
Targeting Signaling Excitability in Cervical and Pancreatic Cancer Cells Through Combined Inhibition of FAK and PI3K

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
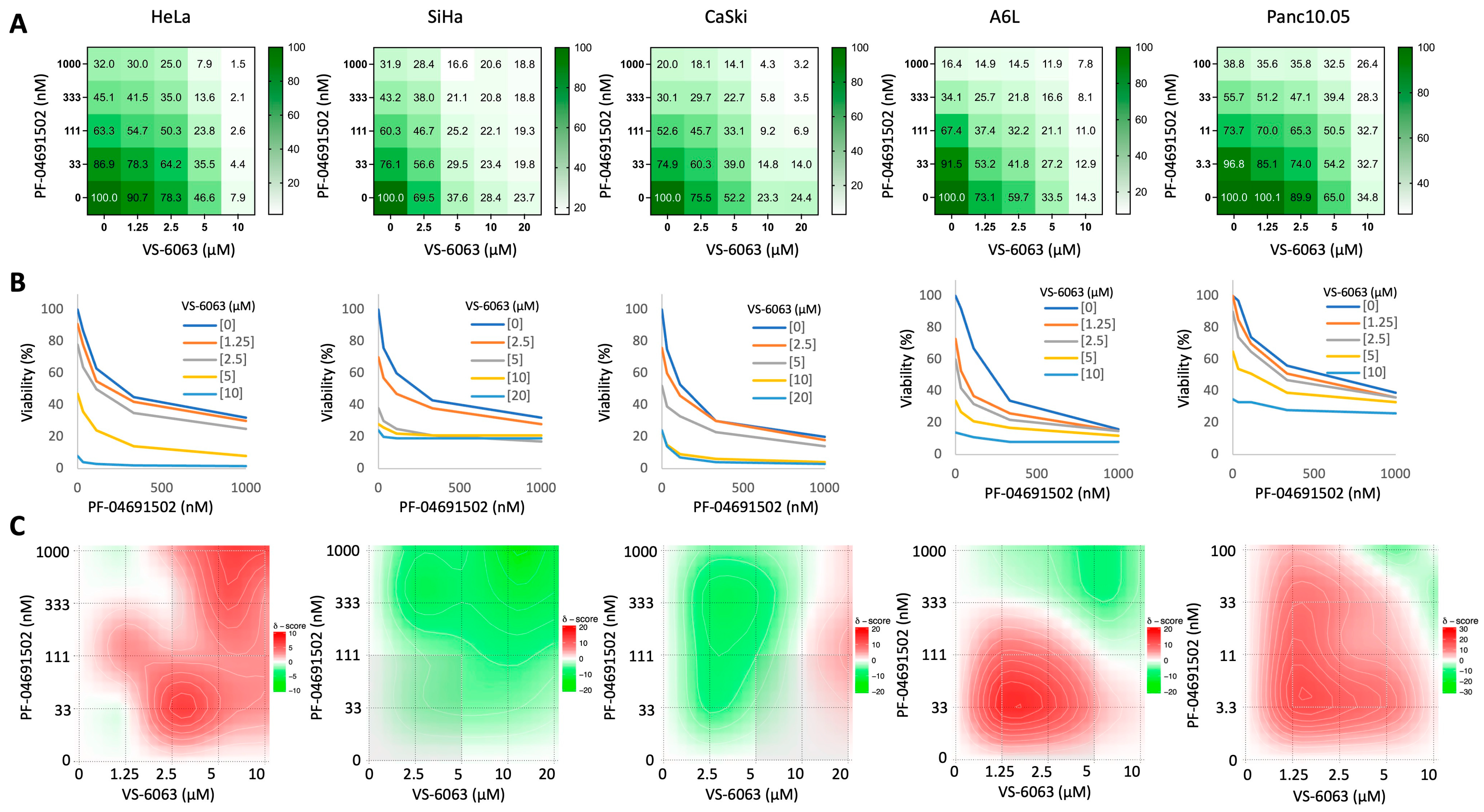
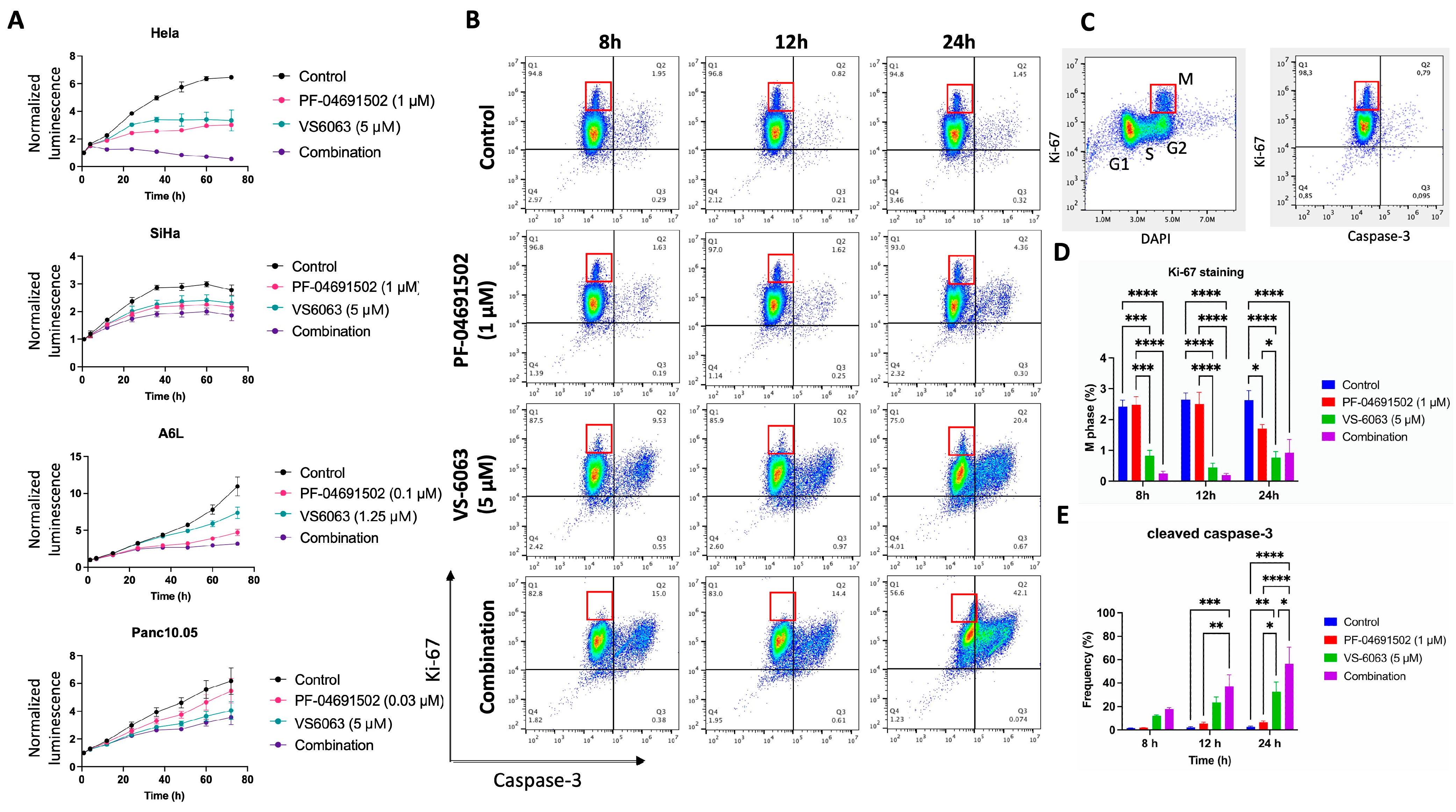
2.1. Synergy Between FAK and PI3K Inhibition on the Growth of Cervical and Pancreatic Cancer Cells
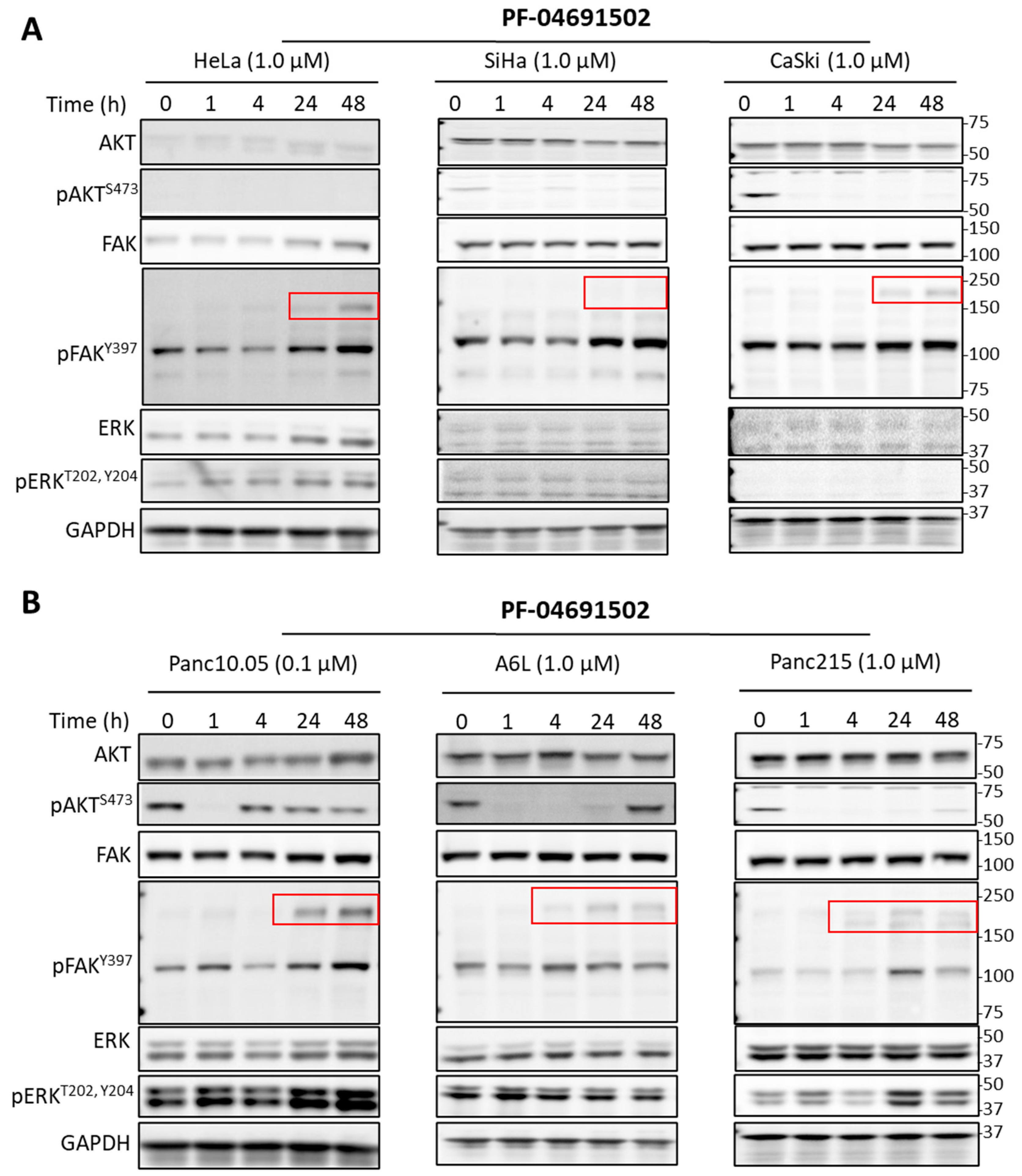
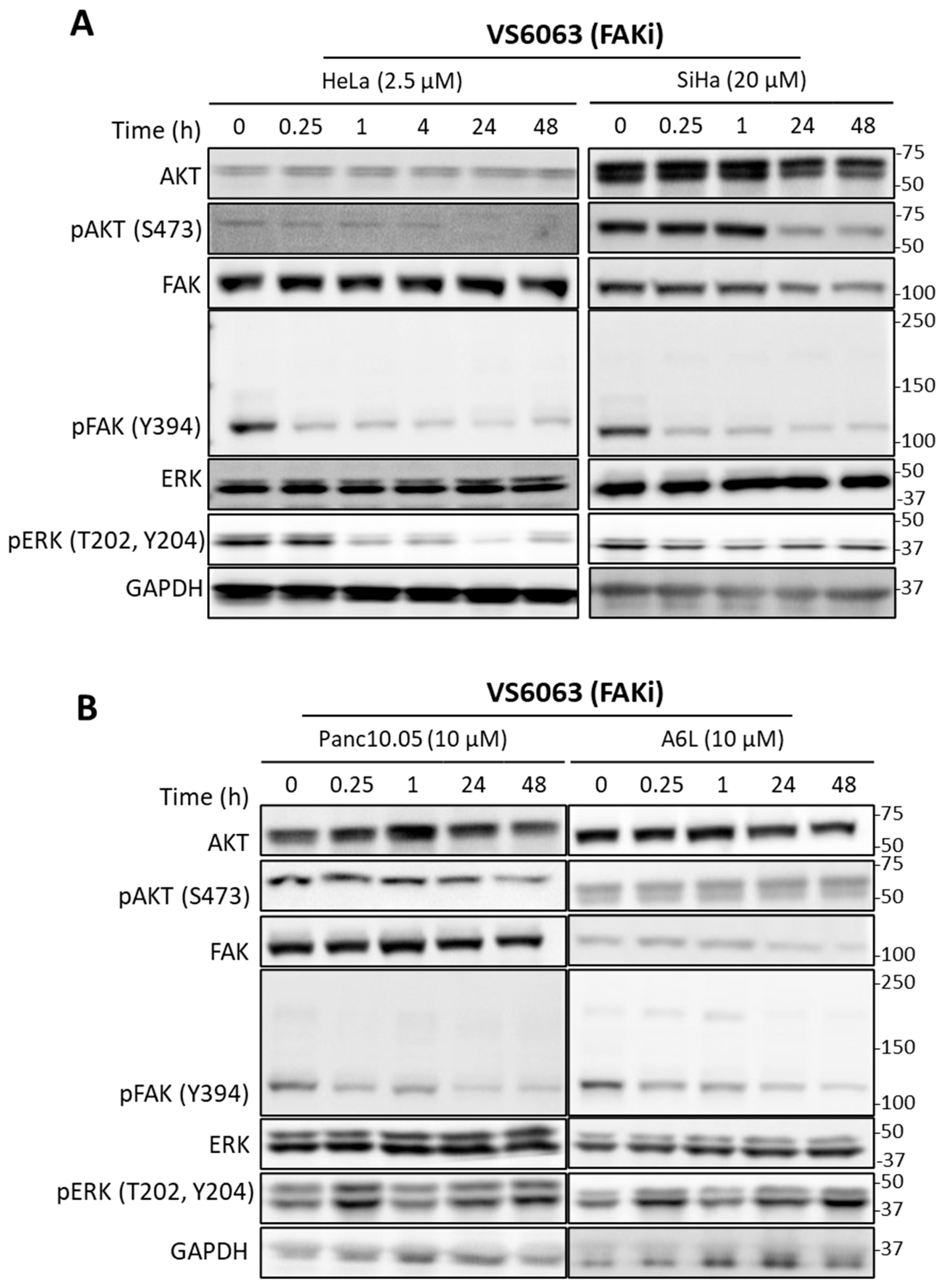
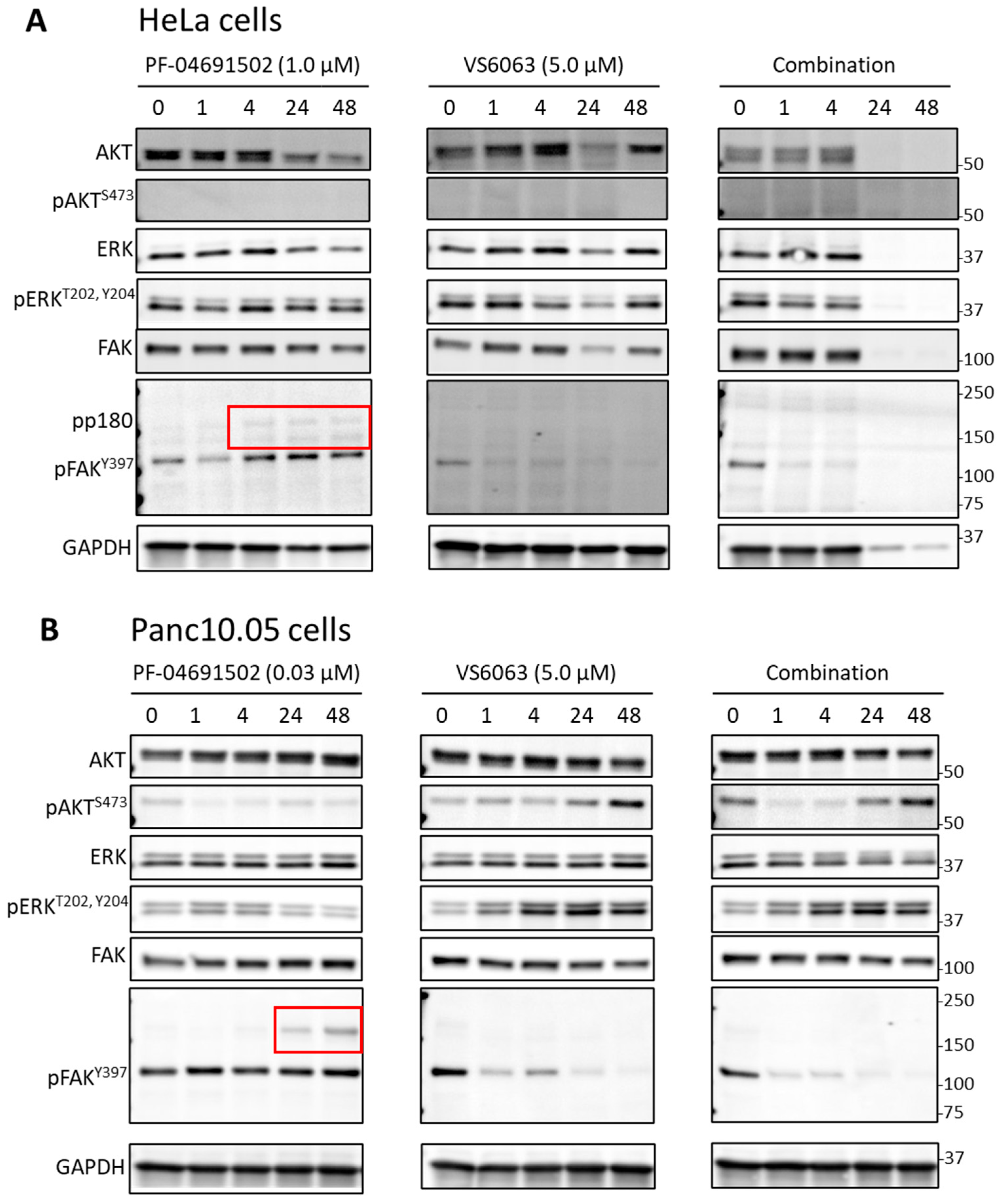
2.2. Signaling Effects of PI3K and FAK Inhibitors in Cervical and Pancreatic Cancer Cells
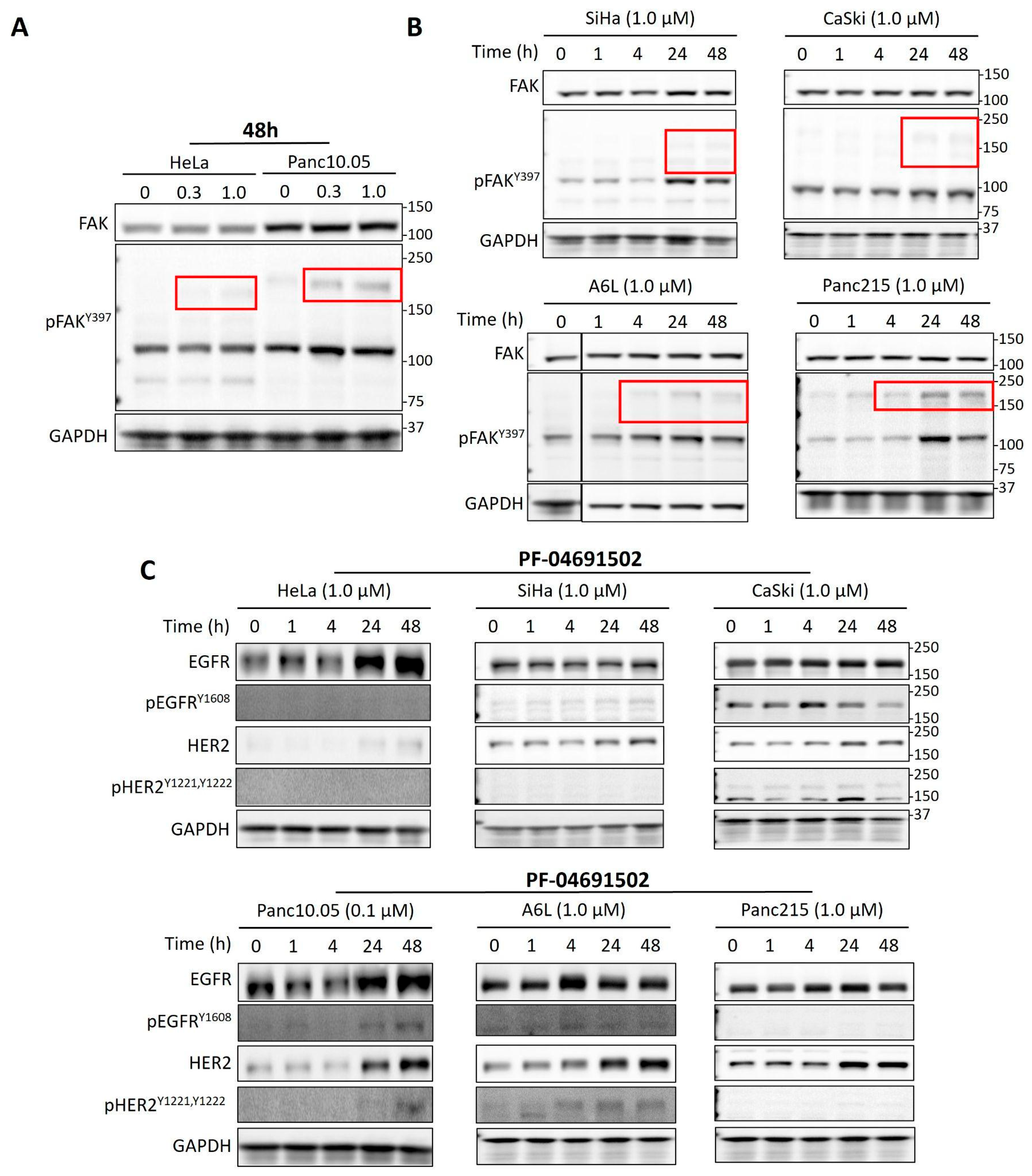
2.3. PI3K Inhibition Induces Activation of Multiple RTKs
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Chemical Reagents
4.3. Cell Viability Assay
4.4. Analysis of Synergy
4.5. RealTime-Glo MT Cell Viability Assay
4.6. Cell Cycle, Cell Growth and Apoptosis Analysis by Flow Cytometry
4.7. Immunoblotting
4.8. Phospho-RTK Array
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, J.B.; Wu, J.S.; Craighead, P.S.; Phan, T.; Köbel, M.; Lees-Miller, S.P.; Ghatage, P.; Magliocco, A.M.; Doll, C.M. PIK3CA mutational status and overall survival in patients with cervical cancer treated with radical chemoradiotherapy. Gynecol. Oncol. 2013, 128, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Miyake, T.; Yoshino, K.; Enomoto, T.; Takata, T.; Ugaki, H.; Kim, A.; Fujiwara, K.; Miyatake, T.; Fujita, M.; Kimura, T. PIK3CA gene mutations and amplifications in uterine cancers, identified by methods that avoid confounding by PIK3CA pseudogene sequences. Cancer Lett. 2008, 261, 120–126. [Google Scholar] [CrossRef]
- Janku, F.; Lee, J.J.; Tsimberidou, A.M.; Hong, D.S.; Naing, A.; Falchook, G.S.; Fu, S.; Luthra, R.; Garrido-Laguna, I.; Kurzrock, R. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS ONE 2011, 6, e22769. [Google Scholar] [CrossRef]
- Jiang, W.; Xiang, L.; Pei, X.; He, T.; Shen, X.; Wu, X.; Yang, H. Mutational analysis of KRAS and its clinical implications in cervical cancer patients. J. Gynecol. Oncol. 2018, 29, e4. [Google Scholar] [CrossRef]
- Meira, D.D.; de Almeida, V.H.; Mororó, J.S.; Nóbrega, I.; Bardella, L.; A Silva, R.L.; Albano, R.M.; Ferreira, C.G. Combination of cetuximab with chemoradiation, trastuzumab or MAPK inhibitors: Mechanisms of sensitisation of cervical cancer cells. Br. J. Cancer 2009, 101, 782–791. [Google Scholar] [CrossRef]
- Wright, A.A.; Howitt, B.E.; Myers, A.P.; Dahlberg, S.E.; Palescandolo, E.; Van Hummelen, P.; MacConaill, L.E.; Shoni, M.; Wagle, N.; Jones, R.T.; et al. Oncogenic mutations in cervical cancer: Genomic differences between adenocarcinomas and squamous cell carcinomas of the cervix. Cancer 2013, 119, 3776–3783. [Google Scholar] [CrossRef]
- Hou, M.-M.; Liu, X.; Wheler, J.; Naing, A.; Hong, D.; Coleman, R.L.; Tsimberidou, A.; Janku, F.; Zinner, R.; Lu, K.; et al. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: A phase I clinical experience. Oncotarget 2014, 5, 11168–11179. [Google Scholar] [CrossRef]
- Hou, M.-M.; Liu, X.; Wheler, J.; Naing, A.; Hong, D.; Bodurka, D.; Schmeler, K.; Tsimberidou, A.M.; Janku, F.; Zinner, R.; et al. Outcomes of patients with metastatic cervical cancer in a phase I clinical trials program. Anticancer Res. 2014, 34, 2349–2355. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Nissim, S.; Leshchiner, I.; Mancias, J.D.; Greenblatt, M.B.; Maertens, O.; Cassa, C.A.; Rosenfeld, J.A.; Cox, A.G.; Hedgepeth, J.; Wucherpfennig, J.I.; et al. Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat. Genet. 2019, 51, 1308–1314. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Paolino, G.; Mattiolo, P.; Piredda, M.L.; Cavaliere, A.; Gaule, M.; Melisi, D.; Salvia, R.; Malleo, G.; Shin, J.I.; et al. KRAS wild-type pancreatic ductal adenocarcinoma: Molecular pathology and therapeutic opportunities. J. Exp. Clin. Cancer Res. 2020, 39, 227. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Janetopoulos, C.; Lee, S.; Charest, P.G.; Takeda, K.; Sundheimer, L.W.; Meili, R.; Devreotes, P.N.; Firtel, R.A. G protein–independent Ras/PI3K/F-actin circuit regulates basic cell motility. J. Cell Biol. 2007, 178, 185–191. [Google Scholar] [CrossRef]
- Asano, Y.; Nagasaki, A.; Uyeda, T.Q. Correlated waves of actin filaments and PIP3 in Dictyostelium cells. Cell Motil. Cytoskelet. 2008, 65, 923–934. [Google Scholar] [CrossRef]
- Weiger, M.C.; Wang, C.-C.; Krajcovic, M.; Melvin, A.T.; Rhoden, J.J.; Haugh, J.M. Spontaneous phosphoinositide 3-kinase signaling dynamics drive spreading and random migration of fibroblasts. J. Cell Sci. 2009, 122, 313–323. [Google Scholar] [CrossRef]
- Arai, Y.; Shibata, T.; Matsuoka, S.; Sato, M.J.; Yanagida, T.; Ueda, M. Self-organization of the phosphatidylinositol lipids signaling system for random cell migration. Proc. Natl. Acad. Sci. USA 2010, 107, 12399–12404. [Google Scholar] [CrossRef]
- Taniguchi, D.; Ishihara, S.; Oonuki, T.; Honda-Kitahara, M.; Kaneko, K.; Sawai, S. Phase geometries of two-dimensional excitable waves govern self-organized morphodynamics of amoeboid cells. Proc. Natl. Acad. Sci. USA 2013, 110, 5016–5021. [Google Scholar] [CrossRef]
- Huang, C.-H.; Tang, M.; Shi, C.; Iglesias, P.A.; Devreotes, P.N. An excitable signal integrator couples to an idling cytoskeletal oscillator to drive cell migration. Nat. Cell Biol. 2013, 15, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Bhattacharya, S.; Cai, H.; Iglesias, P.A.; Huang, C.-H.; Devreotes, P.N. An Excitable Ras/PI3K/ERK Signaling Network Controls Migration and Oncogenic Transformation in Epithelial Cells. Dev. Cell 2020, 54, 608–623.e5. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M.; Bhattacharya, S.; West-Foyle, H.; Hung, C.-F.; Wu, T.-C.; Iglesias, P.A.; Huang, C.-H. Integrating chemical and mechanical signals through dynamic coupling between cellular protrusions and pulsed ERK activation. Nat. Commun. 2018, 9, 4673. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-M.; Chi, W.-Y.; Liang, J.; Takayanagi, S.; Iglesias, P.A.; Huang, C.-H. Deciphering cell signaling networks with massively multiplexed biosensor barcoding. Cell 2021, 184, 6193–6206.e14. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 2013, 49, 249–261. [Google Scholar] [CrossRef]
- Aoki, K.; Kumagai, Y.; Sakurai, A.; Komatsu, N.; Fujita, Y.; Shionyu, C.; Matsuda, M. Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 2013, 52, 529–540. [Google Scholar] [CrossRef]
- Regot, S.; Hughey, J.J.; Bajar, B.T.; Carrasco, S.; Covert, M.W. High-sensitivity measurements of multiple kinase activities in live single cells. Cell 2014, 157, 1724–1734. [Google Scholar] [CrossRef]
- Aoki, K.; Kondo, Y.; Naoki, H.; Hiratsuka, T.; Itoh, R.E.; Matsuda, M. Propagating Wave of ERK Activation Orients Collective Cell Migration. Dev. Cell 2017, 43, 305–317.e5. [Google Scholar] [CrossRef]
- Hino, N.; Rossetti, L.; Marín-Llauradó, A.; Aoki, K.; Trepat, X.; Matsuda, M.; Hirashima, T. ERK-Mediated Mechanochemical Waves Direct Collective Cell Polarization. Dev. Cell 2020, 53, 646–660.e8. [Google Scholar] [CrossRef]
- Hiratsuka, T.; Fujita, Y.; Naoki, H.; Aoki, K.; Kamioka, Y.; Matsuda, M. Japan Intercellular propagation of extracellular signal-regulated kinase activation revealed by in vivo imaging of mouse skin. eLife 2015, 4, e05178. [Google Scholar] [CrossRef]
- Tang, M.; Wang, M.; Shi, C.; Iglesias, P.A.; Devreotes, P.N.; Huang, C.-H. Evolutionarily conserved coupling of adaptive and excitable networks mediates eukaryotic chemotaxis. Nat. Commun. 2014, 5, 5175. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Hörning, M.; Ueda, M.; Shibata, T. Excitable signal transduction induces both spontaneous and directional cell asymmetries in the phosphatidylinositol lipid signaling system for eukaryotic chemotaxis. Biophys. J. 2014, 106, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Chung, M.; Dobrzyński, M.; Fey, D.; Blum, Y.; Lee, S.S.; Peter, M.; Kholodenko, B.N.; Jeon, N.L.; Pertz, O. Frequency modulation of ERK activation dynamics rewires cell fate. Mol. Syst. Biol. 2015, 11, 838. [Google Scholar] [CrossRef]
- Ozkan-Dagliyan, I.; Diehl, J.N.; George, S.D.; Schaefer, A.; Papke, B.; Klotz-Noack, K.; Waters, A.M.; Goodwin, C.M.; Gautam, P.; Pierobon, M.; et al. Low-Dose Vertical Inhibition of the RAF-MEK-ERK Cascade Causes Apoptotic Death of KRAS Mutant Cancers. Cell Rep. 2020, 31, 107764. [Google Scholar] [CrossRef]
- McCormack, S.J.; Brazinski, S.E.; Moore, J.L., Jr.; Werness, B.A.; Goldstein, D.J. Activation of the focal adhesion kinase signal transduction pathway in cervical carcinoma cell lines and human genital epithelial cells immortalized with human papillomavirus type 18. Oncogene 1997, 15, 265–274. [Google Scholar] [CrossRef]
- Golubovskaya, V.M. Focal Adhesion Kinase as a Cancer Therapy Target. Anti-Cancer Agents Med. Chem. 2010, 10, 735–741. [Google Scholar] [CrossRef]
- Dawson, J.C.; Serrels, A.; Stupack, D.G.; Schlaepfer, D.D.; Frame, M.C. Targeting FAK in anticancer combination therapies. Nat. Rev. Cancer 2021, 21, 313–324. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 2.0: Visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Schwock, J.; Dhani, N.; Cao, M.P.-J.; Zheng, J.; Clarkson, R.; Radulovich, N.; Navab, R.; Horn, L.-C.; Hedley, D.W. Targeting focal adhesion kinase with dominant-negative FRNK or Hsp90 inhibitor 17-DMAG suppresses tumor growth and metastasis of SiHa cervical xenografts. Cancer Res. 2009, 69, 4750–4759. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, P. Cancer-stromal interactions: Role in cell survival, metabolism and drug sensitivity. Cancer Biol. Ther. 2011, 11, 150–156. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- García-García, C.; Ibrahim, Y.H.; Serra, V.; Calvo, M.T.; Guzmán, M.; Grueso, J.; Aura, C.; Pérez, J.; Jessen, K.; Liu, Y.; et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to Anti-HER2 therapy. Clin. Cancer Res. 2012, 18, 2603–2612. [Google Scholar] [CrossRef]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; DeFazio-Eli, L.; Arteaga, C.L. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110α inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013, 73, 6013–6023. [Google Scholar] [CrossRef]
- Meister, K.S.; Godse, N.R.; Khan, N.I.; Hedberg, M.L.; Kemp, C.; Kulkarni, S.; Alvarado, D.; LaVallee, T.; Kim, S.; Grandis, J.R.; et al. HER3 targeting potentiates growth suppressive effects of the PI3K inhibitor BYL719 in pre-clinical models of head and neck squamous cell carcinoma. Sci. Rep. 2019, 9, 9130. [Google Scholar] [CrossRef]
- Zhao, J.; Guan, J.-L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Cavazzoni, A.; La Monica, S.; Alfieri, R.; Ravelli, A.; Van Der Steen, N.; Sciarrillo, R.; Madeddu, D.; Lagrasta, C.A.M.; Quaini, F.; Bonelli, M.; et al. Enhanced efficacy of AKT and FAK kinase combined inhibition in squamous cell lung carcinomas with stable reduction in PTEN. Oncotarget 2017, 8, 53068–53083. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, T.A.; Lenzo, F.L.; Figel, S.A.; Grapes, A.T.; Cance, W.G. Oncogenic Receptor Tyrosine Kinases Directly Phosphorylate Focal Adhesion Kinase (FAK) as a Resistance Mechanism to FAK-Kinase Inhibitors. Mol. Cancer Ther. 2016, 15, 3028–3039. [Google Scholar] [CrossRef] [PubMed]
- Sieg, D.J.; Hauck, C.R.; Ilic, D.; Klingbeil, C.K.; Schaefer, E.; Damsky, C.H.; Schlaepfer, D.D. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2000, 2, 249–256. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 2718–2723. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-C.; Wang, S.; Yang, J.-M.; Huang, C.-H. Targeting Signaling Excitability in Cervical and Pancreatic Cancer Cells Through Combined Inhibition of FAK and PI3K. Int. J. Mol. Sci. 2025, 26, 3040. https://doi.org/10.3390/ijms26073040
Chen C-C, Wang S, Yang J-M, Huang C-H. Targeting Signaling Excitability in Cervical and Pancreatic Cancer Cells Through Combined Inhibition of FAK and PI3K. International Journal of Molecular Sciences. 2025; 26(7):3040. https://doi.org/10.3390/ijms26073040
Chicago/Turabian StyleChen, Chao-Cheng, Suyang Wang, Jr-Ming Yang, and Chuan-Hsiang Huang. 2025. "Targeting Signaling Excitability in Cervical and Pancreatic Cancer Cells Through Combined Inhibition of FAK and PI3K" International Journal of Molecular Sciences 26, no. 7: 3040. https://doi.org/10.3390/ijms26073040
APA StyleChen, C.-C., Wang, S., Yang, J.-M., & Huang, C.-H. (2025). Targeting Signaling Excitability in Cervical and Pancreatic Cancer Cells Through Combined Inhibition of FAK and PI3K. International Journal of Molecular Sciences, 26(7), 3040. https://doi.org/10.3390/ijms26073040