
Investigating the Antioxidant Efficiency of Tea Flavonoid Derivatives: A Density Functional Theory Study


Abstract
:1. Introduction
2. Results and Discussion
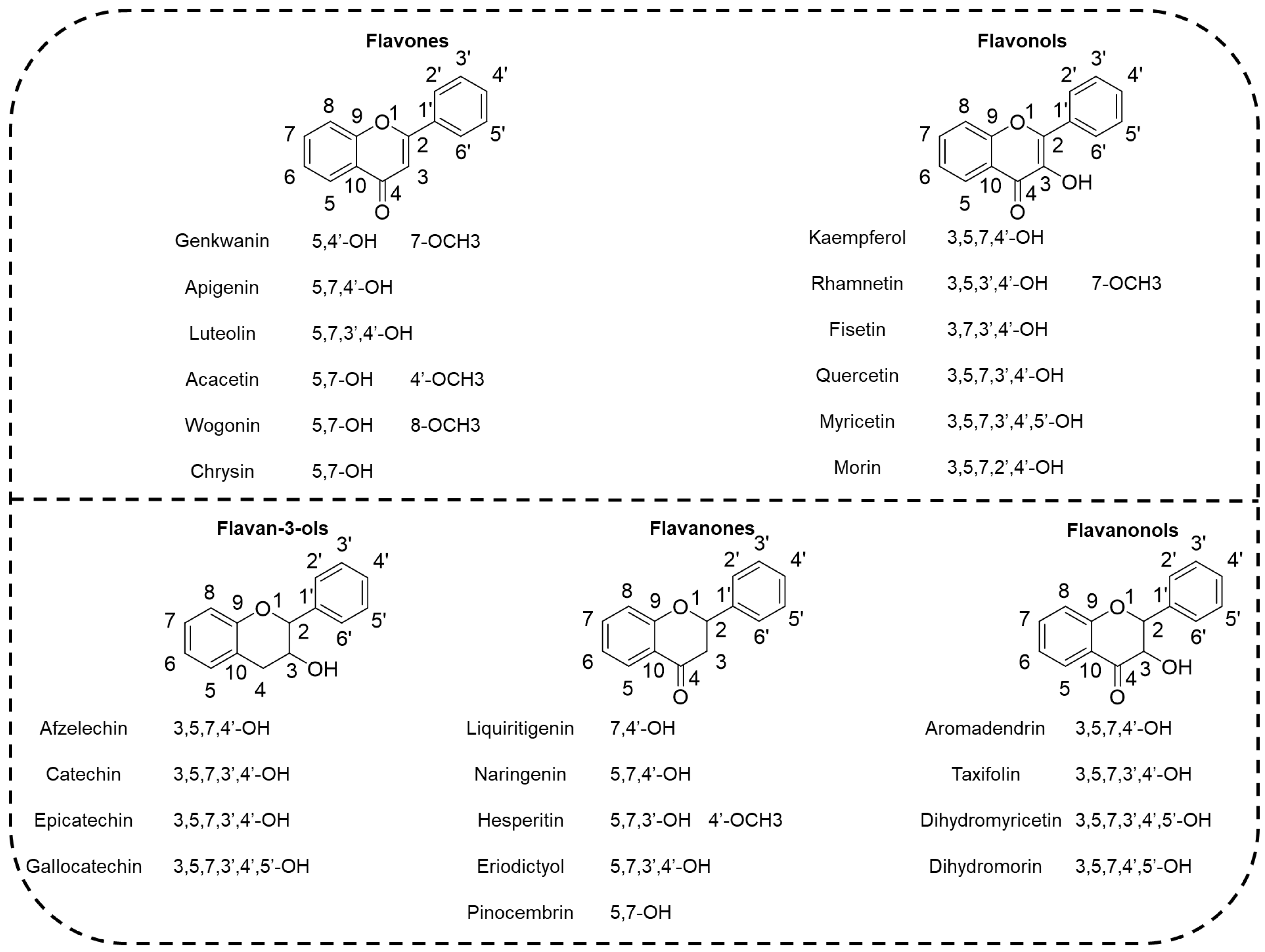
2.1. Geometrical Structures
2.2. Antioxidant Mechanisms
2.2.1. HAT Mechanism
2.2.2. SET-PT Mechanism
2.2.3. SPLET Mechanism
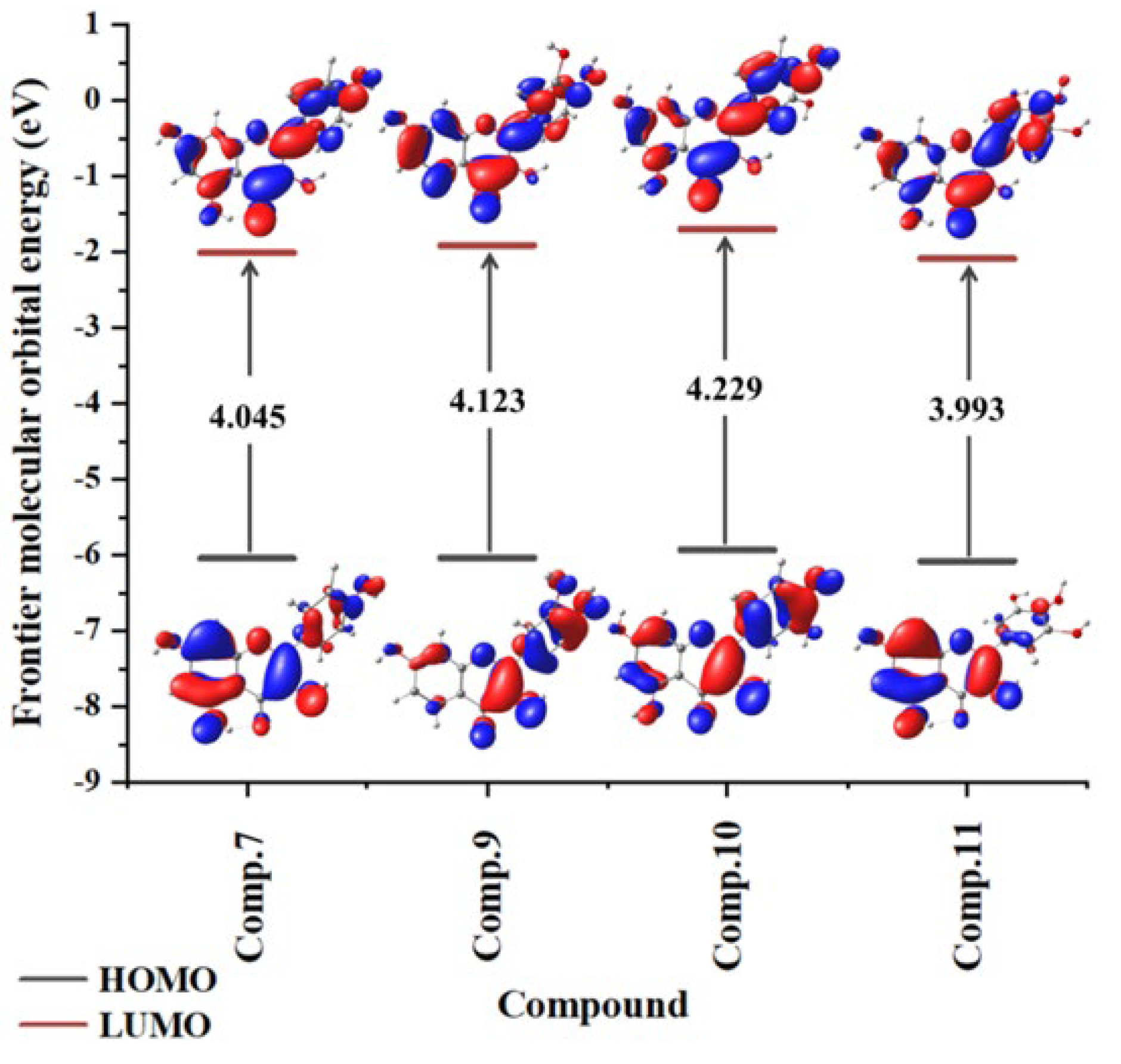
2.3. Frontier Molecular Orbital Theory and Spin Density Distribution
2.4. Electronic Properties
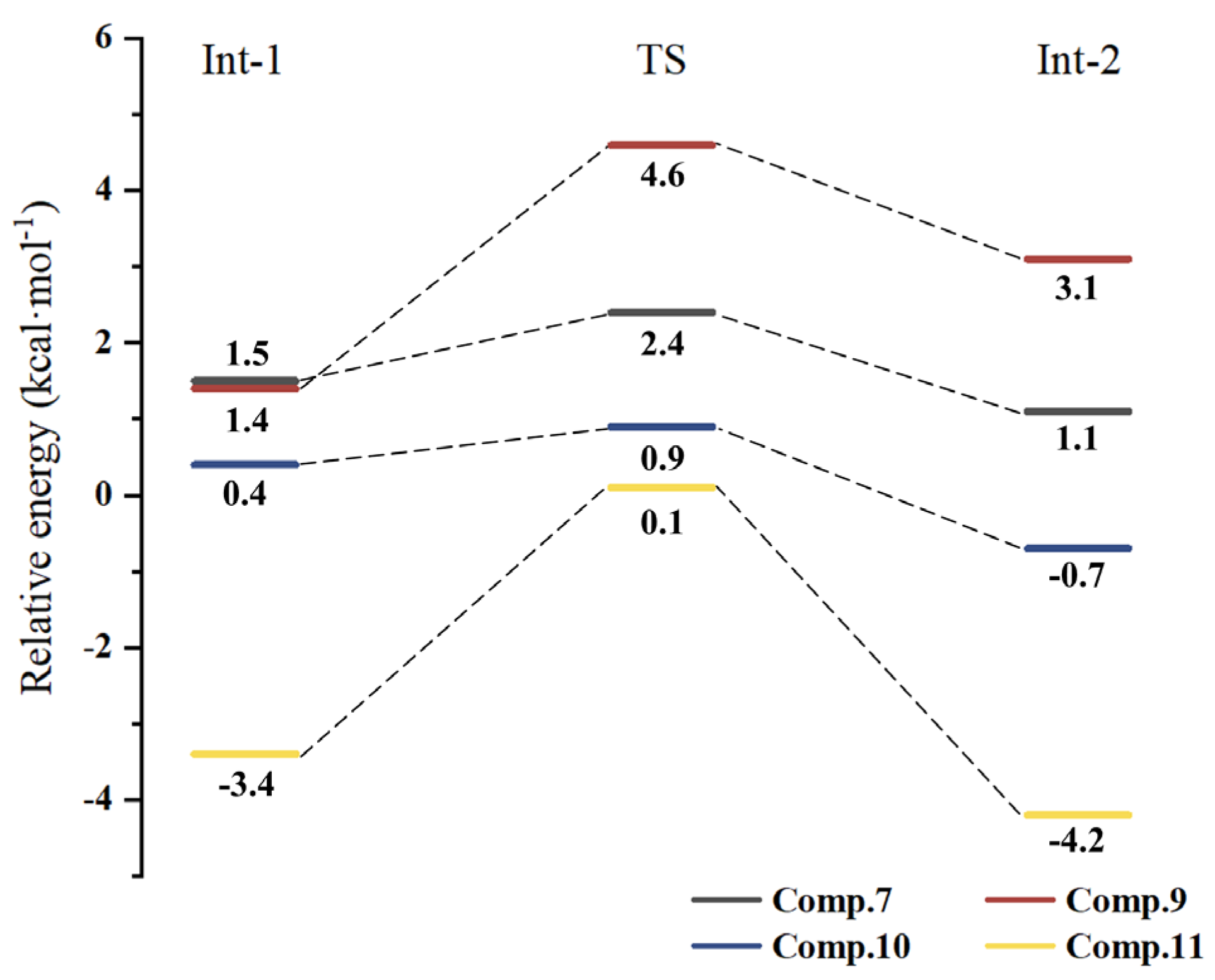
2.5. Potential Energy Surface of HOO· Radical Scavenging
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, R.; Yang, W.; Li, Y.; Sun, C. Multi-Residue Analytical Methods for Pesticides in Teas: A Review. Eur. Food Res. Technol. 2021, 247, 1839–1858. [Google Scholar] [CrossRef]
- Tsuji, P.A.; Stephenson, K.K.; Wade, K.L.; Liu, H.; Fahey, J.W. Structure-Activity Analysis of Flavonoids: Direct and Indirect Antioxidant, and Antiinflammatory Potencies and Toxicities. Nutr. Cancer 2013, 65, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Plumb, G.W.; Price, K.R.; Williamson, G. Antioxidant Properties of Flavonol Glycosides from Tea. Redox Rep. 1999, 4, 13–16. [Google Scholar] [CrossRef]
- Xue, Z.; Wang, J.; Chen, Z.; Ma, Q.; Guo, Q.; Gao, X.; Chen, H. Antioxidant, Antihypertensive, and Anticancer Activities of the Flavonoid Fractions from Green, Oolong, and Black Tea Infusion Waste. J. Food Biochem. 2018, 42, e12690. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, L.; Li, W.; Xing, Y.; Zhang, P.; Wang, Q.; Li, H.; Liu, H.; Yang, H.; Liu, X.; et al. Scented Tartary Buckwheat Tea: Aroma Components and Antioxidant Activity. Molecules 2019, 24, 4368. [Google Scholar] [CrossRef]
- Kaurinovic, B.; Vastag, D.; Kaurinovic, B.; Vastag, D. Flavonoids and Phenolic Acids as Potential Natural Antioxidants. In Antioxidants; IntechOpen: London, UK, 2019; ISBN 978-1-78923-920-1. [Google Scholar]
- He, H.-F.; Wei, K.; Yin, J.; Ye, Y. Insight into Tea Flavonoids: Composition and Chemistry. Food Rev. Int. 2021, 37, 812–823. [Google Scholar] [CrossRef]
- Son, N.T.; Mai Thanh, D.T.; Van Trang, N. Flavone Norartocarpetin and Isoflavone 2′-Hydroxygenistein: A Spectroscopic Study for Structure, Electronic Property and Antioxidant Potential Using DFT (Density Functional Theory). J. Mol. Struct. 2019, 1193, 76–88. [Google Scholar] [CrossRef]
- Koç, E.; Üngördü, A.; Candan, F. Antioxidant Properties of Methanolic Extract of ‘Veronica Multifida’ and DFT and HF Analyses of Its the Major Flavonoid Component. J. Mol. Struct. 2019, 1197, 436–442. [Google Scholar] [CrossRef]
- Shahab, S.; Sheikhi, M. Antioxidant Properties of the Phorbol: A DFT Approach. Russ. J. Phys. Chem. B 2020, 14, 15–18. [Google Scholar] [CrossRef]
- McArdle, S.; Endo, S.; Aspuru-Guzik, A.; Benjamin, S.C.; Yuan, X. Quantum Computational Chemistry. Rev. Mod. Phys. 2020, 92, 015003–015054. [Google Scholar] [CrossRef]
- Tungmunnithum, D.; Garros, L.; Drouet, S.; Cruz-Martins, N.; Hano, C. Extraction Kinetics and Reaction Rates of Sacred Lotus Stamen Tea Infusion-Derived Flavonoids in Relation with Its Antioxidant Capacity. Plants 2022, 11, 2234. [Google Scholar] [CrossRef]
- Ninh The, S.; Do Minh, T.; Nguyen Van, T. Isoflavones and Isoflavone Glycosides: Structural-Electronic Properties and Antioxidant Relations—A Case of DFT Study. J. Chem. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Ali, H.M.; Ali, I.H. Structure-Antioxidant Activity Relationships, QSAR, DFT Calculation, and Mechanisms of Flavones and Flavonols. Med. Chem. Res. 2019, 28, 2262–2269. [Google Scholar] [CrossRef]
- Anbazhakan, K.; Sadasivam, K.; Praveena, R.; Dhandapani, M. Target Prediction and Antioxidant Analysis on Isoflavones of Demethyltexasin: A DFT Study. J. Mol. Model. 2019, 25, 169. [Google Scholar] [CrossRef] [PubMed]
- Fifen, J.J.; Nsangou, M.; Dhaouadi, Z.; Motapon, O.; Jaidane, N. Solvent Effects on the Antioxidant Activity of 3,4-Dihydroxyphenylpyruvic Acid: DFT and TD-DFT Studies. Comput. Theor. Chem. 2011, 966, 232–243. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the Activity of Phenolic Antioxidants: Theoretical Method, Analysis of Substituent Effects, and Application to Major Families of Antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef]
- Nakanishi, I.; Miyazaki, K.; Shimada, T.; Ohkubo, K.; Urano, S.; Ikota, N.; Ozawa, T.; Fukuzumi, S.; Fukuhara, K. Effects of Metal Ions Distinguishing between One-Step Hydrogen- and Electron-Transfer Mechanisms for the Radical-Scavenging Reaction of (+)-Catechin. J. Phys. Chem. A 2002, 106, 11123–11126. [Google Scholar] [CrossRef]
- Bag, S.; Mondal, A.; Majumder, A.; Banik, A. Tea and Its Phytochemicals: Hidden Health Benefits & Modulation of Signaling Cascade by Phytochemicals. Food Chem. 2022, 371, 131098. [Google Scholar] [CrossRef]
- Spiegel, M.; Andruniów, T.; Sroka, Z. Flavones’ and Flavonols’ Antiradical Structure–Activity Relationship—A Quantum Chemical Study. Antioxidants 2020, 9, 461. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Dehkordi, M.M.; Asgarshamsi, M.H. Density Functional Theory Studies of the Antioxidants—A Review. J. Mol. Model. 2021, 27, 271. [Google Scholar] [CrossRef]
- Vo, Q.V.; Nam, P.C.; Thong, N.M.; Trung, N.T.; Phan, C.-T.D.; Mechler, A. Antioxidant Motifs in Flavonoids: O–H versus C–H Bond Dissociation. ACS Omega 2019, 4, 8935–8942. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Qiang, M.; Li, F.; Zhang, H.; Zhang, S. Theoretical Investigation on the Antioxidative Activity of Anthocyanidins: A DFT/B3LYP Study. Dye. Pigment. 2014, 103, 175–182. [Google Scholar] [CrossRef]
- Sun, L.; Lu, B.; Liu, Y.; Wang, Q.; Li, G.; Zhao, L.; Zhao, C. Synthesis, Characterization and Antioxidant Activity of Quercetin Derivatives. Synth. Commun. 2021, 51, 2944–2953. [Google Scholar] [CrossRef]
- Lesjak, M.; Beara, I.; Simin, N.; Pintać, D.; Majkić, T.; Bekvalac, K.; Orčić, D.; Mimica-Dukić, N. Antioxidant and Anti-Inflammatory Activities of Quercetin and Its Derivatives. J. Funct. Foods 2018, 40, 68–75. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A Computational Methodology for Accurate Predictions of Rate Constants in Solution: Application to the Assessment of Primary Antioxidant Activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef]
- Rammohan, A.; Bhaskar, B.V.; Camilo, A.; Gunasekar, D.; Gu, W.; Zyryanov, G.V. In Silico, in Vitro Antioxidant and Density Functional Theory Based Structure Activity Relationship Studies of Plant Polyphenolics as Prominent Natural Antioxidants. Arab. J. Chem. 2020, 13, 3690–3701. [Google Scholar] [CrossRef]
- Tao, Y.; Han, L.; Sun, A.; Sun, K.; Zhang, Q.; Liu, W.; Du, J.; Liu, Z. Crystal Structure and Computational Study on Methyl-3-Aminothiophene-2-Carboxylate. Crystals 2020, 10, 19. [Google Scholar] [CrossRef]
- Cao, H.; Cheng, W.-X.; Li, C.; Pan, X.-L.; Xie, X.-G.; Li, T.-H. DFT Study on the Antioxidant Activity of Rosmarinic Acid. J. Mol. Struct. THEOCHEM 2005, 719, 177–183. [Google Scholar] [CrossRef]
- Sharapov, M.G.; Gudkov, S.V.; Lankin, V.Z.; Novoselov, V.I. Role of Glutathione Peroxidases and Peroxiredoxins in Free Radical-Induced Pathologies. Biokhimiya 2021, 86, 1418–1433. [Google Scholar] [CrossRef]
- Ghiasi, M.; Heravi, M.M. Quantum Mechanical Study of Antioxidative Ability and Antioxidative Mechanism of Rutin (Vitamin P) in Solution. Carbohydr. Res. 2011, 346, 739–744. [Google Scholar] [CrossRef]
- Sulaiman, K.O.; Onawole, A.T. Quantum Chemical Evaluation of the Corrosion Inhibition of Novel Aromatic Hydrazide Derivatives on Mild Steel in Hydrochloric Acid. Comput. Theor. Chem. 2016, 1093, 73–80. [Google Scholar] [CrossRef]
- Fassihi, A.; Hasanzadeh, F.; Attar, A.; Saghaie, L.; Mohammadpour, M. Synthesis and Evaluation of Antioxidant Activity of Some Novel Hydroxypyridinone Derivatives: A DFT Approach for Explanation of Their Radical Scavenging Activity. Res. Pharm. Sci. 2020, 15, 515. [Google Scholar] [CrossRef] [PubMed]
- Sadasivam, K.; Kumaresan, R. Antioxidant Behavior of Mearnsetin and Myricetin Flavonoid Compounds—A DFT Study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Han, R.-M.; Tian, Y.-X.; Liu, Y.; Chen, C.-H.; Ai, X.-C.; Zhang, J.-P.; Skibsted, L.H. Comparison of Flavonoids and Isoflavonoids as Antioxidants. J. Agric. Food Chem. 2009, 57, 3780–3785. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Gauss, J. Calculation of NMR Chemical Shifts at Second-Order Many-Body Perturbation Theory Using Gauge-Including Atomic Orbitals. Chem. Phys. Lett. 1992, 191, 614–620. [Google Scholar] [CrossRef]
- Nakanishi, I.; Ohkubo, K.; Shoji, Y.; Fujitaka, Y.; Shimoda, K.; Matsumoto, K.; Fukuhara, K.; Hamada, H. Relationship between the Radical-Scavenging Activity of Selected Flavonols and Thermodynamic Parameters Calculated by Density Functional Theory. Free Radic. Res. 2020, 54, 535–539. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Deng, G.; Guo, R.; Chen, D.-F.; Fu, Z.-M. Substituent Effects on the Radical Scavenging Activity of Isoflavonoid. Int. J. Mol. Sci. 2019, 20, 397. [Google Scholar] [CrossRef]
- Wang, M.; He, X.; Taylor, M.; Lorpaiboon, W.; Mun, H.; Ho, J. Molecular Geometries and Vibrational Contributions to Reaction Thermochemistry Are Surprisingly Insensitive to the Choice of Basis Sets. J. Chem. Theory Comput. 2023, 19, 5036–5046. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Hansen, A.; Bauer, C.; Ehrlich, S.; Najibi, A.; Grimme, S. A Look at the Density Functional Theory Zoo with the Advanced GMTKN55 Database for General Main Group Thermochemistry, Kinetics and Noncovalent Interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [Google Scholar] [CrossRef] [PubMed]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cammi, R.; Tomasi, J. Remarks on the Use of the Apparent Surface Charges (ASC) Methods in Solvation Problems: Iterative versus Matrix-Inversion Procedures and the Renormalization of the Apparent Charges. J. Comput. Chem. 1995, 16, 1449–1458. [Google Scholar] [CrossRef]
- Bartmess, J.E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Liu, Y.; Ke, Z.; Cui, J.; Chen, W.-H.; Ma, L.; Wang, B. Synthesis, Inhibitory Activities, and QSAR Study of Xanthone Derivatives as α-Glucosidase Inhibitors. Bioorg. Med. Chem. 2008, 16, 7185–7192. [Google Scholar] [CrossRef]
- Chattaraj, P.; Sarkar, U.; Roy, D. Electrophilicity Index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- Marković, Z.; Tošović, J.; Milenković, D.; Marković, S. Revisiting the Solvation Enthalpies and Free Energies of the Proton and Electron in Various Solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name | O-H Position | BDEs | ||
|---|---|---|---|---|---|
| Gas Phase | Water | Ethanol | |||
| 1 | Genkwanin | 4′-OH | 83.1 | 113.3 | 113.3 |
| 2 | Apigenin | 4′-OH | 83.2 | 112.1 | 83.3 |
| 3 | Luteolin | 4′-OH | 74.2 | 107.2 | 75.8 |
| 4 | Acacetin | 7-OH | 81.3 | 117.5 | 117.6 |
| 5 | Wogonin | 7-OH | 74.2 | 109.8 | 109.8 |
| 6 | Chrysin | 7-OH | 88.3 | 114.4 | 114.4 |
| 7 | Kaempferol | 3-OH | 74.4 | 103.9 | 103.9 |
| 8 | Rhamnetin | 4′-OH | 54.6 | 109.3 | 109.3 |
| 9 | Fisetin | 3-OH | 72.9 | 103.7 | 103.8 |
| 10 | Quercetin | 3-OH | 70.5 | 105.1 | 105.1 |
| 11 | Myricetin | 4′-OH | 65.9 | 106.2 | 106.0 |
| 12 | Morin | 3-OH | 75.5 | 109.2 | 109.3 |
| 13 | Afzelechin | 5-OH | 77.6 | 107.9 | 108.0 |
| 14 | Catechin | 3′-OH | 74.5 | 104.8 | 104.8 |
| 15 | Epicatechin | 4′-OH | 79.4 | 107.1 | 107.3 |
| 16 | Gallocatechin | 4′-OH | 75.6 | 72.8 | 105.2 |
| 17 | Liquiritigenin | 4′-OH | 83.1 | 107.5 | 107.6 |
| 18 | Naringenin | 4′-OH | 82.9 | 110.3 | 82.6 |
| 19 | Hesperitin | 3′-OH | 71.3 | 109.4 | 109.4 |
| 20 | Eriodictyol | 3′-OH | 75.0 | 106.0 | 106.0 |
| 21 | Pinocembrin | 5-OH | 85.0 | 109.4 | 109.5 |
| 22 | Aromadendrin | 4′-OH | 75.5 | 113.2 | 113.3 |
| 23 | Taxifolin | 4′-OH | 74.4 | 108.4 | 108.4 |
| 24 | Dihydromyricetin | 4′-OH | 75.8 | 109.3 | 109.5 |
| 25 | Dihydromorin | 2′-OH | 81.5 | 114.5 | 114.5 |
| Compound | Name | IPs | O-H Position | PDEs | SPT-ET (IP+PDE) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas Phase | Water | Ethanol | Gas Phase | Water | Ethanol | Gas Phase | Water | Ethanol | |||
| 1 | Genkwanin | 141.5 | 138.3 | 139.2 | 4′-OH | 256.0 | 289.5 | 288.5 | 397.5 | 427.8 | 427.7 |
| 2 | Apigenin | 143.4 | 138.0 | 110.0 | 4’-OH | 254.3 | 288.6 | 287.7 | 397.7 | 426.6 | 397.7 |
| 3 | Luteolin | 138.8 | 135.5 | 105.1 | 4’-OH | 249.9 | 286.2 | 285.1 | 388.7 | 421.7 | 390.2 |
| 4 | Acacetin | 140.1 | 137.2 | 138.1 | 7-OH | 262.1 | 294.8 | 294.0 | 402.2 | 432.0 | 432.1 |
| 5 | Wogonin | 133.2 | 128.9 | 129.9 | 7-OH | 260.6 | 295.3 | 294.4 | 393.8 | 424.2 | 424.3 |
| 6 | Chrysin | 150.4 | 140.4 | 141.5 | 7-OH | 252.3 | 288.4 | 287.4 | 402.7 | 428.8 | 428.9 |
| 7 | Kaempferol | 135.0 | 132.3 | 133.3 | 3-OH | 251.8 | 286.0 | 285.0 | 386.8 | 418.3 | 418.3 |
| 8 | Rhamnetin | 125.1 | 127.6 | 128.5 | 4’-OH | 263.0 | 296.1 | 295.2 | 388.1 | 423.7 | 423.7 |
| 9 | Fisetin | 134.5 | 131.2 | 132.2 | 3-OH | 252.9 | 286.9 | 286.0 | 387.4 | 418.1 | 418.2 |
| 10 | Quercetin | 128.7 | 129.5 | 130.5 | 3-OH | 256.2 | 290.1 | 289.1 | 384.9 | 419.6 | 419.6 |
| 11 | Myricetin | 124.8 | 129.1 | 129.9 | 4′-OH | 255.5 | 291.6 | 290.6 | 380.3 | 420.7 | 420.5 |
| 12 | Morin | 130.2 | 132.1 | 133.0 | 3-OH | 259.9 | 291.6 | 290.7 | 390.1 | 423.7 | 423.7 |
| 13 | Afzelechin | 139.5 | 132.7 | 133.7 | 5-OH | 256.9 | 289.7 | 288.8 | 396.4 | 422.4 | 422.5 |
| 14 | Catechin | 134.4 | 133.1 | 134.2 | 3′-OH | 254.6 | 286.1 | 285.0 | 389.0 | 419.2 | 419.2 |
| 15 | Epicatechin | 135.7 | 131.8 | 132.7 | 4′-OH | 258.1 | 289.7 | 289.0 | 393.8 | 421.5 | 421.7 |
| 16 | Gallocatechin | 133.8 | 132.7 | 133.6 | 4′-OH | 256.4 | 254.6 | 286.0 | 390.2 | 387.3 | 419.6 |
| 17 | Liquiritigenin | 151.9 | 140.7 | 141.9 | 4′-OH | 245.7 | 281.2 | 280.1 | 397.6 | 421.9 | 422.0 |
| 18 | Naringenin | 145.0 | 140.0 | 113.3 | 4′-OH | 252.3 | 284.7 | 283.7 | 397.3 | 424.7 | 397.0 |
| 19 | Hesperitin | 134.4 | 132.2 | 133.2 | 3′-OH | 256.9 | 291.7 | 290.7 | 391.3 | 423.9 | 423.9 |
| 20 | Eriodictyol | 138.8 | 133.9 | 135.0 | 3′-OH | 250.6 | 286.6 | 285.4 | 389.4 | 420.5 | 420.4 |
| 21 | Pinocembrin | 154.1 | 141.5 | 142.7 | 5-OH | 245.4 | 282.4 | 281.3 | 399.5 | 423.9 | 424.0 |
| 22 | Aromadendrin | 146.9 | 142.0 | 143.2 | 4′-OH | 250.9 | 285.7 | 284.6 | 397.8 | 427.7 | 427.8 |
| 23 | Taxifolin | 139.6 | 135.4 | 136.6 | 4′-OH | 249.2 | 287.4 | 286.3 | 388.8 | 422.8 | 422.9 |
| 24 | Dihydromyricetin | 135.2 | 134.5 | 135.6 | 4′-OH | 255.1 | 289.2 | 288.3 | 390.3 | 423.7 | 423.9 |
| 25 | Dihydromorin | 140.5 | 137.8 | 138.9 | 5′-OH | 255.5 | 291.2 | 290.1 | 396.0 | 429.0 | 429.0 |
| Compound | Name | O-H Position | PAs | ETEs | SPLET (PA+ETE) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas Phase | Water | Ethanol | Gas Phase | Water | Ethanol | Gas Phase | Water | Ethanol | |||
| 1 | Genkwanin | 4′-OH | 321.6 | 315.1 | 316.2 | 73.0 | 109.7 | 109.7 | 394.6 | 424.8 | 425.9 |
| 2 | Apigenin | 4′-OH | 321.6 | 313.8 | 286.1 | 73.2 | 109.9 | 109.9 | 394.8 | 423.7 | 396.0 |
| 3 | Luteolin | 4′-OH | 314.1 | 311.4 | 281.0 | 71.7 | 107.3 | 107.3 | 385.8 | 418.7 | 388.3 |
| 4 | Acacetin | 7-OH | 329.6 | 315.6 | 317.0 | 69.7 | 113.5 | 113.5 | 399.3 | 429.1 | 430.5 |
| 5 | Wogonin | 7-OH | 324.3 | 313.6 | 314.9 | 66.6 | 107.7 | 107.7 | 390.9 | 421.3 | 422.6 |
| 6 | Chrysin | 7-OH | 328.2 | 311.3 | 312.6 | 71.6 | 114.6 | 114.6 | 399.8 | 425.9 | 427.2 |
| 7 | Kaempferol | 3-OH | 325.1 | 316.2 | 317.6 | 58.8 | 99.2 | 99.2 | 383.9 | 415.4 | 416.8 |
| 8 | Rhamnetin | 4′-OH | 319.4 | 317.2 | 318.3 | 65.7 | 103.6 | 103.6 | 385.1 | 420.8 | 421.9 |
| 9 | Fisetin | 3-OH | 329.2 | 317.7 | 319.2 | 55.2 | 97.5 | 97.5 | 384.4 | 415.2 | 416.7 |
| 10 | Quercetin | 3-OH | 327.9 | 320.2 | 321.6 | 54.1 | 96.5 | 96.5 | 382.0 | 416.7 | 418.1 |
| 11 | Myricetin | 4′-OH | 307.0 | 313.1 | 314.0 | 70.4 | 104.6 | 104.6 | 377.4 | 417.7 | 418.6 |
| 12 | Morin | 3-OH | 331.7 | 322.7 | 324.3 | 55.5 | 98.0 | 98.0 | 387.2 | 420.7 | 422.3 |
| 13 | Afzelechin | 5-OH | 338.6 | 318.7 | 320.3 | 54.9 | 100.7 | 100.7 | 393.5 | 419.4 | 421.0 |
| 14 | Catechin | 3′-OH | 326.9 | 314.4 | 315.7 | 59.2 | 101.9 | 101.9 | 386.1 | 416.3 | 417.6 |
| 15 | Epicatechin | 4′-OH | 338.3 | 321.1 | 322.7 | 52.6 | 97.5 | 97.5 | 390.9 | 418.6 | 420.2 |
| 16 | Gallocatechin | 4′-OH | 332.7 | 320.2 | 321.6 | 54.4 | 64.2 | 64.2 | 387.1 | 384.4 | 385.8 |
| 17 | Liquiritigenin | 4′-OH | 336.2 | 316.3 | 317.8 | 58.4 | 102.7 | 102.7 | 394.6 | 419.0 | 420.5 |
| 18 | Naringenin | 4′-OH | 337.3 | 319.4 | 293.1 | 57.1 | 102.5 | 102.5 | 394.4 | 421.9 | 395.6 |
| 19 | Hesperitin | 3′-OH | 335.7 | 322.2 | 323.3 | 52.8 | 98.7 | 98.7 | 388.5 | 420.9 | 422.0 |
| 20 | Eriodictyol | 3′-OH | 330.0 | 316.8 | 318.2 | 56.6 | 100.7 | 100.7 | 386.6 | 417.5 | 418.9 |
| 21 | Pinocembrin | 5-OH | 331.1 | 311.0 | 312.5 | 65.4 | 110.0 | 110.0 | 396.5 | 421.0 | 422.5 |
| 22 | Aromadendrin | 4′-OH | 335.4 | 321.0 | 322.5 | 59.4 | 103.7 | 103.7 | 394.8 | 424.7 | 426.2 |
| 23 | Taxifolin | 4′-OH | 326.8 | 318.0 | 319.4 | 59.1 | 101.9 | 101.9 | 385.9 | 419.9 | 421.3 |
| 24 | Dihydromyricetin | 4′-OH | 332.4 | 322.4 | 324.1 | 55.0 | 98.4 | 98.4 | 387.4 | 420.8 | 422.5 |
| 25 | Dihydromorin | 5′-OH | 330.7 | 321.2 | 322.6 | 62.3 | 104.8 | 104.8 | 393.0 | 426.0 | 427.4 |
| Compound | Name | Medium | η(eV) | μ (eV) | χ (eV) | ω(eV) | ||
|---|---|---|---|---|---|---|---|---|
| ω− | ω+ | ω | ||||||
| 7 | Kaempferol | Gas phase | 2.018 | −4.029 | 4.029 | 6.288 | 2.260 | 4.022 |
| Water | 1.992 | −4.060 | 4.060 | 6.416 | 2.356 | 4.137 | ||
| Ethanol | 1.994 | −4.054 | 4.054 | 6.398 | 2.343 | 4.121 | ||
| 9 | Fisetin | Gas phase | 2.062 | −3.975 | 3.975 | 6.076 | 2.102 | 3.831 |
| Water | 2.007 | −4.034 | 4.034 | 6.322 | 2.288 | 4.054 | ||
| Ethanol | 2.010 | −4.027 | 4.027 | 6.300 | 2.272 | 4.035 | ||
| 10 | Quercetin | Gas phase | 2.115 | −3.818 | 3.818 | 5.619 | 1.802 | 3.446 |
| Water | 2.047 | −3.906 | 3.906 | 5.934 | 2.029 | 3.726 | ||
| Ethanol | 2.051 | −3.897 | 3.897 | 5.907 | 2.010 | 3.702 | ||
| 11 | Myricetin | Gas phase | 1.997 | −4.083 | 4.083 | 6.467 | 2.384 | 4.176 |
| Water | 1.995 | −4.139 | 4.139 | 6.613 | 2.474 | 4.294 | ||
| Ethanol | 1.996 | −4.132 | 4.132 | 6.592 | 2.461 | 4.277 | ||
| Dipole moment (debye) | IP0 (eV) | EA (eV) | ELUMO-HOMO (eV) | Energy (kcal/mol) | ||||
| 7 | Kaempferol | Gas phase | 4.832 | 6.046 | 2.011 | 4.035 | −2.011 | |
| Water | 6.819 | 6.052 | 2.068 | 3.985 | −645,951.4 | |||
| Ethanol | 6.731 | 6.049 | 2.060 | 3.989 | −645,997.4 | |||
| 9 | Fisetin | Gas phase | 5.627 | 6.036 | 1.913 | 4.123 | −645,996.8 | |
| Water | 8.329 | 6.041 | 2.027 | 4.014 | −645,943.0 | |||
| Ethanol | 8.819 | 6.037 | 2.017 | 4.020 | −645,990.4 | |||
| 10 | Quercetin | Gas phase | 4.465 | 5.932 | 1.703 | 4.229 | −645,989.7 | |
| Water | 6.804 | 5.952 | 1.859 | 4.094 | −693,159.5 | |||
| Ethanol | 6.673 | 5.948 | 1.846 | 4.102 | −693,213.7 | |||
| 11 | Myricetin | Gas phase | 6.909 | 6.080 | 2.087 | 3.993 | −693,212.8 | |
| Water | 9.732 | 6.134 | 2.144 | 3.989 | −740,235.3 | |||
| Ethanol | 9.588 | 6.127 | 2.136 | 3.991 | −740,288.9 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.; Wang, Y.; Tan, X.; Wang, Y.; Li, W.; Li, X. Investigating the Antioxidant Efficiency of Tea Flavonoid Derivatives: A Density Functional Theory Study. Int. J. Mol. Sci. 2025, 26, 2587. https://doi.org/10.3390/ijms26062587
Hou Y, Wang Y, Tan X, Wang Y, Li W, Li X. Investigating the Antioxidant Efficiency of Tea Flavonoid Derivatives: A Density Functional Theory Study. International Journal of Molecular Sciences. 2025; 26(6):2587. https://doi.org/10.3390/ijms26062587
Chicago/Turabian StyleHou, Yingmin, Yuxi Wang, Xiaofei Tan, Yi Wang, Wenzhi Li, and Xianzhen Li. 2025. "Investigating the Antioxidant Efficiency of Tea Flavonoid Derivatives: A Density Functional Theory Study" International Journal of Molecular Sciences 26, no. 6: 2587. https://doi.org/10.3390/ijms26062587
APA StyleHou, Y., Wang, Y., Tan, X., Wang, Y., Li, W., & Li, X. (2025). Investigating the Antioxidant Efficiency of Tea Flavonoid Derivatives: A Density Functional Theory Study. International Journal of Molecular Sciences, 26(6), 2587. https://doi.org/10.3390/ijms26062587