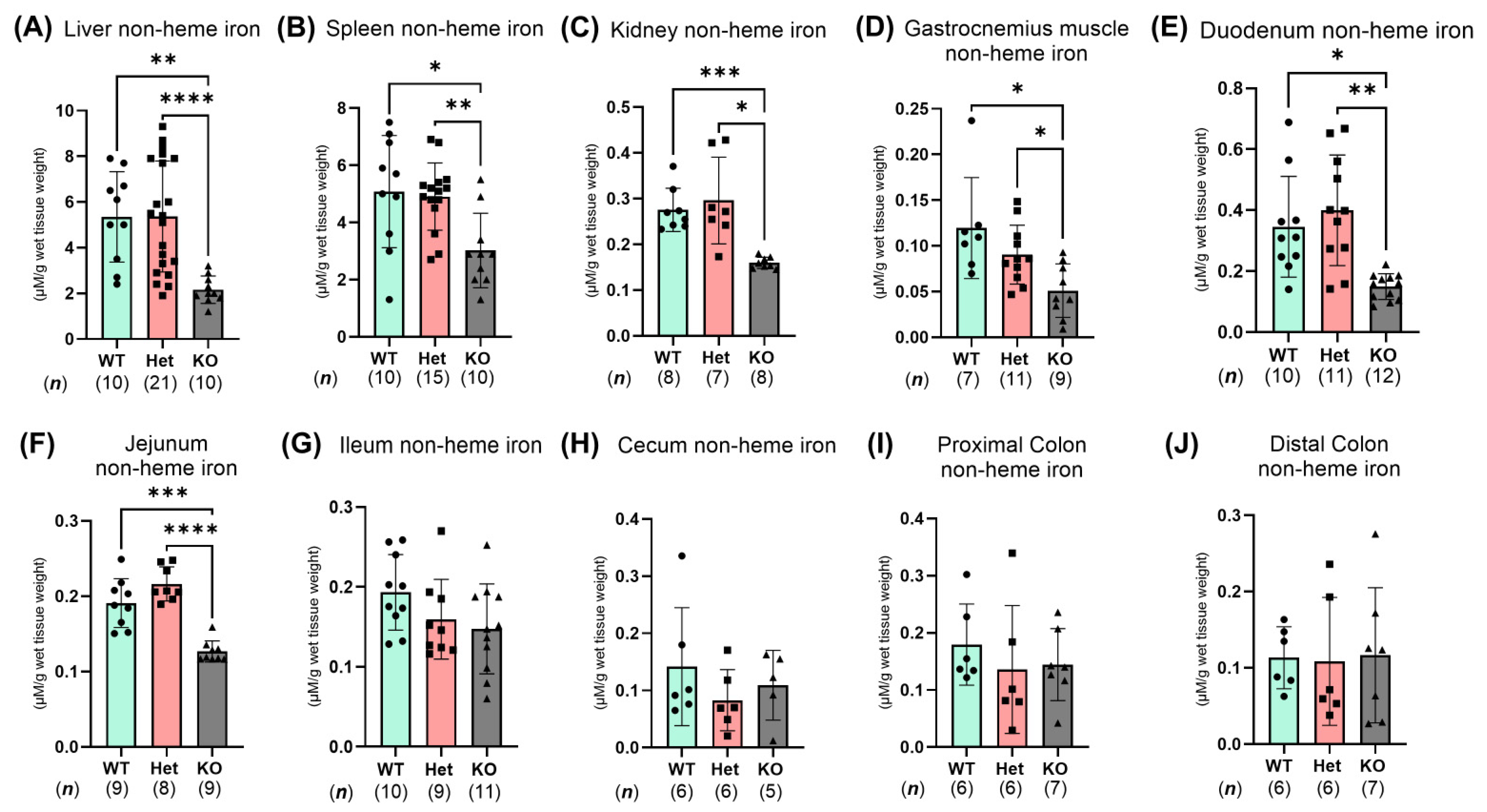
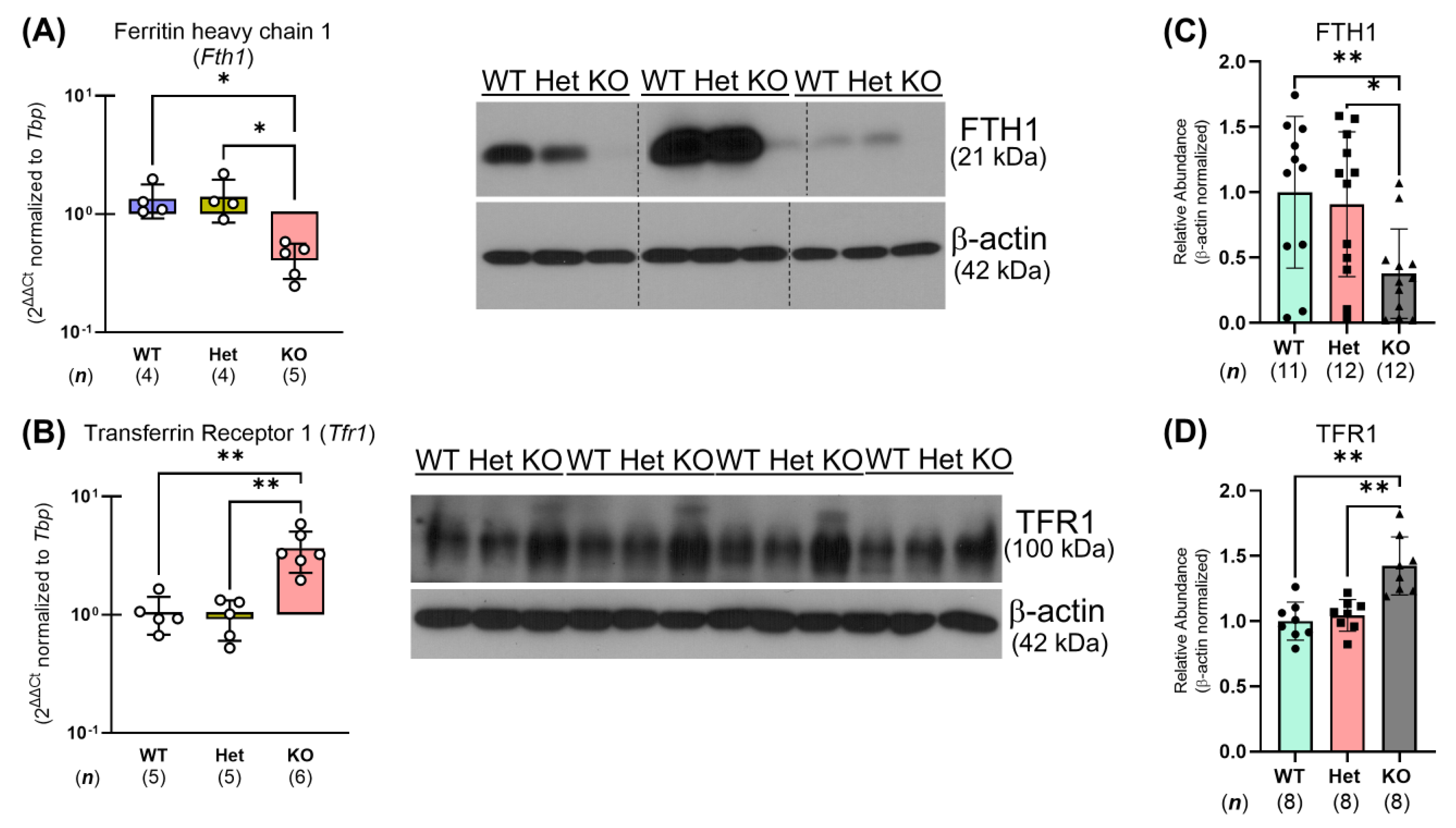
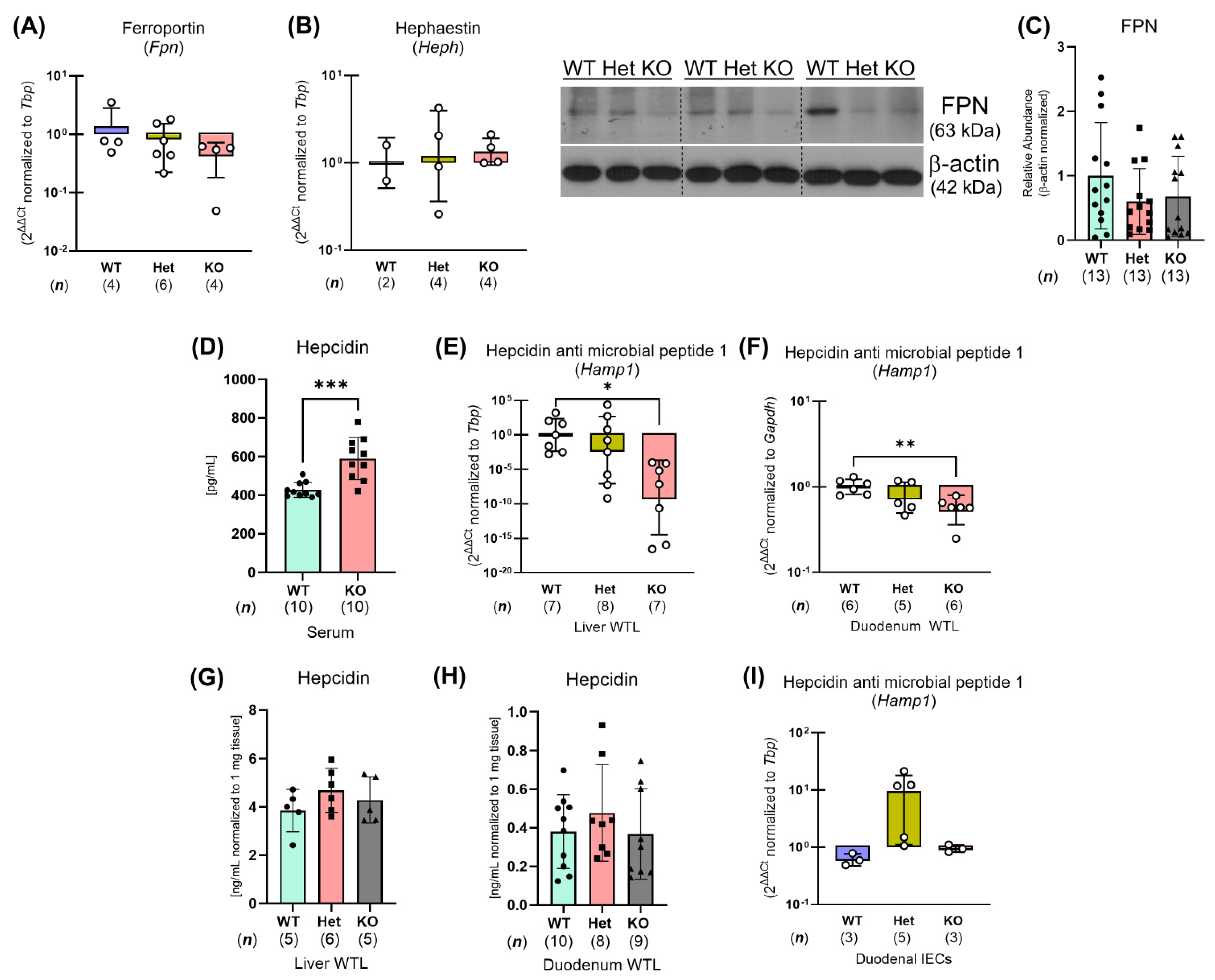
2.3. Ptpn2-KO Mice Have Reduced Iron Storage and Impaired Small Intestinal Iron Homeostasis
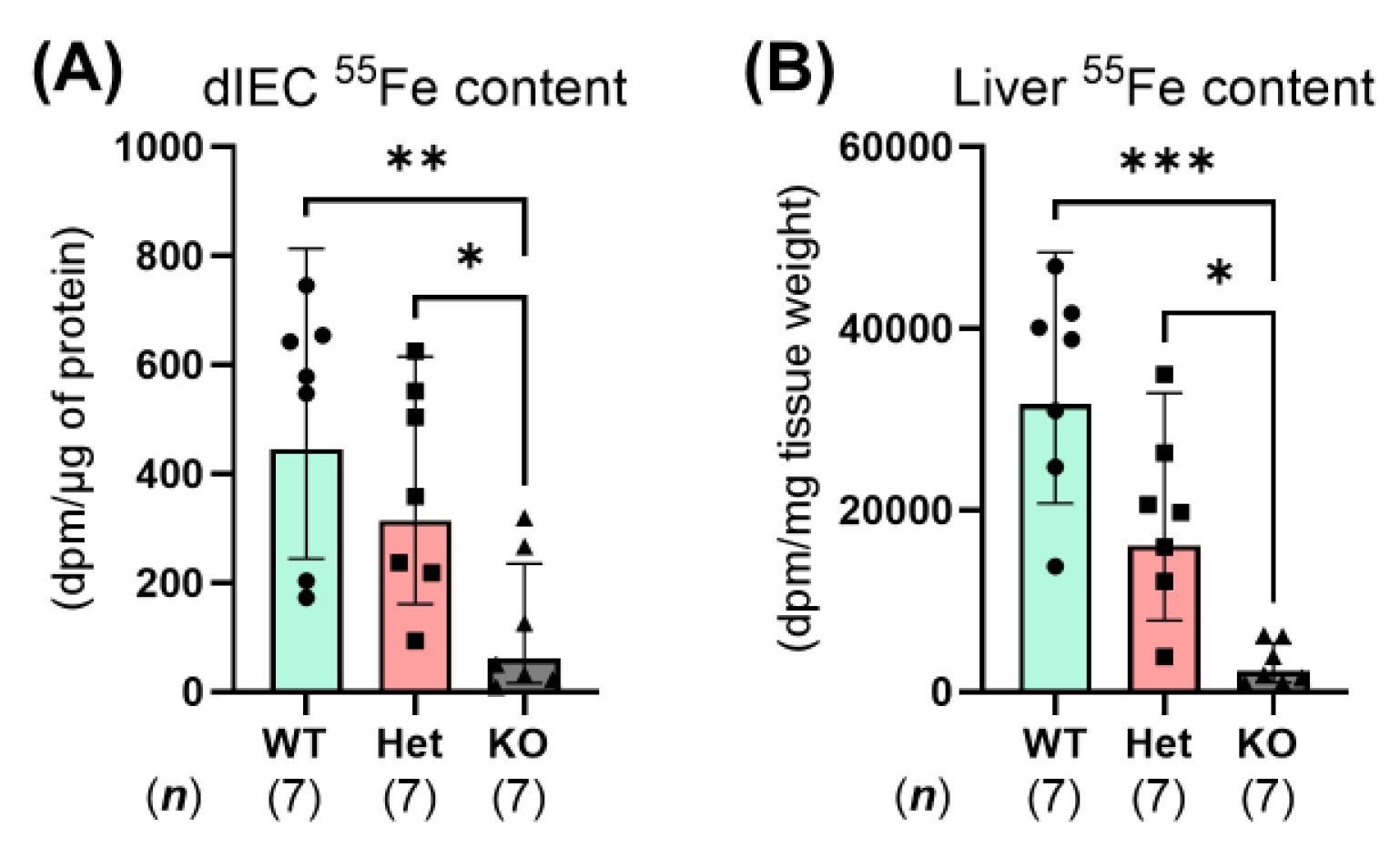
2.4. Constitutive Ptpn2 Deficiency in Mice Reduces Duodenal Iron Absorption
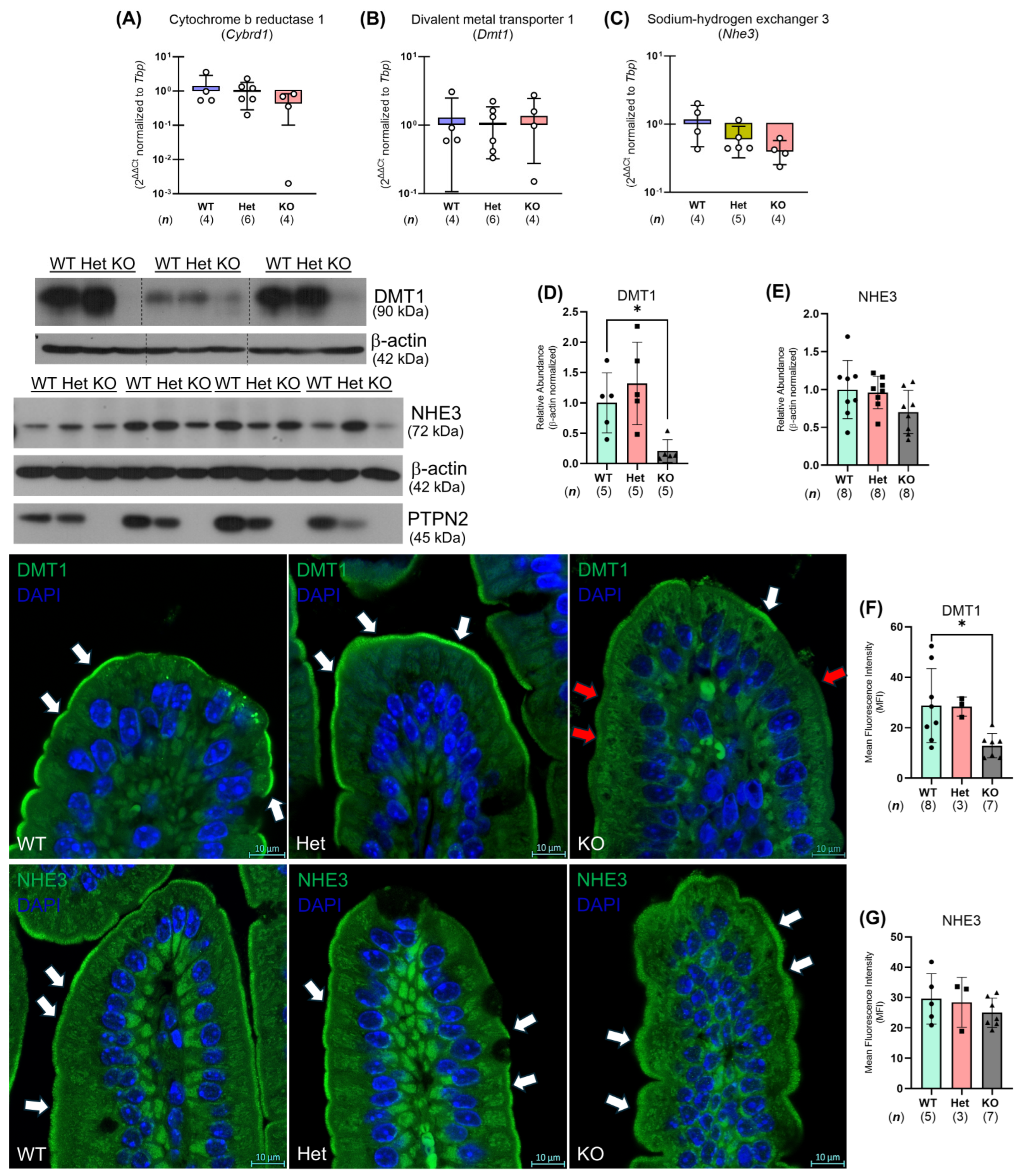
2.5. Ptpn2-KO Mouse Duodenal Enterocytes Show Alterations in Iron Handling Proteins
3. Discussion
4. Materials and Methods
4.1. Swiss IBD Cohort Patient Sample Analysis
4.2. Animals
4.2.1. Animal Experiments
4.2.2. Mouse Housing and Husbandry
4.2.3. Blood and Tissue Analyses
4.2.4. Duodenal Epithelial Cell Isolation
4.3. qPCR
4.4. Western Blot
4.5. Imaging
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. In Vivo Radiotracer Iron Assays
4.8. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Michelle, K.; Christoph, J. Management of Iron Deficiency Anaemia in Inflammatory Bowel Disease. Acta Haematol. 2019, 142, 30–36. [Google Scholar] [CrossRef]
- Blaney, H.; Vu, P.; Mathew, A.; Snelling, R.; England, J.; Duong, C.; Hammonds, K.; Johnson, C. Anemia Severity Associated with Increased Healthcare Utilization and Costs in Inflammatory Bowel Disease. Dig. Dis. Sci. 2021, 66, 2555–2563. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.; Aksan, A.; Farrag, K.; Dignass, A.; Radeke, H.H. Management of inflammatory bowel disease-related anemia and iron deficiency with specific reference to the role of intravenous iron in current practice. Expert Opin. Pharmacother. 2017, 18, 1721–1737. [Google Scholar] [CrossRef]
- Kaitha, S.; Bashir, M.; Ali, T. Iron deficiency anemia in inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2015, 6, 62. [Google Scholar] [CrossRef]
- Loveikyte, R.; Boer, M.; van der Meulen, C.N.; ter Steege, R.W.F.; Tack, G.; Kuyvenhoven, J.; Jharap, B.; Vu, M.K.; Vogelaar, L.; West, R.L.; et al. Anemia and Iron Deficiency in Outpatients with Inflammatory Bowel Disease: Ubiquitous Yet Suboptimally Managed. J. Clin. Med. 2022, 11, 6843. [Google Scholar] [CrossRef]
- Mücke, V.; Mücke, M.M.; Raine, T.; Bettenworth, D. Diagnosis and treatment of anemia in patients with inflammatory bowel disease. Ann. Gastroenterol. Q. Publ. Hell. Soc. Gastroenterol. 2017, 30, 15. [Google Scholar] [CrossRef]
- Katsarou, A.; Pantopoulos, K. Basics and principles of cellular and systemic iron homeostasis. Mol. Aspects Med. 2020, 75, 100866. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1434–1443. [Google Scholar] [CrossRef]
- Wiciński, M.; Liczner, G.; Cadelski, K.; Kołnierzak, T.; Nowaczewska, M.; Malinowski, B. Anemia of Chronic Diseases: Wider Diagnostics—Better Treatment? Nutrients 2020, 12, 1784. [Google Scholar] [CrossRef]
- Basseri, R.J.; Nemeth, E.; Vassilaki, M.E.; Basseri, B.; Enayati, P.; Shaye, O.; Bourikas, L.A.; Ganz, T.; Papadakis, K.A. Hepcidin is a key mediator of anemia of inflammation in Crohn’s disease. J. Crohn’s Colitis 2012, 7, e286–e291. [Google Scholar] [CrossRef]
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C.J. The T Cell Protein Tyrosine Phosphatase Is a Negative Regulator of Janus Family Kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef] [PubMed]
- de Jesús Ibarra-Sánchez, M.; Simoncic, P.D.; Nestel, F.R.; Duplay, P.; Lapp, W.S.; Tremblay, M.L. The T-cell protein tyrosine phosphatase. Semin. Immunol. 2000, 12, 379–386. [Google Scholar] [CrossRef]
- Glas, J.; Wagner, J.; Seiderer, J.; Olszak, T.; Wetzke, M.; Beigel, F.; Tillack, C.; Stallhofer, J.; Friedrich, M.; Steib, C.; et al. PTPN2 Gene Variants Are Associated with Susceptibility to Both Crohn’s Disease and Ulcerative Colitis Supporting a Common Genetic Disease Background. PLoS ONE 2012, 7, e33682. [Google Scholar] [CrossRef]
- Marcil, V.; Mack, D.R.; Kumar, V.; Faure, C.; Carlson, C.S.; Beaulieu, P.; Israel, D.; Krupoves, A.; Costea, I.; Lambrette, P.; et al. Association between the PTPN2 gene and Crohn’s disease: Dissection of potential causal variants. Inflamm. Bowel Dis. 2013, 19, 1149–1155. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Voegelin, M.; Biedermann, L.; Zeitz, J.; Rossel, J.B.; Sulz, M.C.; Frei, P.; Scharl, S.; Vavricka, S.R.; Fried, M.; et al. The clinical relevance of the IBD-associated variation within the risk gene locus encoding protein tyrosine phosphatase non-receptor type 2 in patients of the Swiss IBD cohort. Digestion 2016, 93, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.; Brandenburg, V.; Brunner-La Rocca, H.P. How to diagnose iron deficiency in chronic disease: A review of current methods and potential marker for the outcome. Eur. J. Med. Res. 2023, 28, 15. [Google Scholar] [CrossRef]
- Garcia-Casal, M.N.; Dary, O.; Jefferds, M.E.; Pasricha, S.R. Diagnosing anemia: Challenges selecting methods, addressing underlying causes, and implementing actions at the public health level. Ann. N. Y. Acad. Sci. 2023, 1524, 37–50. [Google Scholar] [CrossRef]
- Svenson, N.; Bailey, J.; Durairaj, S.; Dempsey-Hibbert, N. A simplified diagnostic pathway for the differential diagnosis of iron deficiency anaemia and anaemia of chronic disease. Int. J. Lab. Hematol. 2021, 43, 1644–1652. [Google Scholar] [CrossRef]
- You-Ten, K.E.; Muise, E.S.; Itié, A.; Michaliszyn, E.; Wagner, J.; Jothy, S.; Lapp, W.S.; Tremblay, M.L. Impaired Bone Marrow Microenvironment and Immune Function in T Cell Protein Tyrosine Phosphatase–deficient Mice. J. Exp. Med. 1997, 186, 683. [Google Scholar] [CrossRef]
- Chapman, J.; Goyal, A.; Azevedo, A.M. Splenomegaly. In Select 5-Minute Pediatrics Topics, 8th ed.; StatPearls Publishing: Treasure Island, FL, USA, 2023; pp. 870–871. [Google Scholar]
- Marchelletta, R.R.; Krishnan, M.; Spalinger, M.R.; Placone, T.W.; Alvarez, R.; Sayoc-Becerra, A.; Canale, V.; Shawki, A.; Park, Y.S.; Bernts, L.H.P.; et al. T cell protein tyrosine phosphatase protects intestinal barrier function by restricting epithelial tight junction remodeling. J. Clin. Investig. 2021, 131, 17. [Google Scholar] [CrossRef] [PubMed]
- Piskin, E.; Cianciosi, D.; Gulec, S.; Tomas, M.; Capanoglu, E. Iron Absorption: Factors, Limitations, and Improvement Methods. ACS Omega 2022, 7, 20441–20456. [Google Scholar] [CrossRef] [PubMed]
- Shawki, A.; Engevik, M.A.; Kim, R.S.; Knight, P.B.; Baik, R.A.; Anthony, S.R.; Worrell, R.T.; Shull, G.E.; Mackenzie, B. Intestinal brush-border Na+/H+ exchanger-3 drives H+-coupled iron absorption in the mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G423–G430. [Google Scholar] [CrossRef] [PubMed]
- Shawki, A.; Anthony, S.R.; Nose, Y.; Engevik, M.A.; Niespodzany, E.J.; Barrientos, T.; Öhrvik, H.; Worrell, R.T.; Thiele, D.J.; Mackenzie, B. Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G635–G647. [Google Scholar] [CrossRef]
- Maio, N.; Zhang, D.L.; Ghosh, M.C.; Jain, A.; SantaMaria, A.M.; Rouault, T.A. Mechanisms of cellular iron sensing, regulation of erythropoiesis and mitochondrial iron utilization. Semin. Hematol. 2021, 58, 161. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Musallam, K.M.; Taher, A.T. Iron deficiency anaemia revisited. J. Intern. Med. 2020, 287, 153–170. [Google Scholar] [CrossRef]
- Matusiewicz, M.; Neubauer, K.; Lewandowska, P.; Gamian, A.; Krzystek-Korpacka, M. Reduced transferrin levels in active inflammatory bowel disease. BioMed Res. Int. 2017, 2017, 9541370. [Google Scholar] [CrossRef]
- Oustamanolakis, P.; Koutroubakis, I.E.; Kouroumalis, E.A. Diagnosing anemia in inflammatory bowel disease: Beyond the established markers. J. Crohn’s Colitis 2011, 5, 381–391. [Google Scholar] [CrossRef]
- Ruan, B.; Paulson, R.F. Metabolic regulation of stress erythropoiesis, outstanding questions, and possible paradigms. Front. Physiol. 2023, 13, 1063294. [Google Scholar] [CrossRef]
- Marques, O.; Weiss, G.; Muckenthaler, M.U. The role of iron in chronic inflammatory diseases: From mechanisms to treatment options in anemia of inflammation. Blood 2022, 140, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Guo, L.; Gao, W.; Tang, T.L.; Yan, M. Interaction between macrophages and ferroptosis. Cell Death Dis. 2022, 13, 355. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Sayoc-Becerra, A.; Santos, A.N.; Shawki, A.; Canale, V.; Krishnan, M.; Niechcial, A.; Obialo, N.; Scharl, M.; Li, J.; et al. PTPN2 Regulates Interactions Between Macrophages and Intestinal Epithelial Cells to Promote Intestinal Barrier Function. Gastroenterology 2020, 159, 1763–1777.e14. [Google Scholar] [CrossRef]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 146258. [Google Scholar] [CrossRef]
- Scharl, M.; McCole, D.F.; Weber, A.; Vavricka, S.R.; Frei, P.; Kellermeier, S.; Pesch, T.; Fried, M.; Rogler, G. Protein tyrosine phosphatase N2 regulates TNFα-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut 2011, 60, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Scharl, M.; Hruz, P.; McCole, D.F. Protein tyrosine phosphatase non-receptor Type 2 regulates IFN-γ-induced cytokine signaling in THP-1 monocytes. Inflamm. Bowel Dis. 2010, 16, 2055–2064. [Google Scholar] [CrossRef]
- Delanghe, J.R.; Langlois, M.R. Hemopexin: A review of biological aspects and the role in laboratory medicine. Clin. Chim. Acta 2001, 312, 13–23. [Google Scholar] [CrossRef]
- Peslova, G.; Petrak, J.; Kuzelova, K.; Hrdy, I.; Halada, P.; Kuchel, P.W.; Soe-Lin, S.; Ponka, P.; Sutak, R.; Becker, E.; et al. Hepcidin, the hormone of iron metabolism, is bound specifically to alpha-2-macroglobulin in blood. Blood 2009, 113, 6225–6236. [Google Scholar] [CrossRef]
- Fernández, B.; Fdez, E.; Gómez-Suaga, P.; Gil, F.; Molina-Villalba, I.; Ferrer, I.; Patel, S.; Churchill, G.C.; Hilfiker, S. Iron overload causes endolysosomal deficits modulated by NAADP-regulated 2-pore channels and RAB7A. Autophagy 2016, 12, 1487. [Google Scholar] [CrossRef]
- Ma, J.; Guo, Q.; Shen, M.Q.; Li, W.; Zhong, Q.X.; Qian, Z.M. Apolipoprotein E is required for brain iron homeostasis in mice. Redox Biol. 2023, 64, 102779. [Google Scholar] [CrossRef]
- Wei, Y.H.; He, Y.Z.; Guo, X.Y.; Lin, X.Y.; Zhu, H.B.; Guo, X.J. Investigation and Analysis of Iron-Deficiency Anemia Complicated by Splenomegaly. Int. J. Gen. Med. 2021, 14, 4155. [Google Scholar] [CrossRef] [PubMed]
- Cesta, M.F. Normal Structure, Function, and Histology of the Spleen. Toxicol. Pathol. 2006, 34, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Fillebeen, C.; Gkouvatsos, K.; Fragoso, G.; Calvé, A.; Garcia-Santos, D.; Buffler, M.; Becker, C.; Schümann, K.; Ponka, P.; Santos, M.M.; et al. Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 2015, 100, e334. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.; Anderson, G.J.; Collins, J.F. Mechanistic and regulatory aspects of intestinal iron absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, 397–409. [Google Scholar] [CrossRef]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2023, 25, 133–155. [Google Scholar] [CrossRef]
- D’Angelo, G. Role of hepcidin in the pathophysiology and diagnosis of anemia. Blood Res. 2013, 48, 10. [Google Scholar] [CrossRef]
- Heinonen, K.M.; Nestel, F.P.; Newell, E.W.; Charette, G.; Seemayer, T.A.; Tremblay, M.L.; Lapp, W.S. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 2004, 103, 3457–3464. [Google Scholar] [CrossRef]
- Dincer, M.T.; Dincer, Z.T.; Bakkaloglu, O.K.; Yalin, S.F.; Trabulus, S.; Celik, A.F.; Seyahi, N.; Altiparmak, M.R. Renal Manifestations in Inflammatory Bowel Disease: A Cohort Study During the Biologic Era. Med. Sci. Monit. 2022, 28, e936497-1–e936497-10. [Google Scholar] [CrossRef]
- Dignass, A.U.; Gasche, C.; Bettenworth, D.; Birgegård, G.; Danese, S.; Gisbert, J.P.; Gomollon, F.; Iqbal, T.; Katsanos, K.; Koutroubakis, I.; et al. European consensus on the diagnosis and management of iron deficiency and anaemia in inflammatory bowel diseases. J. Crohn’s Colitis 2015, 9, 211–222. [Google Scholar] [CrossRef]
- Foot, N.J.; Dalton, H.E.; Shearwin-Whyatt, L.M.; Dorstyn, L.; Tan, S.S.; Yang, B.; Kumar, S. Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2. Blood 2008, 112, 4268–4275. [Google Scholar] [CrossRef]
- Foot, N.J.; Leong, Y.A.; Dorstyn, L.E.; Dalton, H.E.; Ho, K.; Zhao, L.; Garrick, M.D.; Yang, B.; Hiwase, D.; Kumar, S. Ndfip1-deficient mice have impaired DMT1 regulation and iron homeostasis. Blood 2011, 117, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Song, Y.; He, C.; Liu, C.; Wu, R.; Fang, L.; Cong, Y.; Miao, Y.; Liu, Z. Divalent metal-ion transporter 1 is decreased in intestinal epithelial cells and contributes to the anemia in inflammatory bowel disease. Sci. Rep. 2015, 5, 16344. [Google Scholar] [CrossRef]
- Johnson, D.; Bayele, H.; Johnston, K.; Tennant, J.; Srai, S.K.; Sharp, P. Tumour necrosis factor alpha regulates iron transport and transporter expression in human intestinal epithelial cells. FEBS Lett. 2004, 573, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, T.H.; Roos, N.; Lifschitz, M.L. A Method for Measuring the Storage Iron Content of Formalinised Tissues. S. Afr. J. Med. Sci. 1964, 29, 21–26. [Google Scholar] [PubMed]
- Talbot, C.; Lytle, C. Segregation of Na/H exchanger-3 and Cl/HCO3 exchanger SLC26A3 (DRA) in rodent cecum and colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G358–G367. [Google Scholar] [CrossRef] [PubMed]
- Biemesderfer, D.; Rutherford, P.A.; Nagy, T.; Pizzonia, J.H.; Abu-Alfa, A.K.; Aronson, P.S. Monoclonal antibodies for high-resolution localization of NHE3 in adult and neonatal rat kidney. Am. J. Physiol. 1997, 273, F289–F299. [Google Scholar] [CrossRef]
- Donowitz, M.; Li, X. Regulatory binding partners and complexes of NHE3. Physiol. Rev. 2007, 87, 825–872. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007, 447, 661. [Google Scholar] [CrossRef]
- Festen, E.A.M.; Goyette, P.; Green, T.; Boucher, G.; Beauchamp, C.; Trynka, G.; Dubois, P.C.; Lagacé, C.; Stokkers, P.C.F.; Hommes, D.W.; et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn’s disease and celiac disease. PLoS Genet. 2011, 7, e1001283. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||||
| Crohn’s Disease | |||||
| Active | Quiescent | Total | |||
| CC allele | 3 | 7 | 10 | ||
| CT allele (Het) | 6 | 4 | 10 | ||
| TT allele (WT) | 6 | 4 | 10 | ||
| Sub-total | 15 | 15 | 30 | ||
| Ulcerative Colitis | |||||
| Active | Quiescent | Total | |||
| CC allele | 2 | 8 | 10 | ||
| CT allele (Het) | 6 | 4 | 10 | ||
| TT allele (WT) | 6 | 4 | 10 | ||
| Sub-total | 14 | 16 | 30 | ||
| (B) | |||||
| Variant | Diagnosis | Severity | Sex | Age | Montreal Classification |
| AA/TT | CD | Severe | M | 66 | B2p |
| AA/TT | CD | Severe | F | 29 | B1 |
| AA/TT | CD | Severe | F | 20 | B2 |
| AA/TT | CD | Severe | M | 21 | B2p |
| AA/TT | CD | Severe | F | 25 | B1 |
| AA/TT | CD | Moderate | F | 40 | B3 |
| AA/TT | CD | Quiescent | F | 66 | B1 |
| AA/TT | CD | Quiescent | M | 55 | B1 |
| AA/TT | CD | Quiescent | F | 34 | B2 |
| AA/TT | CD | Quiescent | F | 16 | B1 |
| GA/CT | CD | Severe | F | 24 | B1 |
| GA/CT | CD | Moderate | F | 35 | B2p |
| GA/CT | CD | Moderate | F | 60 | B1 |
| GA/CT | CD | Moderate | M | 36 | B1p |
| GA/CT | CD | Moderate | F | 31 | B2 |
| GA/CT | CD | Mild | M | 42 | B2 |
| GA/CT | CD | Quiescent | F | 50 | B1 |
| GA/CT | CD | Quiescent | M | 40 | B1 |
| GA/CT | CD | Quiescent | F | 62 | B1 |
| GA/CT | CD | Quiescent | F | 67 | B2p |
| GG/CC | CD | Severe | M | 67 | B2 |
| GG/CC | CD | Moderate | F | 47 | B2 |
| GG/CC | CD | Moderate | F | 41 | B1p |
| GG/CC | CD | Quiescent | M | 34 | B1p |
| GG/CC | CD | Quiescent | M | 25 | B1p |
| GG/CC | CD | Quiescent | M | 76 | B1 |
| GG/CC | CD | Quiescent | F | 42 | B1p |
| GG/CC | CD | Quiescent | M | 70 | B2 |
| GG/CC | CD | Quiescent | F | 13 | B1 |
| GG/CC | CD | Quiescent | M | 14 | B1 |
| (C) | |||||
| Variant | Diagnosis | Severity | Sex | Age | |
| AA/TT | UC | Severe | M | 31 | |
| AA/TT | UC | Severe | M | 30 | |
| AA/TT | UC | Severe | M | 54 | |
| AA/TT | UC | Severe | F | 20 | |
| AA/TT | UC | Moderate | M | 35 | |
| AA/TT | UC | Moderate | M | 40 | |
| AA/TT | UC | Quiescent | M | 47 | |
| AA/TT | UC | Quiescent | F | 15 | |
| AA/TT | UC | Quiescent | F | 26 | |
| AA/TT | UC | Quiescent | M | 50 | |
| GA/CT | UC | Moderate | F | 26 | |
| GA/CT | UC | Moderate | M | 18 | |
| GA/CT | UC | Moderate | F | 53 | |
| GA/CT | UC | Moderate | M | 32 | |
| GA/CT | UC | Mild | F | 52 | |
| GA/CT | UC | Mild | M | 51 | |
| GA/CT | UC | Quiescent | F | 45 | |
| GA/CT | UC | Quiescent | M | 57 | |
| GA/CT | UC | Quiescent | M | 25 | |
| GA/CT | UC | Quiescent | M | 13 | |
| GG/CC | UC | Moderate | M | 47 | |
| GG/CC | UC | Moderate | M | 43 | |
| GG/CC | UC | Quiescent | F | 10 | |
| GG/CC | UC | Quiescent | M | 50 | |
| GG/CC | UC | Quiescent | F | 48 | |
| GG/CC | UC | Quiescent | F | 45 | |
| GG/CC | UC | Quiescent | M | 52 | |
| GG/CC | UC | Quiescent | M | 49 | |
| GG/CC | UC | Quiescent | M | 31 | |
| GG/CC | UC | Quiescent | F | 16 | |
| Gene | Primer Sequence | GC% | Length | Tm | RefSeq |
|---|---|---|---|---|---|
| Cybrd1 | F: CAGTGATTGCGACGGTTCTC | 55% | 20 | 56.1 °C | AF354666.1 |
| R: ATGGTACGAGGGGTGTTTCA | 50% | 20 | 56.0 °C | ||
| Dmt1 | F: CGCTCGGTAAGCATCTCGAA | 55% | 20 | 57.2 °C | NM_008732.2 |
| R: TGTTGCCACCGCTGGTATCT | 55% | 20 | 59.2 °C | ||
| Fpn | F: CATTGCTAGAATCGGTCTT | 42.1% | 19 | 49.6 °C | NM_016917.2 |
| R: GCAACTGTGTCACCGTCAAAT | 57.6% | 21 | 56.1 °C | ||
| Fth1 | F: TGGAGTTGTATGCCTCCTACG | 52.4% | 21 | 56.3 °C | NM_010239.2 |
| R: TGGAGAAAGTATTTGGCAAAGTT | 34.8% | 23 | 53.0 °C | ||
| Hamp1 | F: AAGCAGGGCAGACATTGCGAT | 52.4% | 21 | 59.8 °C | AF503444.1 |
| R: CAGGATGTGGCTCTAGGCTATGT | 52.2% | 23 | 58.2 °C | ||
| Heph | F:TAATCCCCGCCAGACAGGA | 57.9% | 19 | 58.1 °C | NM_001159627.1 and NM_181273.4 |
| R:GCCTTCAGCTTATTTACCTTGTT | 39.1% | 23 | 53.5 °C | ||
| Nhe3 | F: GAGGAGGAACCGAGCAGTGA | 60% | 20 | 58.8 °C | NM_001081060.2 and XM_006517022.5 |
| R: GTGGGACAGGTGAAAGACGATT | 50% | 22 | 57.1 °C | ||
| Tfr1 | F: TGGAATCCCAGCAGTTTCTT | 45% | 20 | 54.3 °C | NM_001357298.1 |
| R: GCTGCTGTACGAACCATTTG | 50% | 20 | 54.5 °C | ||
| Gapdh (RG) | F: GTTTGTGATGGGTGTGAACCACG | 52.2% | 23 | 58.8 °C | NM_001289726.1 |
| R: GTGGCAGTGATGGCATGGAC | 60% | 20 | 59 °C | ||
| Tbp (RG) | F: CCTTGTACCCTTCACCAATGAC | 50% | 22 | 55.6 °C | NM_013684.3 |
| R: ACAGCCAAGATTCACGGTAGA | 47.6% | 21 | 56.0 °C |
| Antibody | Source | Catalog # |
|---|---|---|
| DMT1 | Dr. Francois Canonne-Hergaux (INSERM) | N/A |
| FPN | Thermofisher | PA5-22993 |
| FTH1 | Thermofisher | MA5-32244 |
| NHE3 | EMD Millipore (Burlington, MA, USA) | AB3085 |
| PTPN2 | Cell Signaling (Danvers, MA, USA) | 58935 |
| TFR1 | Thermofisher | PA5-110480 |
| β-actin | Sigma-Aldrich | A5316 |
| Antibody | Source | Catalog # | Antigen Retrieval Buffer | Primary Concentration |
|---|---|---|---|---|
| DMT1 | Dr. Francois Canonne-Hergaux (INSERM) | N/A | Tris-EGTA pH 9 | 1:100 |
| NHE3 | EMD Millipore | AB3085 | Tris-EGTA pH 9 | 1:100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, H.; Shawki, A.; Santos, A.N.; Canale, V.; Manz, S.; Crawford, M.S.; Chatterjee, P.; Spalinger, M.R.; Scharl, M.; McCole, D.F. PTPN2 Regulates Iron Handling Protein Expression in Inflammatory Bowel Disease Patients and Prevents Iron Deficiency in Mice. Int. J. Mol. Sci. 2025, 26, 3356. https://doi.org/10.3390/ijms26073356
Lei H, Shawki A, Santos AN, Canale V, Manz S, Crawford MS, Chatterjee P, Spalinger MR, Scharl M, McCole DF. PTPN2 Regulates Iron Handling Protein Expression in Inflammatory Bowel Disease Patients and Prevents Iron Deficiency in Mice. International Journal of Molecular Sciences. 2025; 26(7):3356. https://doi.org/10.3390/ijms26073356
Chicago/Turabian StyleLei, Hillmin, Ali Shawki, Alina N. Santos, Vinicius Canale, Salomon Manz, Meli’sa S. Crawford, Pritha Chatterjee, Marianne R. Spalinger, Michael Scharl, and Declan F. McCole. 2025. "PTPN2 Regulates Iron Handling Protein Expression in Inflammatory Bowel Disease Patients and Prevents Iron Deficiency in Mice" International Journal of Molecular Sciences 26, no. 7: 3356. https://doi.org/10.3390/ijms26073356
APA StyleLei, H., Shawki, A., Santos, A. N., Canale, V., Manz, S., Crawford, M. S., Chatterjee, P., Spalinger, M. R., Scharl, M., & McCole, D. F. (2025). PTPN2 Regulates Iron Handling Protein Expression in Inflammatory Bowel Disease Patients and Prevents Iron Deficiency in Mice. International Journal of Molecular Sciences, 26(7), 3356. https://doi.org/10.3390/ijms26073356