
Cannabinerol Restores mRNA Splicing Defects Induced by β-Amyloid in an In Vitro Model of Alzheimer’s Disease: A Transcriptomic Study

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. CBNR Pre-Treatment Regulates Splicing-Related Biological Processes
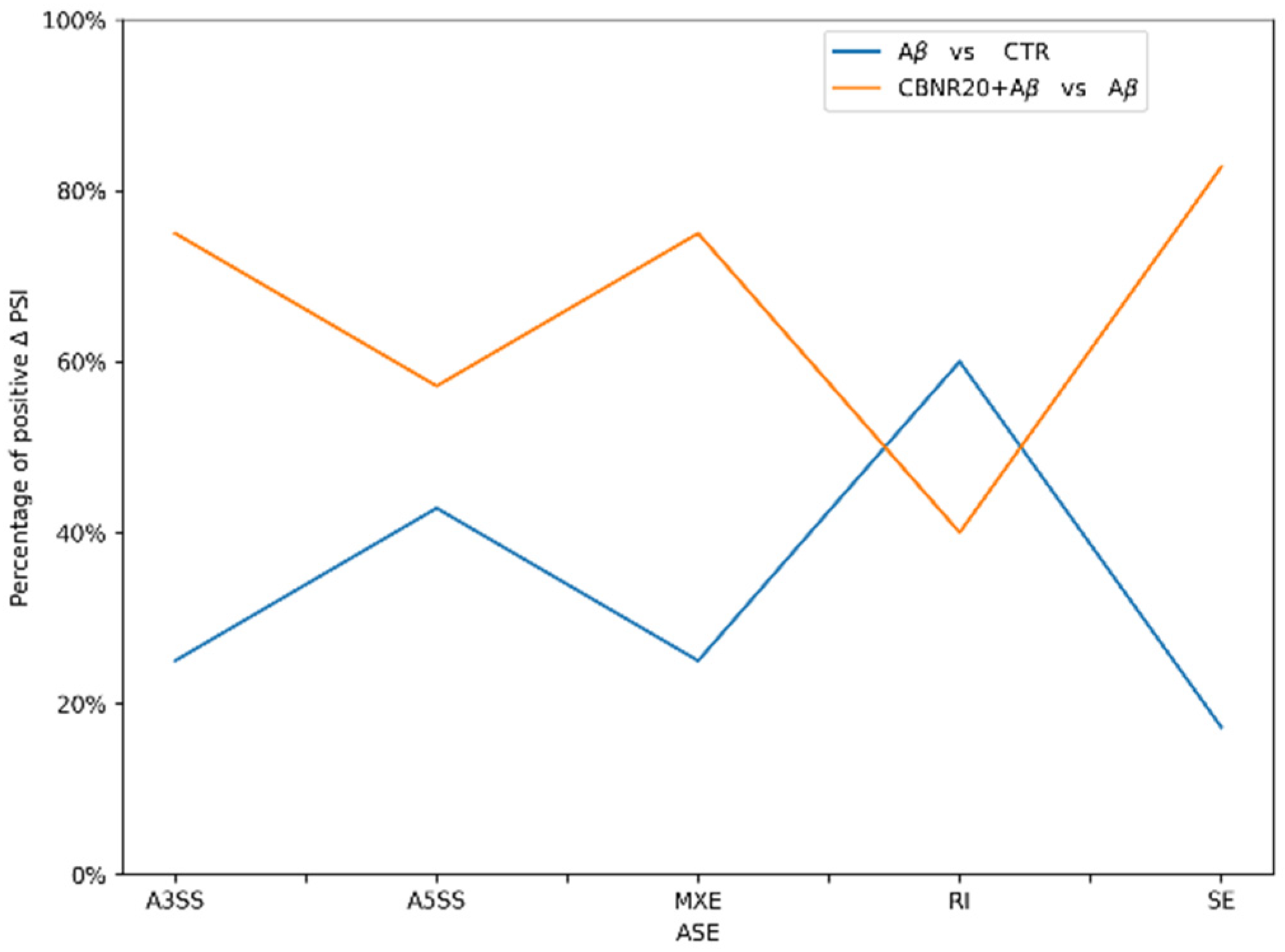
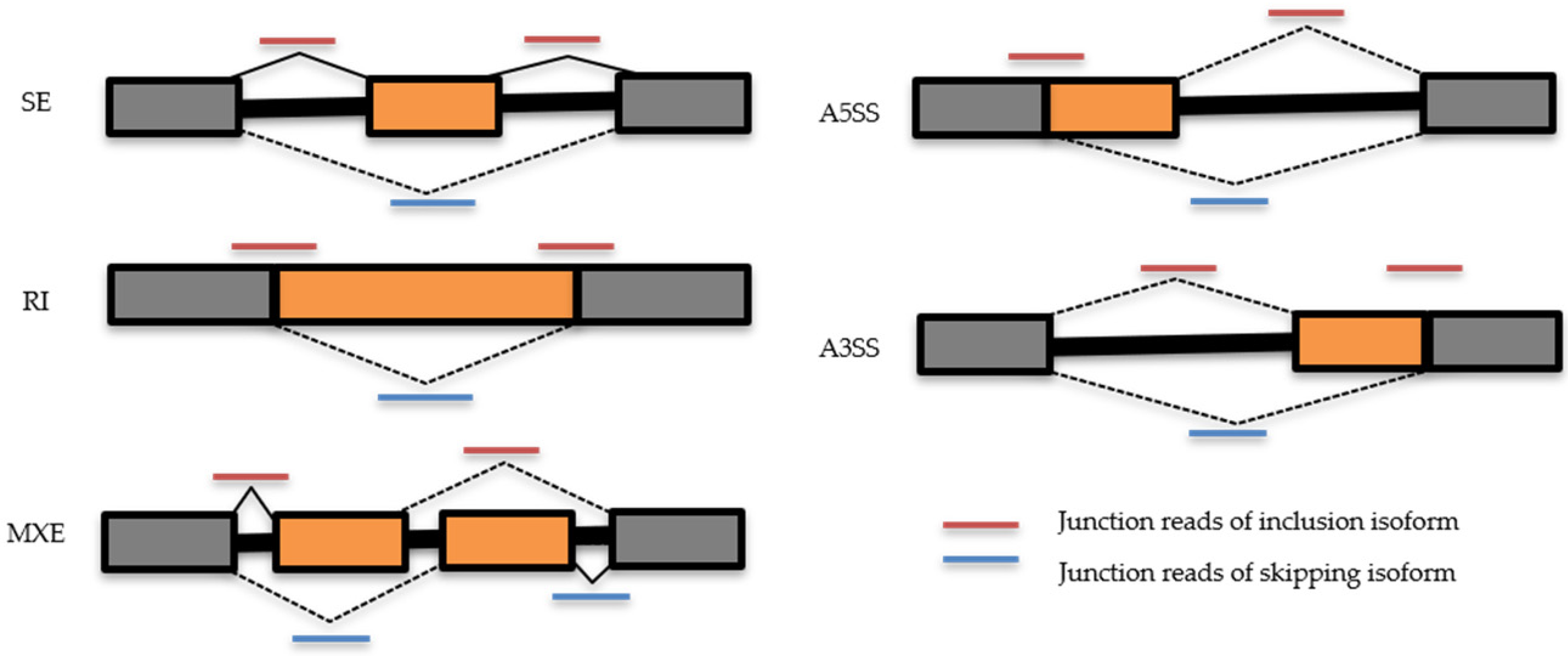
2.2. Effects of CBNR Pre-Treatment on Alternative mRNA Splicing
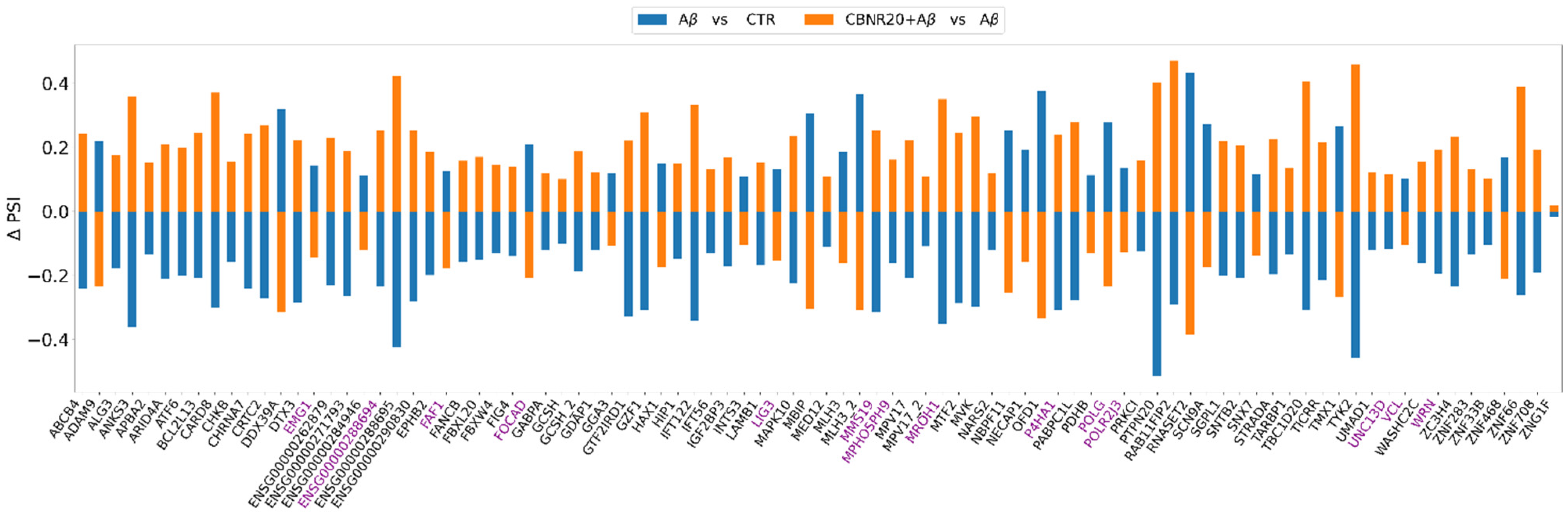
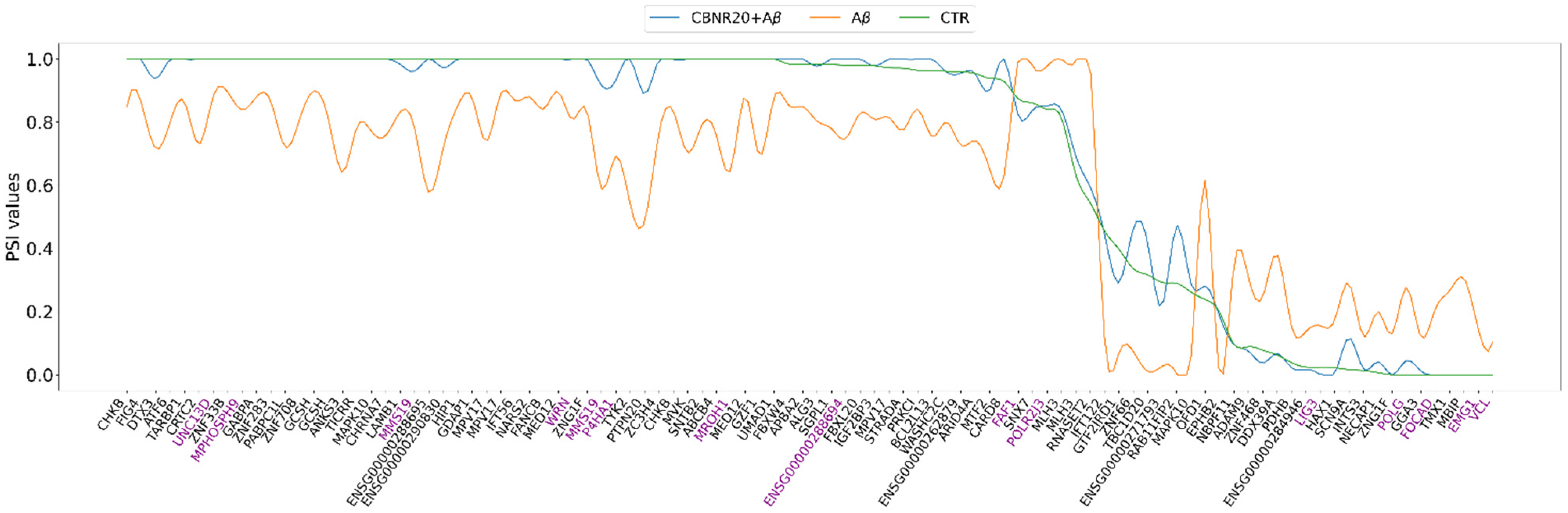
2.3. CBNR Pre-Treatment Counteracts Aβ-Induced mRNA Splicing Defects
2.4. RI Events of Shared DASEs Include Premature Stop Codons
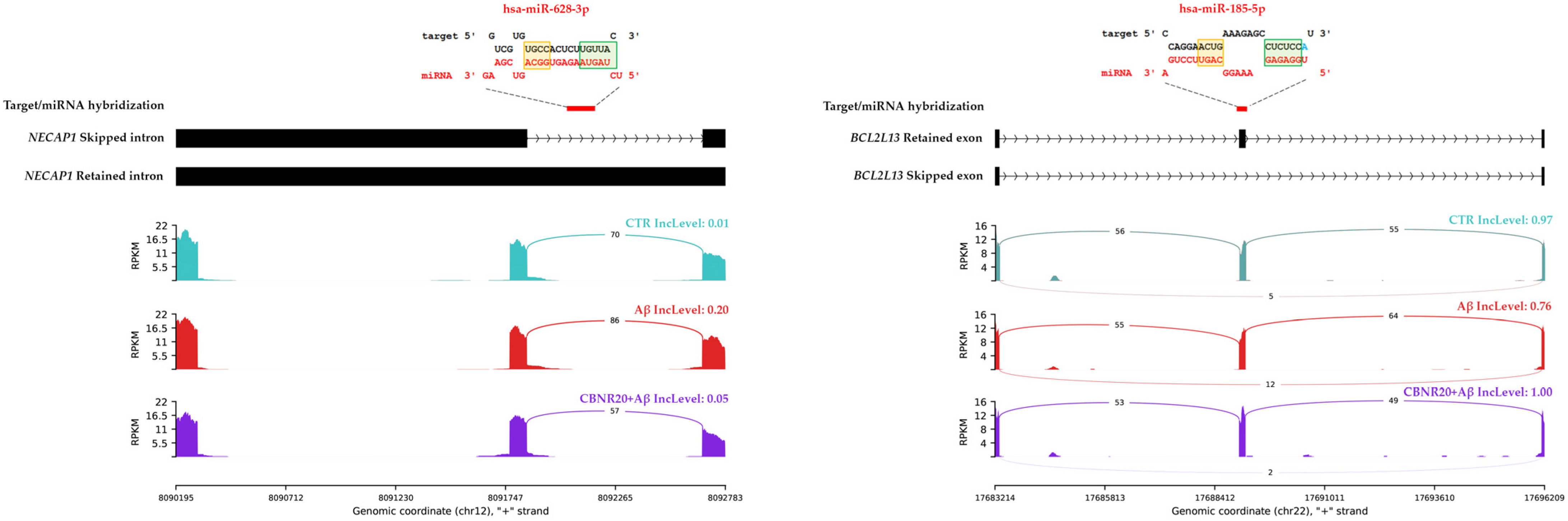
2.5. DASE Regions Results to Be Targeted by Brain- and AD-Associated miRNAs
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Treatment and Transcriptomic Analysis
4.2. Gene Ontology Analysis of DEGs
4.3. Differential Alternative Splicing Events Analysis
4.4. RI Premature Stop Codon Identification
4.5. DASE/miRNA Hybridization Analysis
miRNA-DASE Hybrids Classification and Filtering
4.6. lncRNA Mapping to DASE
4.7. Sashimi Plot Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| A3SS | alternative 3′ splice site |
| A5SS | alternative 5′ splice site |
| AS | alternative splicing |
| Aβ | β-amyloid |
| BP | biological process |
| CBD | cannabidiol |
| CBG | cannabigerol |
| CBNR | cannabinerol |
| DAG | directed acyclic graph |
| DASE | differential alternative splicing event |
| DEG | differentially expressed gene |
| FDR | false discovery rate |
| GO | Gene Ontology |
| GWAS | genome-wide association studies |
| IGV | Integrative Genomics Viewer |
| lncRNA | long non-coding RNA |
| MFE | Minimum Free Energy |
| miRNA | microRNA |
| MXE | mutually exclusive exon |
| ncRNA | non-coding RNA |
| NMD | nonsense-mediated decay |
| ORA | over-representation analysis |
| ORF | open reading frame |
| PSI | Percent Spliced In |
| RA | retinoic acid |
| RI | retained intron |
| rMATS | replicate Multivariate Analysis of Transcript Splicing |
| SE | skipped exon |
| sncRNAs | small non-coding RNAs |
| Δ8-THC | ∆8-tetrahydrocannabinol |
| Δ9-THC | Δ9-tetrahydrocannabinol |
References
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Lee, H.; Hsieh, Y.C.; Chou, V.; Selkoe, D.J. Moving beyond amyloid and tau to capture the biological heterogeneity of Alzheimer’s disease. Trends Neurosci. 2023, 46, 426–444. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef]
- Heyd, F.; Lynch, K.W. Degrade, move, regroup: Signaling control of splicing proteins. Trends Biochem. Sci. 2011, 36, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Braunschweig, U.; Gonatopoulos-Pournatzis, T.; Weatheritt, R.J.; Hirsch, C.L.; Ha, K.C.H.; Radovani, E.; Nabeel-Shah, S.; Sterne-Weiler, T.; Wang, J.; et al. Multilayered Control of Alternative Splicing Regulatory Networks by Transcription Factors. Mol. Cell 2017, 65, 539–553.e7. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zhang, Q.; Wang, H.; Yang, X.; Mu, H. Alternative splicing and related RNA binding proteins in human health and disease. Signal Transduct. Target. Ther. 2024, 9, 26. [Google Scholar] [CrossRef]
- Orengo, J.P.; Cooper, T.A. Alternative splicing in disease. Adv. Exp. Med. Biol. 2007, 623, 212–223. [Google Scholar] [CrossRef]
- Ishunina, T.A. Alternative splicing in aging and Alzheimer’s disease: Highlighting the role of tau and estrogen receptor alpha isoforms in the hypothalamus. Handb. Clin. Neurol. 2021, 182, 177–189. [Google Scholar] [CrossRef]
- Nikom, D.; Zheng, S. Alternative splicing in neurodegenerative disease and the promise of RNA therapies. Nat. Rev. Neurosci. 2023, 24, 457–473. [Google Scholar] [CrossRef]
- Bai, B.; Hales, C.M.; Chen, P.C.; Gozal, Y.; Dammer, E.B.; Fritz, J.J.; Wang, X.; Xia, Q.; Duong, D.M.; Street, C.; et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16562–16567. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Dammer, E.B.; Diner, I.; Yi, H.; Seyfried, N.T.; Gearing, M.; Glass, J.D.; Montine, T.J.; Levey, A.I.; Lah, J.J. Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 2014, 24, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Bishof, I.; Dammer, E.B.; Duong, D.M.; Kundinger, S.R.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. RNA-binding proteins with basic-acidic dipeptide (BAD) domains self-assemble and aggregate in Alzheimer’s disease. J. Biol. Chem. 2018, 293, 11047–11066. [Google Scholar] [CrossRef]
- Hsieh, Y.C.; Guo, C.; Yalamanchili, H.K.; Abreha, M.; Al-Ouran, R.; Li, Y.; Dammer, E.B.; Lah, J.J.; Levey, A.I.; Bennett, D.A.; et al. Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep. 2019, 29, 301–316.e10. [Google Scholar] [CrossRef]
- Adusumalli, S.; Ngian, Z.K.; Lin, W.Q.; Benoukraf, T.; Ong, C.T. Increased intron retention is a post-transcriptional signature associated with progressive aging and Alzheimer’s disease. Aging Cell 2019, 18, e12928. [Google Scholar] [CrossRef]
- Ngian, Z.K.; Tan, Y.Y.; Choo, C.T.; Lin, W.Q.; Leow, C.Y.; Mah, S.J.; Lai, M.K.; Chen, C.L.; Ong, C.T. Truncated Tau caused by intron retention is enriched in Alzheimer’s disease cortex and exhibits altered biochemical properties. Proc. Natl. Acad. Sci. USA 2022, 119, e2204179119. [Google Scholar] [CrossRef]
- Tan, S.; Guo, J.; Huang, Q.; Chen, X.; Li-Ling, J.; Li, Q.; Ma, F. Retained introns increase putative microRNA targets within 3′ UTRs of human mRNA. FEBS Lett. 2007, 581, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.G.; Smith, C.W.J. Intron retention as a component of regulated gene expression programs. Human. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef]
- Abate, G.; Uberti, D.; Tambaro, S. Potential and Limits of Cannabinoids in Alzheimer’s Disease Therapy. Biology 2021, 10, 542. [Google Scholar] [CrossRef]
- Stone, N.L.; Murphy, A.J.; England, T.J.; O’Sullivan, S.E. A systematic review of minor phytocannabinoids with promising neuroprotective potential. Br. J. Pharmacol. 2020, 177, 4330–4352. [Google Scholar] [CrossRef] [PubMed]
- Voicu, V.; Brehar, F.M.; Toader, C.; Covache-Busuioc, R.A.; Corlatescu, A.D.; Bordeianu, A.; Costin, H.P.; Bratu, B.G.; Glavan, L.A.; Ciurea, A.V. Cannabinoids in Medicine: A Multifaceted Exploration of Types, Therapeutic Applications, and Emerging Opportunities in Neurodegenerative Diseases and Cancer Therapy. Biomolecules 2023, 13, 1388. [Google Scholar] [CrossRef]
- Gugliandolo, A.; Blando, S.; Salamone, S.; Caprioglio, D.; Pollastro, F.; Mazzon, E.; Chiricosta, L. Delta(8)-THC Protects against Amyloid Beta Toxicity Modulating ER Stress In Vitro: A Transcriptomic Analysis. Int. J. Mol. Sci. 2023, 24, 6598. [Google Scholar] [CrossRef] [PubMed]
- Chiricosta, L.; Minuti, A.; Gugliandolo, A.; Salamone, S.; Pollastro, F.; Mazzon, E.; Artimagnella, O. Cannabinerol Prevents Endoplasmic Reticulum and Mitochondria Dysfunctions in an In Vitro Model of Alzheimer’s Disease: A Network-Based Transcriptomic Analysis. Cells 2024, 13, 1012. [Google Scholar] [CrossRef]
- Scherma, M.; Qvist, J.S.; Asok, A.; Huang, S.C.; Masia, P.; Deidda, M.; Wei, Y.B.; Soni, R.K.; Fratta, W.; Fadda, P.; et al. Cannabinoid exposure in rat adolescence reprograms the initial behavioral, molecular, and epigenetic response to cocaine. Proc. Natl. Acad. Sci. USA 2020, 117, 9991–10002. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Z.; Kutschera, E.; Adams, J.I.; Kadash-Edmondson, K.E.; Xing, Y. rMATS-turbo: An efficient and flexible computational tool for alternative splicing analysis of large-scale RNA-seq data. Nat. Protoc. 2024, 19, 1083–1104. [Google Scholar] [CrossRef] [PubMed]
- Belgrader, P.; Cheng, J.; Zhou, X.; Stephenson, L.S.; Maquat, L.E. Mammalian nonsense codons can be cis effectors of nuclear mRNA half-life. Mol. Cell. Biol. 1994, 14, 8219–8228. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Rosler, T.W.; Tayaranian Marvian, A.; Brendel, M.; Nykanen, N.P.; Hollerhage, M.; Schwarz, S.C.; Hopfner, F.; Koeglsperger, T.; Respondek, G.; Schweyer, K.; et al. Four-repeat tauopathies. Progress. Neurobiol. 2019, 180, 101644. [Google Scholar] [CrossRef]
- Lejeune, F. Nonsense-Mediated mRNA Decay, a Finely Regulated Mechanism. Biomedicines 2022, 10, 141. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.B.; Fu, Q.; Guo, M.; Du, Y.; Chen, Y.; Cheng, Y. MicroRNAs: Pioneering regulators in Alzheimer’s disease pathogenesis, diagnosis, and therapy. Transl. Psychiatry 2024, 14, 367. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Y.; Chen, C.; Sun, Z.; Liu, E.; Wei, N.; Liu, X.; Shu, J.; Zhao, N.; Sun, M. Long Non-Coding RNAs: Crucial Regulators in Alzheimer’s Disease Pathogenesis and Prospects for Precision Medicine. Mol. Neurobiol. 2025; Online ahead of print. [Google Scholar] [CrossRef]
- Chipman, L.B.; Pasquinelli, A.E. miRNA Targeting: Growing beyond the Seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Riolo, G.; Cantara, S.; Marzocchi, C.; Ricci, C. miRNA Targets: From Prediction Tools to Experimental Validation. Methods Protoc. 2020, 4, 1. [Google Scholar] [CrossRef]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical Aspects of microRNA Target Prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef]
- Iwakawa, H.O.; Tomari, Y. Molecular insights into microRNA-mediated translational repression in plants. Mol. Cell 2013, 52, 591–601. [Google Scholar] [CrossRef]
- Madan, S.; Kuhnel, L.; Frohlich, H.; Hofmann-Apitius, M.; Fluck, J. Dataset of miRNA-disease relations extracted from textual data using transformer-based neural networks. Database J. Biol. Databases Curation 2024, 2024, baae066. [Google Scholar] [CrossRef]
- Chouery, E.; Mehawej, C.; Sabbagh, S.; Bleik, J.; Megarbane, A. Early infantile epileptic encephalopathy related to NECAP1: Clinical delineation of the disease and review. Eur. J. Neurol. 2022, 29, 2486–2492. [Google Scholar] [CrossRef]
- Lopez-Cerdan, A.; Andreu, Z.; Hidalgo, M.R.; Soler-Saez, I.; de la Iglesia-Vaya, M.; Mikozami, A.; Guerini, F.R.; Garcia-Garcia, F. An integrated approach to identifying sex-specific genes, transcription factors, and pathways relevant to Alzheimer’s disease. Neurobiol. Dis. 2024, 199, 106605. [Google Scholar] [CrossRef]
- Melberg, A.; Nennesmo, I.; Moslemi, A.R.; Kollberg, G.; Luoma, P.; Suomalainen, A.; Holme, E.; Oldfors, A. Alzheimer pathology associated with POLG1 mutation, multiple mtDNA deletions, and APOE4/4: Premature ageing or just coincidence? Acta Neuropathol. 2005, 110, 315–316. [Google Scholar] [CrossRef]
- Chen, A.; Yangzom, T.; Hong, Y.; Lundberg, B.C.; Sullivan, G.J.; Tzoulis, C.; Bindoff, L.A.; Liang, K.X. Hallmark Molecular and Pathological Features of POLG Disease are Recapitulated in Cerebral Organoids. Adv. Sci. 2024, 11, e2307136. [Google Scholar] [CrossRef]
- Borgione, E.; Lo Giudice, M.; Santa Paola, S.; Giuliano, M.; Lanza, G.; Cantone, M.; Ferri, R.; Scuderi, C. The Y831C Mutation of the POLG Gene in Dementia. Biomedicines 2023, 11, 1172. [Google Scholar] [CrossRef] [PubMed]
- Gillis, A.; Berry, S. Global control of RNA polymerase II. Biochim. Biophys. Acta. Gene Regul. Mech. 2024, 1867, 195024. [Google Scholar] [CrossRef]
- Kim, J.; Tadros, B.; Liang, Y.H.; Kim, Y.; Lasagna-Reeves, C.; Sonn, J.Y.; Chung, D.C.; Hyman, B.; Holtzman, D.M.; Zoghbi, H.Y. TYK2 regulates tau levels, phosphorylation and aggregation in a tauopathy mouse model. Nat. Neurosci. 2024, 27, 2417–2429. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Wang, L.; Chen, X.; Lan, T.; Wang, C.; Chen, S.; Yu, S. FBXL20 promotes synaptic impairment in depression disorder via degrading vesicle-associated proteins. J. Affect. Disord. 2024, 349, 132–144. [Google Scholar] [CrossRef]
- Meng, F.; Sun, N.; Liu, D.; Jia, J.; Xiao, J.; Dai, H. BCL2L13: Physiological and pathological meanings. Cell. Mol. Life Sci. 2021, 78, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Ren, H.; Fan, Y. Microglia specific alternative splicing alterations in multiple sclerosis. Aging 2024, 16, 11656–11667. [Google Scholar] [CrossRef]
- Lu, Y.; Tan, L.; Xie, J.; Cheng, L.; Wang, X. Distinct microglia alternative splicing in Alzheimer’s disease. Aging 2022, 14, 6554–6566. [Google Scholar] [CrossRef]
- Farhadieh, M.E.; Ghaedi, K. Analyzing alternative splicing in Alzheimer’s disease postmortem brain: A cell-level perspective. Front. Mol. Neurosci. 2023, 16, 1237874. [Google Scholar] [CrossRef]
- Lu, Y.; Yue, D.; Xie, J.; Cheng, L.; Wang, X. Ontology Specific Alternative Splicing Changes in Alzheimer’s Disease. Front. Genet. 2022, 13, 926049. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef]
- Fromm, B.; Hoye, E.; Domanska, D.; Zhong, X.; Aparicio-Puerta, E.; Ovchinnikov, V.; Umu, S.U.; Chabot, P.J.; Kang, W.; Aslanzadeh, M.; et al. MirGeneDB 2.1: Toward a complete sampling of all major animal phyla. Nucleic Acids Res. 2022, 50, D204–D210. [Google Scholar] [CrossRef]
- Alves, L., Jr.; Niemeier, S.; Hauenschild, A.; Rehmsmeier, M.; Merkle, T. Comprehensive prediction of novel microRNA targets in Arabidopsis thaliana. Nucleic Acids Res. 2009, 37, 4010–4021. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Groger, L.; Tschernig, T.; Solomon, J.; Laham, O.; Schaum, N.; Wagner, V.; Kern, F.; Schmartz, G.P.; Li, Y.; et al. miRNATissueAtlas2: An update to the human miRNA tissue atlas. Nucleic Acids Res. 2022, 50, D211–D221. [Google Scholar] [CrossRef]
- Rishik, S.; Hirsch, P.; Grandke, F.; Fehlmann, T.; Keller, A. miRNATissueAtlas 2025: An update to the uniformly processed and annotated human and mouse non-coding RNA tissue atlas. Nucleic Acids Res. 2025, 53, D129–D137. [Google Scholar] [CrossRef]
- Xie, C.; Yuan, J.; Li, H.; Li, M.; Zhao, G.; Bu, D.; Zhu, W.; Wu, W.; Chen, R.; Zhao, Y. NONCODEv4: Exploring the world of long non-coding RNA genes. Nucleic Acids Res. 2014, 42, D98–D103. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Biological Process (BP) | CBNR20+Aβ vs. Aβ Fold Enrichment | CBNR20+Aβ vs. Aβ FDR | Aβ vs. CTR Fold Enrichment | Aβ vs. CTR FDR |
|---|---|---|---|---|
| regulation of DNA-templated transcription elongation | 3.1 | 0.00108 | 2.25 | 0.0118 |
| RNA splicing, via transesterification reactions | 2.08 | 0.00985 | 1.85 | 0.00272 |
| mRNA splicing, via spliceosome | 2.05 | 0.0147 | 1.88 | 0.00236 |
| RNA splicing, via transesterification reactions with bulged adenosine as nucleophile | 2.05 | 0.0145 | 1.88 | 0.00235 |
| mRNA processing | 1.98 | 0.000156 | 1.78 | 0.0000267 |
| RNA splicing | 1.92 | 0.00386 | 1.82 | 0.0000962 |
| RNA processing | 1.75 | 0.0000119 | 1.52 | 0.0000751 |
| PANTHER Pathways | Genes | Fold Enrichment | Raw p Value | FDR |
|---|---|---|---|---|
| Alzheimer disease-amyloid secretase pathway | ADAM9, APBA2, CHRNA7, MAPK10, PRKCI | 19.25 | 6.17 × 10−6 | 9.87 × 10−4 |
| Gene | RI Coordinates | Frame of ORF | n. of Premature Stop Codons | Position of the First Stop Codon |
|---|---|---|---|---|
| ADAM9 | chr8:39018918-39021642 (+) | Frame 1 | 59 | 73 |
| DDX39A | chr19:14409447-14409535 (−) | Frame 2 | 2 | 2 |
| GGA3 | chr17:75240108-75240341 (−) | Frame 1 | 3 | 19 |
| HAX1 | chr1:154273598-154273773 (+) | Frame 3 | 2 | 57 |
| LIG3 | chr17:34991837-34991957 (+) | Frame 2 | 1 | 41 |
| MAPK10 | chr4:86098595-86101051 (−) | Frame 3 | 57 | 39 |
| MROH1 | chr8:144260544-144260676 (+) | Frame 1 | No stop codons | --- |
| NECAP1 | chr12:8091850-8092675 (+) | Frame 2 | 12 | 2 |
| PDHB | chr3:58431793-58431876 (−) | Frame 1 | No stop codons | --- |
| POLG | chr15:89318749-89318930 (−) | Frame 1 | 1 | 130 |
| TBC1D20 | chr20:441689-441856 (−) | Frame 2 | 3 | 8 |
| ENSG00000271793 | chr6:85547397-85547505 (−) | Frame 1 | 1 | 40 |
| ENSG00000284946 | chr15:90981033-90981498 (−) | Frame 1 | 16 | 25 |
| ASE Class | Gene | miRNA | ΔPSI Aβ vs. CTR | ΔPSI CBNR20+Aβ vs. Aβ | MFE (kcal/mol) |
|---|---|---|---|---|---|
| A5SS | POLR2J3 | hsa-miR-134-5p | 0.136 | −0.127 | −30.3 |
| RI | ENSG00000284946 | hsa-miR-877-5p | 0.113 | −0.12 | −31.9 |
| RI | NECAP1 | hsa-miR-628-3p | 0.194 | −0.159 | −32.5 |
| RI | MAPK10 | hsa-miR-744-5p | −0.217 | 0.244 | −40.5 |
| RI | POLG | hsa-miR-328-3p | 0.278 | −0.233 | −37.1 |
| RI | POLG | hsa-miR-423-3p | 0.278 | −0.233 | −38.4 |
| RI | POLG | hsa-miR-874-3p | 0.278 | −0.233 | −35.7 |
| RI | POLG | hsa-miR-1249-3p | 0.278 | −0.233 | −32.8 |
| RI | MROH1 | hsa-miR-328-3p | −0.351 | 0.351 | −34.8 |
| RI | MROH1 | hsa-miR-331-3p | −0.351 | 0.351 | −34.8 |
| RI | MROH1 | hsa-miR-874-3p | −0.351 | 0.351 | −35.5 |
| A3SS | TYK2 | hsa-miR-328-3p | −0.458 | 0.458 | −34.1 |
| A3SS | TYK2 | hsa-miR-874-3p | −0.458 | 0.458 | −42.4 |
| A3SS | TYK2 | hsa-miR-1249-3p | −0.458 | 0.458 | −32.9 |
| A3SS | TYK2 | hsa-miR-3200-3p | −0.458 | 0.458 | −31.3 |
| A3SS | GTF2IRD1 | hsa-miR-185-5p | −0.328 | 0.221 | −30.7 |
| SE | FBXL20 | hsa-miR-671-5p | −0.15 | 0.171 | −32.6 |
| SE | APBA2 | hsa-miR-145-5p | −0.134 | 0.151 | −30 |
| SE | APBA2 | hsa-miR-328-3p | −0.134 | 0.151 | −31.7 |
| SE | APBA2 | hsa-miR-370-3p | −0.134 | 0.151 | −41 |
| SE | APBA2 | hsa-miR-744-5p | −0.134 | 0.151 | −39.6 |
| SE | APBA2 | hsa-miR-1301-3p | −0.134 | 0.151 | −30.9 |
| SE | CHRNA7 | hsa-miR-149-5p | −0.242 | 0.242 | −33.3 |
| SE | FBXW4 | hsa-miR-874-3p | −0.13 | 0.146 | −35.4 |
| SE | HIP1 | hsa-miR-145-5p | −0.149 | 0.149 | −32 |
| SE | HIP1 | hsa-miR-423-3p | −0.149 | 0.149 | −35.3 |
| SE | HIP1 | hsa-miR-1180-3p | −0.149 | 0.149 | −32.5 |
| SE | LAMB1 | hsa-miR-744-5p | −0.167 | 0.151 | −31.1 |
| SE | BCL2L13 | hsa-miR-185-5p | −0.209 | 0.246 | −31.4 |
| SE | ZC3H4 | hsa-miR-149-5p | −0.234 | 0.234 | −31 |
| SE | ZC3H4 | hsa-miR-328-3p | −0.234 | 0.234 | −31.1 |
| SE | MVK | hsa-miR-652-3p | −0.297 | 0.297 | −30.2 |
| SE | MVK | hsa-miR-874-3p | −0.297 | 0.297 | −34.3 |
| ASE Class | DASE Gene Name | ΔPSI Aβ vs. CTR | ΔPSI CBNR20+Aβ vs. Aβ | lncRNA Gene_ID |
|---|---|---|---|---|
| MXE | MAPK10 | −0.229 | 0.229 | HSALNG0035653 |
| RI | ENSG00000284946 | 0.113 | −0.12 | HSALNG0108169 |
| RI | PDHB | 0.114 | −0.131 | HSALNG0026424 |
| SE | ZNF468 | 0.168 | −0.21 | HSALNG0127347 |
| SE | CHRNA7 | −0.242 | 0.242 | HSALNG0104832 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lui, M.; Salamone, S.; Pollastro, F.; Mazzon, E.; Artimagnella, O. Cannabinerol Restores mRNA Splicing Defects Induced by β-Amyloid in an In Vitro Model of Alzheimer’s Disease: A Transcriptomic Study. Int. J. Mol. Sci. 2025, 26, 3113. https://doi.org/10.3390/ijms26073113
Lui M, Salamone S, Pollastro F, Mazzon E, Artimagnella O. Cannabinerol Restores mRNA Splicing Defects Induced by β-Amyloid in an In Vitro Model of Alzheimer’s Disease: A Transcriptomic Study. International Journal of Molecular Sciences. 2025; 26(7):3113. https://doi.org/10.3390/ijms26073113
Chicago/Turabian StyleLui, Maria, Stefano Salamone, Federica Pollastro, Emanuela Mazzon, and Osvaldo Artimagnella. 2025. "Cannabinerol Restores mRNA Splicing Defects Induced by β-Amyloid in an In Vitro Model of Alzheimer’s Disease: A Transcriptomic Study" International Journal of Molecular Sciences 26, no. 7: 3113. https://doi.org/10.3390/ijms26073113
APA StyleLui, M., Salamone, S., Pollastro, F., Mazzon, E., & Artimagnella, O. (2025). Cannabinerol Restores mRNA Splicing Defects Induced by β-Amyloid in an In Vitro Model of Alzheimer’s Disease: A Transcriptomic Study. International Journal of Molecular Sciences, 26(7), 3113. https://doi.org/10.3390/ijms26073113