
Comparative Transcriptome Analysis of Arabidopsis Seedlings Under Heat Stress on Whole Plants, Shoots, and Roots Reveals New HS-Regulated Genes, Organ-Specific Responses, and Shoots-Roots Communication
 ,
,
Abstract
1. Introduction
2. Results
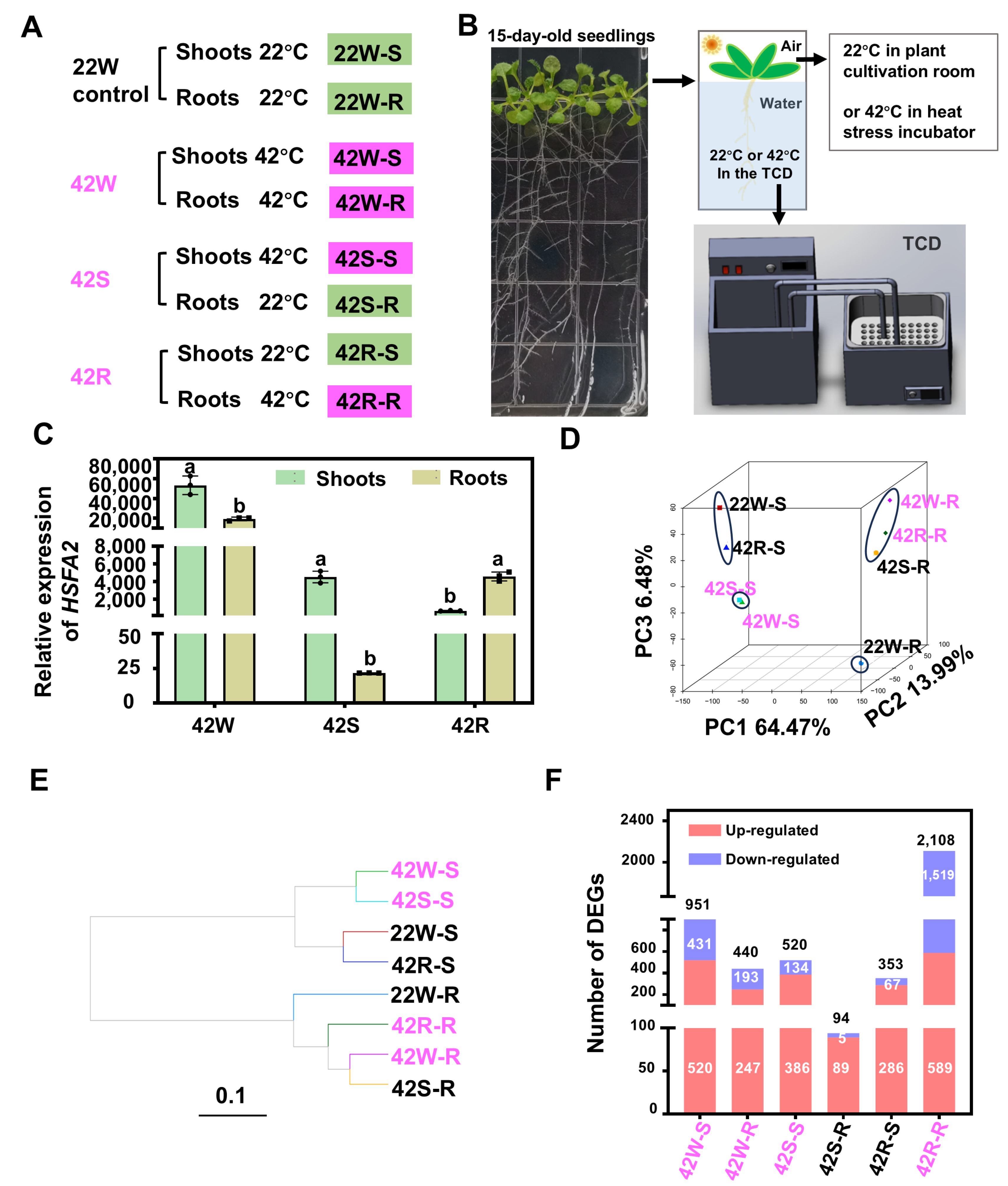
2.1. Transcriptomic Data Were Obtained from the Aboveground Shoots and Roots of Arabidopsis Seedlings Following Heat Shock Treatment of the Whole Plant, Shoots, or Roots
2.2. The Transcriptomic Profiles of the Aboveground Shoots Were Significantly Different from Those of the Roots in Whole Plants Subjected to Early HS Treatment
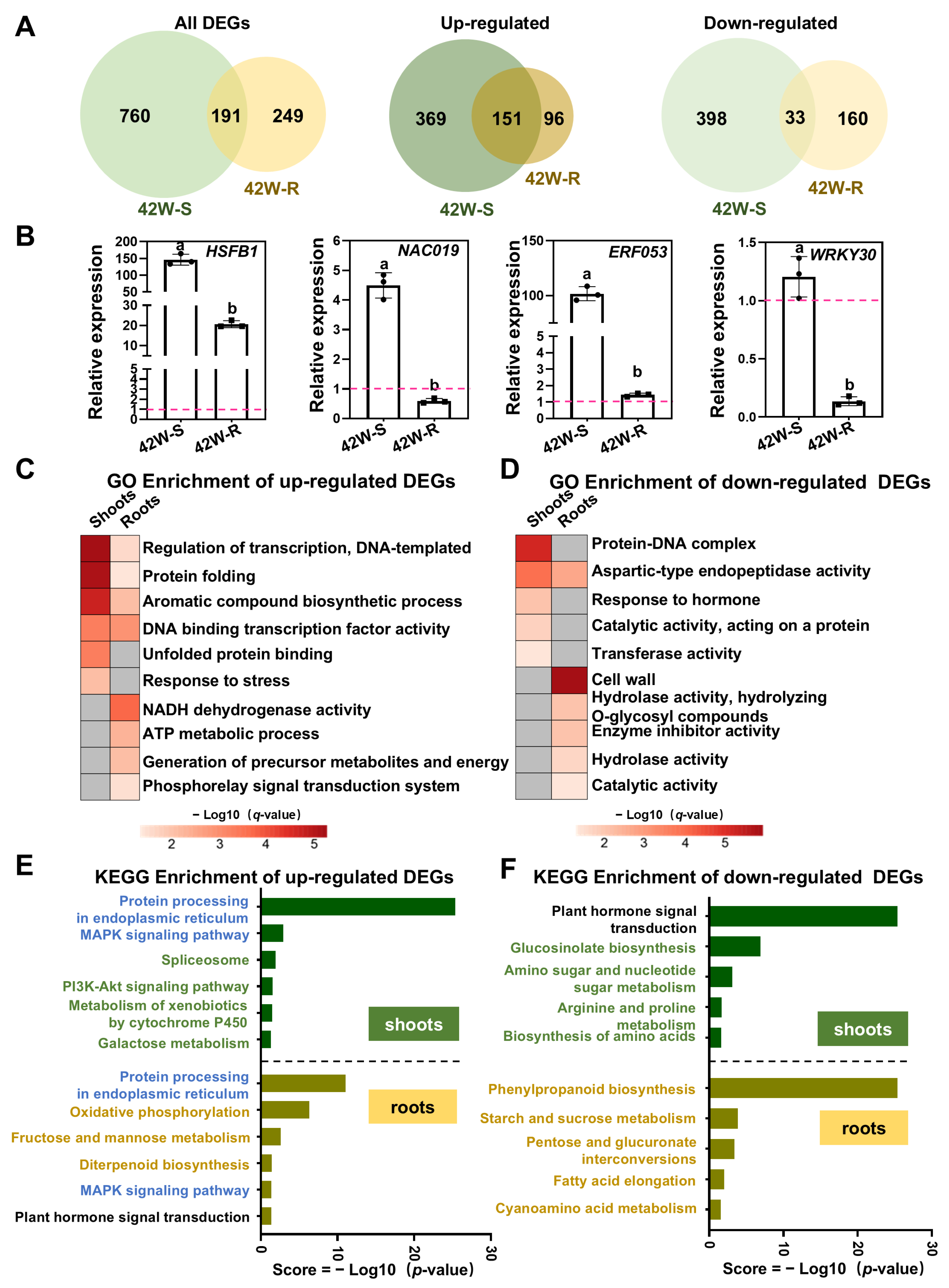
2.3. 62 DEGs Were Specifically Expressed Following HS Treatment of Shoots
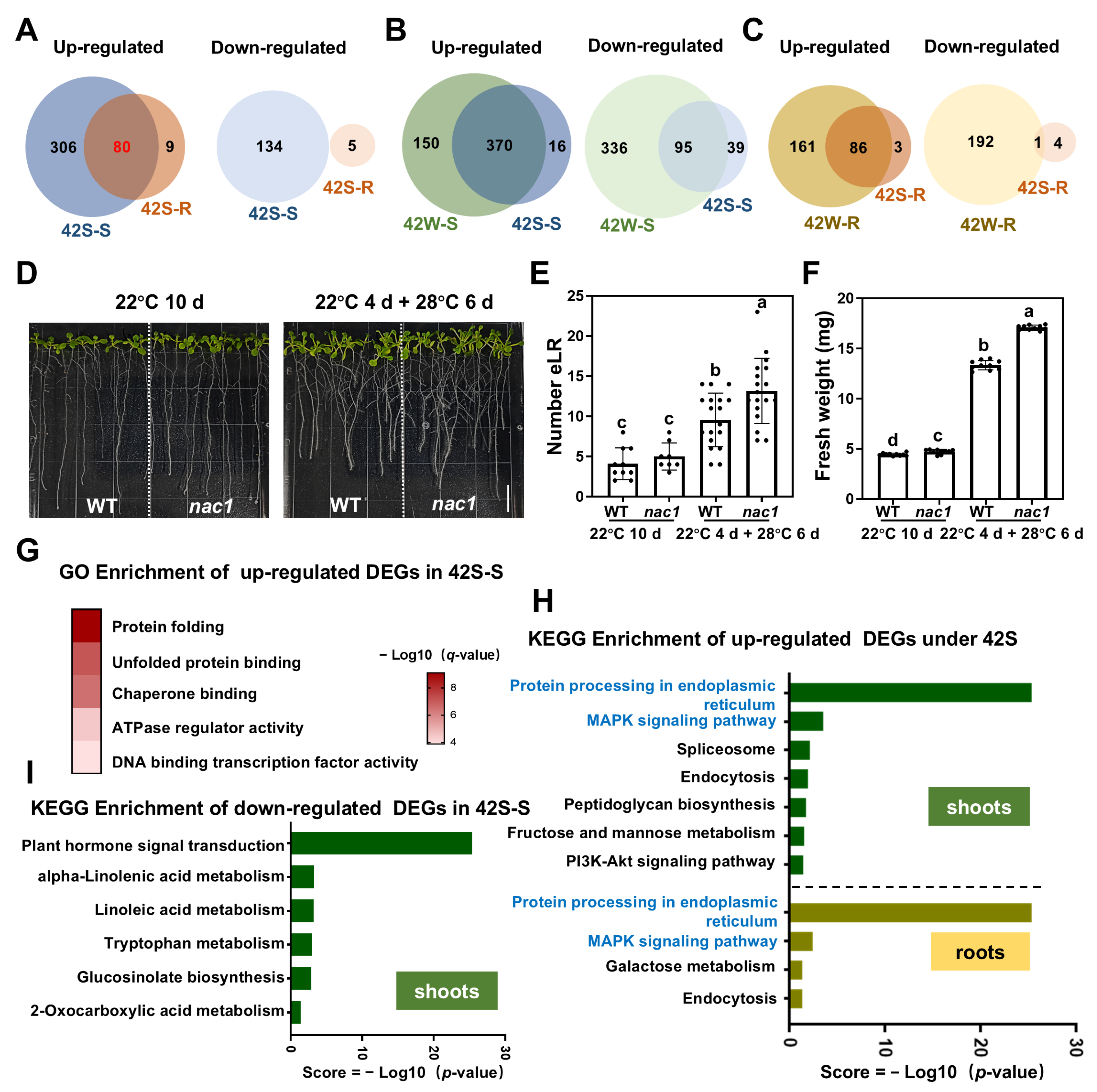
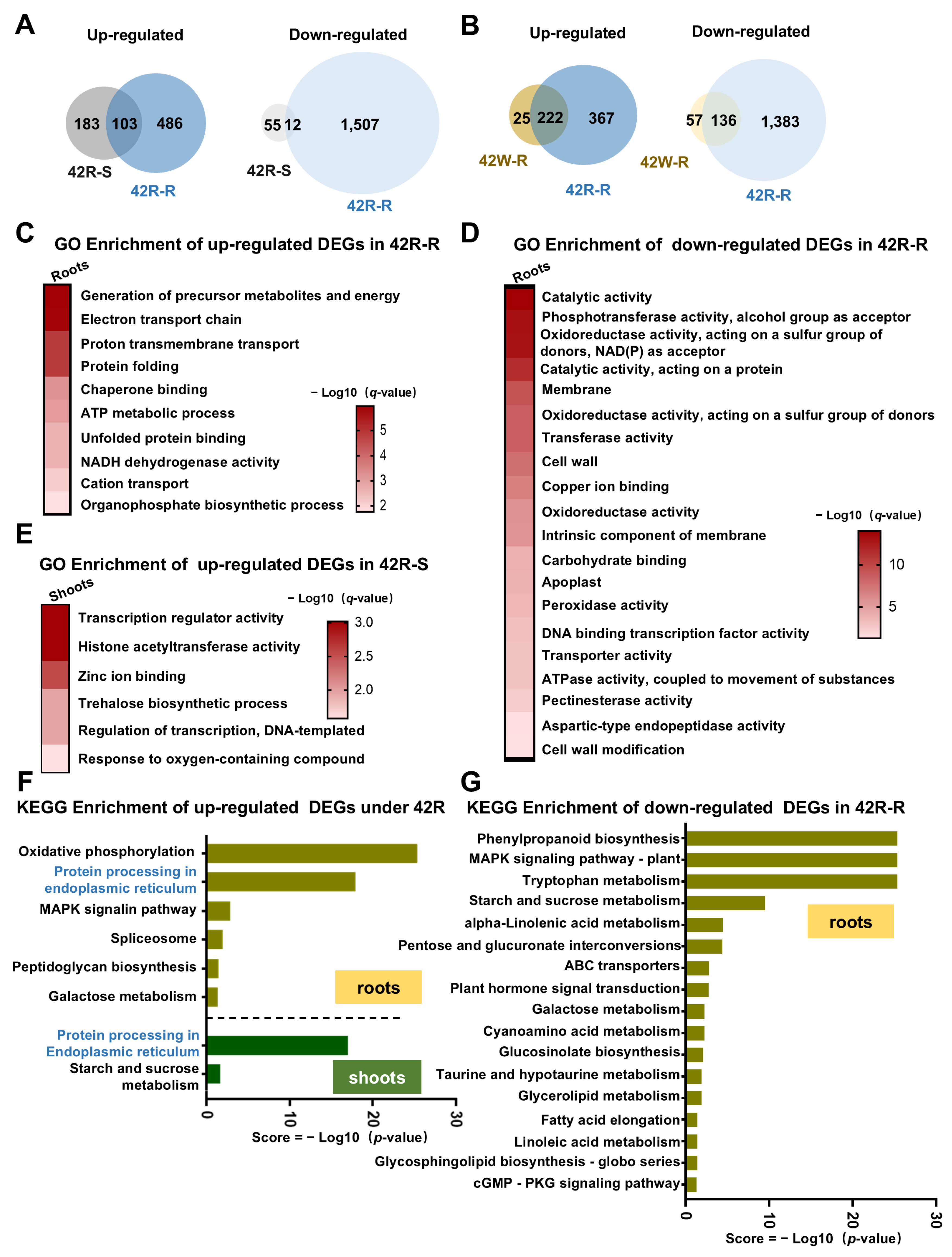
2.4. The Transcriptional Response in Roots of Plants Exposed to Root-Specific HS Was Much More Pronounced than That in Roots from Whole Plants Exposed to HS
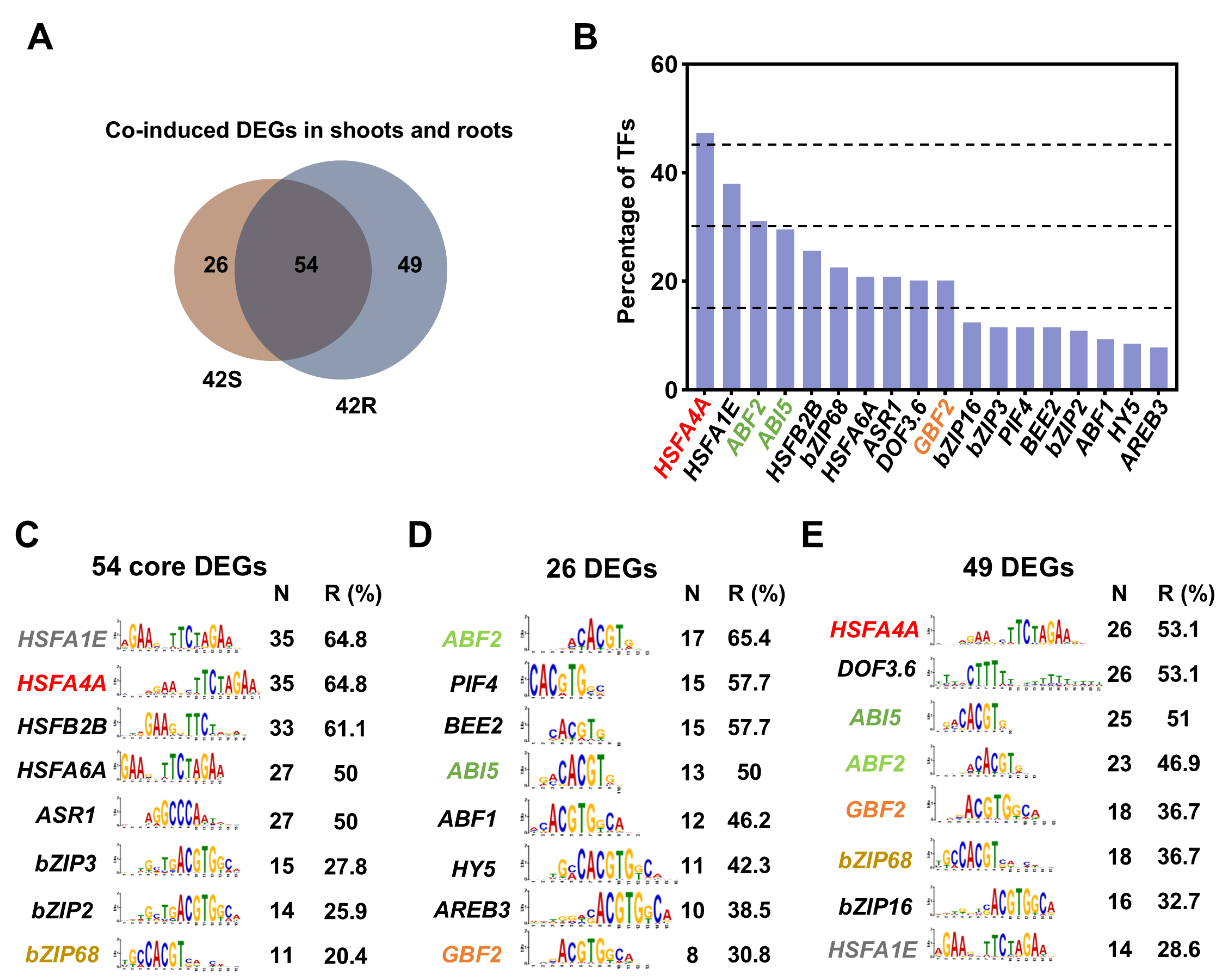
2.5. The Co-Induced DEGs in Shoots and Roots Following Shoot- or Root-Specific HS Treatment May Be Regulated by Transcription Factors Acting Downstream of ROS, ABA, and BR Signaling
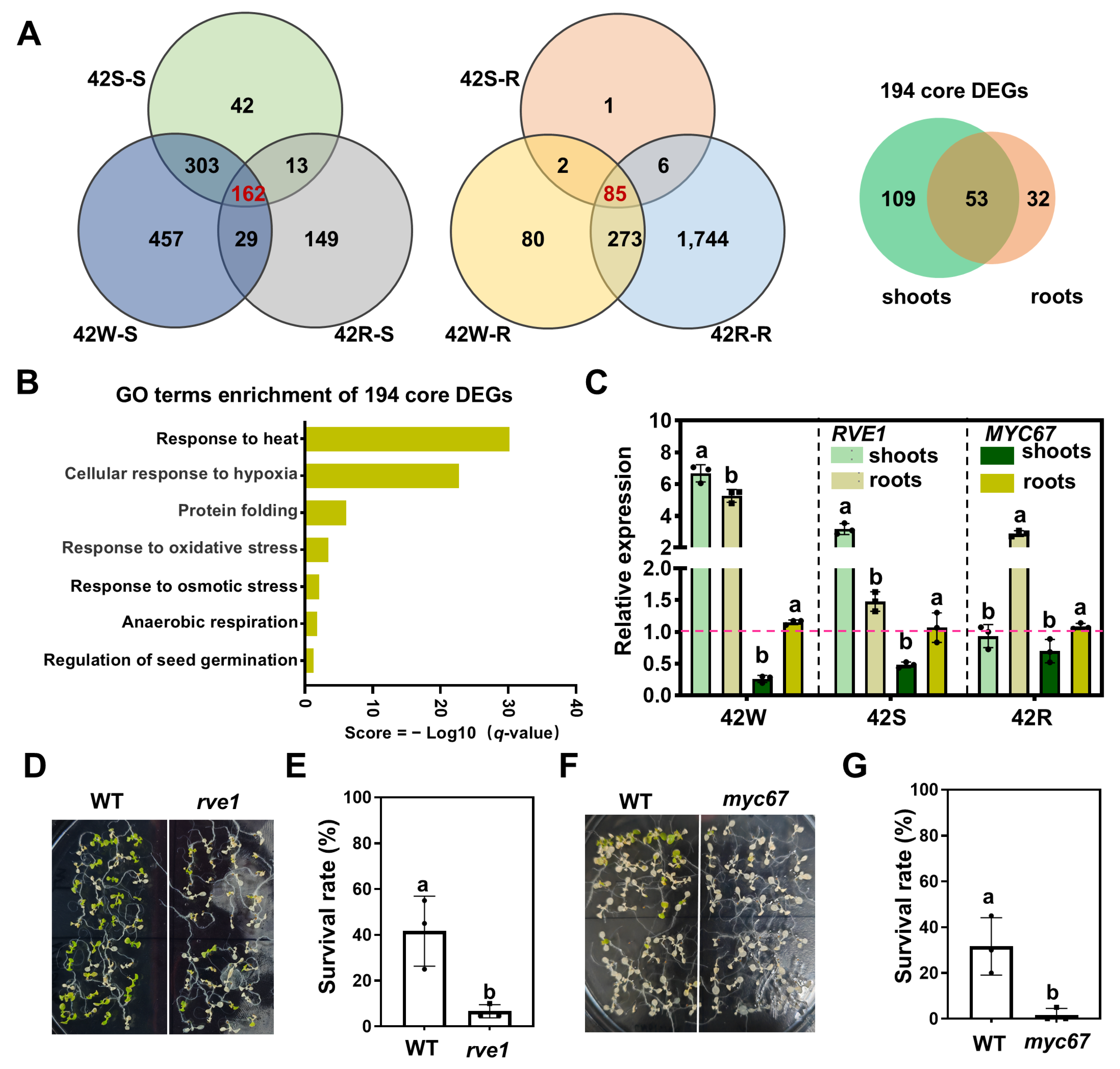
2.6. A Total of 194 Core DEGs Responsive to Early HS Were Identified Using Three HS Treatments
3. Discussion
3.1. Comparative Analysis of the Transcriptome Between HS-Treated Whole Plants and HS-Treated Aboveground Shoots or Roots
3.2. Different Response Strategies to Heat Shock Are Employed in Aboveground Shoots and Roots
3.3. The Reciprocal Influence and Intercommunication Between Aboveground Shoots and Roots in Response to Heat Shock
4. Methods
4.1. Plant Materials
4.2. Stress Treatment and Sample Collection
4.3. RNA Isolation and Assessment
4.4. Illumina Transcriptome Library Preparation, Sequencing, and Data Analysis
4.5. Quantification of Gene Expression Levels and DEG Analysis
4.6. Functional Enrichment Among the DEGs
4.7. Differential Gene Cis-Element Enrichment
4.8. RT-qPCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HS | Heat shock; |
| 22W | Whole-plant treatment at 22 °C; |
| 42W | Whole-plant treatment at 42 °C; |
| 42S | Aboveground shoot-specific treatment at 42 °C; |
| 42R | Root-specific treatment at 42 °C; |
| TCD | Temperature control device; |
| RT-qPCR | Reverse transcription-quantitative polymerase chain reaction; |
| RNA-Seq | RNA sequencing; |
| TPM | Million mapped reads; |
| PCA | Principal component analysis; |
| DEGs | Differentially expressed genes; |
| GO | Gene Ontology; |
| KEGG | Kyoto Encyclopedia of Genes and Genomes; |
| ABA | Abscisic acid; |
| BR | Brassinosteroid; |
| SA | Salicylic acid; |
| ET | ethylene; |
| ROS | Reactive oxygen species; |
| eLR | Emerged lateral roots; |
| MEME | Multiple Em for Motif Elicitation; |
| HSF/HSPs | Heat shock factors/proteins; |
| TFs | Transcription factors; |
| FC | Fold change; |
| min | Minutes; |
| s | Seconds. |
References
- Ibanez, C.; Poeschl, Y.; Peterson, T.; Bellstadt, J.; Denk, K.; Gogol-Doring, A.; Quint, M.; Delker, C. Ambient temperature and genotype differentially affect developmental and phenotypic plasticity in Arabidopsis thaliana. BMC Plant Biol. 2017, 17, 114. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, Y.; Xie, Y.; Gao, Y.; Li, W.; Lang, S. Transcriptomic and Metabolomic Analyses of the Effects of Exogenous Trehalose on Heat Tolerance in Wheat. Int. J. Mol. Sci. 2022, 23, 5194. [Google Scholar] [CrossRef]
- Li, H.; Xue, M.D.; Zhang, H.R.; Zhao, F.Y.; Li, X.Y.; Yu, S.C.; Jiang, D.H. A warm temperature-released negative feedback loop fine-tunes PIF4-mediated thermomorphogenesis in Arabidopsis. Plant Commun. 2024, 5, 100833. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, R.; Xu, X.; Fowler, J.C.; Miller, T.E.X.; Dong, T. Effect of summer warming on growth, photosynthesis and water status in female and male Populus cathayana: Implications for sex-specific drought and heat tolerances. Tree Physiol. 2020, 40, 1178–1191. [Google Scholar] [CrossRef]
- Raja, V.; Qadir, S.U.; Alyemeni, M.N.; Ahmad, P. Impact of drought and heat stress individually and in combination on physio-biochemical parameters, antioxidant responses, and gene expression in Solanum lycopersicum. 3 Biotech 2020, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.B.; Brady, S.M. Plant developmental responses to climate change. Dev. Biol. 2016, 419, 64–77. [Google Scholar] [CrossRef]
- Hedhly, A.; Hormaza, J.I.; Herrero, M. Global warming and sexual plant reproduction. Trends Plant Sci. 2009, 14, 30–36. [Google Scholar] [CrossRef]
- Li, B.J.; Gao, Z.H.; Liu, X.Y.; Sun, D.Y.; Tang, W.Q. Transcriptional Profiling Reveals a Time-of-Day-Specific Role of REVEILLE 4/8 in Regulating the First Wave of Heat Shock-Induced Gene Expression in Arabidopsis. Plant Cell 2019, 31, 2353–2369. [Google Scholar] [CrossRef]
- Hayes, S.; Schachtschabel, J.; Mishkind, M.; Munnik, T.; Arisz, S.A. Hot topic: Thermosensing in plants. Plant Cell Environ. 2021, 44, 2018–2033. [Google Scholar] [CrossRef]
- Zhao, J.G.; Lu, Z.G.; Wang, L.; Jin, B. Plant Responses to Heat Stress: Physiology, Transcription, Noncoding RNAs, and Epigenetics. Int. J. Mol. Sci. 2021, 22, 117. [Google Scholar] [CrossRef]
- Li, Y.Q.; Liu, Y.A.; Jin, L.B.; Peng, R.Y. Crosstalk between Ca2+ and Other Regulators Assists Plants in Responding to Abiotic Stress. Plants 2022, 11, 1351. [Google Scholar] [CrossRef] [PubMed]
- Castroverde, C.D.M.; Dina, D. Temperature regulation of plant hormone signaling during stress and development. J. Exp. Bot. 2021, 72, 7436–7458. [Google Scholar] [CrossRef]
- Vives-Peris, V.; López-Climent, M.F.; Pérez-Clemente, R.M.; Gómez-Cadenas, A. Root Involvement in Plant Responses to Adverse Environmental Conditions. Agronomy 2020, 10, 942. [Google Scholar] [CrossRef]
- Bellstaedt, J.; Trenner, J.; Lippmann, R.; Poeschl, Y.; Zhang, X.; Friml, J.; Quint, M.; Delker, C. A Mobile Auxin Signal Connects Temperature Sensing in Cotyledons with Growth Responses in Hypocotyls. Plant Physiol. 2019, 180, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, M.P.; Conesa, C.M.; Lozano-Enguita, A.; Baca-Gonzalez, V.; Simancas, B.; Navarro-Neila, S.; Sanchez-Bermudez, M.; Salas-Gonzalez, I.; Caro, E.; Castrillo, G.; et al. Temperature changes in the root ecosystem affect plant functionality. Plant Commun. 2023, 4, 100514. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Wang, W.L.; Huq, E. Spatial regulation of thermomorphogenesis by HY5 and PIF4 in Arabidopsis. Nat. Commun. 2021, 12, 3656. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Kieffer, M.; Yu, H.; Kepinski, S.; Estelle, M. HSP90 regulates temperature-dependent seedling growth in Arabidopsis by stabilizing the auxin co-receptor F-box protein TIR1. Nat. Commun. 2016, 7, 10269. [Google Scholar] [CrossRef]
- Feraru, E.; Feraru, M.I.; Barbez, E.; Waidmann, S.; Sun, L.; Gaidora, A.; Kleine-Vehn, J. PILS6 is a temperature-sensitive regulator of nuclear auxin input and organ growth in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2019, 116, 3893–3898. [Google Scholar] [CrossRef]
- Valdés-López, O.; Batek, J.; Gomez-Hernandez, N.; Nguyen, C.T.; Isidra-Arellano, M.C.; Zhang, N.; Joshi, T.; Xu, D.; Hixson, K.K.; Weitz, K.K.; et al. Soybean Roots Grown under Heat Stress Show Global Changes in Their Transcriptional and Proteomic Profiles. Front. Plant Sci. 2016, 7, 517. [Google Scholar] [CrossRef]
- Estravis-Barcala, M.; Heer, K.; Marchelli, P.; Ziegenhagen, B.; Arana, M.V.; Bellora, N. Deciphering the transcriptomic regulation of heat stress responses in Nothofagus pumilio. PLoS ONE 2021, 16, e0246615. [Google Scholar] [CrossRef]
- Wahid, A. Physiological implications of metabolite biosynthesis for net assimilation and heat-stress tolerance of sugarcane (Saccharum officinarum) sprouts. J. Plant Res. 2007, 120, 219–228. [Google Scholar] [CrossRef]
- Scharf, K.D.; Berberich, T.; Ebersberger, I.; Nover, L. The plant heat stress transcription factor (Hsf) family: Structure, function and evolution. Biochim. Biophys. Acta 2012, 1819, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- David, L.C.; Lee, S.K.; Bruderer, E.; Abt, M.R.; Fischer-Stettler, M.; Tschopp, M.A.; Solhaug, E.M.; Sanchez, K.; Zeeman, S.C. BETA-AMYLASE9 is a plastidial nonenzymatic regulator of leaf starch degradation. Plant Physiol. 2022, 188, 191–207. [Google Scholar] [CrossRef]
- Ying, S.; Scheible, W.R. A novel calmodulin-interacting Domain of Unknown Function 506 protein represses root hair elongation in Arabidopsis. Plant Cell Environ. 2022, 45, 1796–1812. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.T.; Wang, D.; Zhang, S.Z.; Ehlting, J.; Ni, F.; Jakab, S.; Zheng, C.C.; Zhong, Y. Genome-wide and expression analysis of protein phosphatase 2C in rice and Arabidopsis. BMC Genom. 2008, 9, 550. [Google Scholar] [CrossRef]
- Ren, B.; Liang, Y.; Deng, Y.; Chen, Q.G.; Zhang, J.; Yang, X.H.; Zuo, J.R. Genome-wide comparative analysis of type-A genes by overexpression studies reveals their diverse roles and regulatory mechanisms in cytokinin signaling. Cell Res. 2009, 19, 1178–1190. [Google Scholar] [CrossRef]
- Li, W.Q.; Sun, Y.R.; Li, K.; Tian, H.T.; Jia, J.T.; Zhang, H.Y.; Wang, Y.P.; Wang, H.; Bi, B.D.; Guo, J.G.; et al. Sinapate Esters Mediate UV-B-Induced Stomatal Closure by Regulating Nitric Oxide, Hydrogen Peroxide, and Malate Accumulation in Arabidopsis thaliana. Plant Cell Physiol. 2023, 63, 1890–1899. [Google Scholar] [CrossRef]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b Act as Repressors of the Expression of Heat-Inducible But Positively Regulate the Acquired Thermotolerance. Plant Physiol. 2011, 157, 1243–1254. [Google Scholar] [CrossRef]
- Guan, Q.M.; Yue, X.L.; Zeng, H.T.; Zhu, J.H. The Protein Phosphatase RCF2 and Its Interacting Partner NAC019 Are Critical for Heat Stress-Responsive Gene Regulation and Thermotolerance in Arabidopsis. Plant Cell 2014, 26, 438–453. [Google Scholar] [CrossRef]
- El-Esawi, M.A.; Al-Ghamdi, A.A.; Ali, H.M.; Ahmad, M. Overexpression of Transcription Factor Enhances Heat and Drought Stress Tolerance in Wheat (Triticum aestivum L.). Genes. 2019, 10, 163. [Google Scholar] [CrossRef]
- Li, S.J.; Fu, Q.T.; Chen, L.G.; Huang, W.D.; Yu, D.Q. Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta 2011, 233, 1237–1252. [Google Scholar] [CrossRef] [PubMed]
- Luhua, S.; Hegie, A.; Suzuki, N.; Shulaev, E.; Luo, X.; Cenariu, D.; Ma, V.; Kao, S.; Lim, J.; Gunay, M.B.; et al. Linking genes of unknown function with abiotic stress responses by high-throughput phenotype screening. Physiol. Plant. 2013, 148, 322–333. [Google Scholar] [CrossRef]
- Lee, J.; He, K.; Stolc, V.; Lee, H.; Figueroa, P.; Gao, Y.; Tongprasit, W.; Zhao, H.Y.; Lee, I.; Deng, X. Analysis of transcription factor HY5 genomic binding sites revealed its hierarchical role in light regulation of development. Plant Cell 2007, 19, 731–749. [Google Scholar] [CrossRef] [PubMed]
- So, H.A.; Chung, E.; Lee, J.H. Arabidopsis encoding a type III J-protein plays a negative role in heat shock tolerance. Genes. Genom. 2014, 36, 733–744. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, W.H.; Berkowitz, O.; Radomiljac, J.D.; Law, S.R.; Murcha, M.W.; Carrie, C.; Teixeira, P.F.; Kmiec, B.; Duncan, O.; et al. Inactivation of Mitochondrial Complex I Induces the Expression of a Twin Cysteine Protein that Targets and Affects Cytosolic, Chloroplastidic and Mitochondrial Function. Mol. Plant 2016, 9, 696–710. [Google Scholar] [CrossRef]
- Lee, T.A.; Bailey-Serres, J. Integrative Analysis from the Epigenome to Translatome Uncovers Patterns of Dominant Nuclear Regulation during Transient Stress. Plant Cell 2019, 31, 2573–2595. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wang, Y.; Ding, Y.Q.; Qiu, C.; Sun, L.T.; Gai, Z.S.; Gu, H.L.; Ding, Z.T. Global Ubiquitome Profiling Revealed the Roles of Ubiquitinated Proteins in Metabolic Pathways of Tea Leaves in Responding to Drought Stress. Sci. Rep. 2019, 9, 4286. [Google Scholar] [CrossRef]
- Piacentini, D.; Della Rovere, F.; Bertoldi, I.; Massimi, L.; Sofo, A.; Altamura, M.M.; Falasca, G. Peroxisomal PEX7 Receptor Affects Cadmium-Induced ROS and Auxin Homeostasis in Arabidopsis Root System. Antioxidants 2021, 10, 1494. [Google Scholar] [CrossRef]
- Cao, Y.Y.; Yang, J.F.; Liu, T.Y.; Su, Z.F.; Zhu, F.Y.; Chen, M.X.; Fan, T.; Ye, N.H.; Feng, Z.; Wang, L.J.; et al. A Phylogenetically Informed Comparison of GH1 Hydrolases between Arabidopsis and Rice Response to Stressors. Front. Plant Sci. 2017, 8, 350. [Google Scholar] [CrossRef]
- De Lucia, F.; Crevillen, P.; Jones, A.M.E.; Greb, T.; Dean, C. A PHD-Polycomb Repressive Complex 2 triggers the epigenetic silencing of during vernalization. Proc. Natl. Acad. Sci. USA 2008, 105, 16831–16836. [Google Scholar] [CrossRef] [PubMed]
- Meiri, D.; Breiman, A. Arabidopsis ROF1 (FKBP62) modulates thermotolerance by interacting with HSP90.1 and affecting the accumulation of HsfA2-regulated sHSPs. Plant J. 2009, 59, 387–399. [Google Scholar] [CrossRef]
- Prasad, B.D.; Goel, S.; Krishna, P. Identification of Carboxylate Clamp Type Tetratricopeptide Repeat Proteins in and Rice As Putative Co-Chaperones of Hsp90/Hsp70. PLoS ONE 2010, 5, e12761. [Google Scholar] [CrossRef]
- Xie, C.T.; Ding, Z.J. NAC1 Maintains Root Meristem Activity by Repressing the Transcription of E2Fa in Arabidopsis. Int. J. Mol. Sci. 2022, 23, 12258. [Google Scholar] [CrossRef] [PubMed]
- Thieme, C.J.; Rojas-Triana, M.; Stecyk, E.; Schudoma, C.; Zhang, W.N.; Yang, L.; Miñambres, M.; Walther, D.; Schulze, W.X.; Paz-Ares, J.; et al. Endogenous Arabidopsis messenger RNAs transported to distant tissues. Nat. Plants 2016, 2, 15025. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.E.; Bauerle, T.L. Long-distance plant signaling pathways in response to multiple stressors: The gap in knowledge. J. Exp. Bot. 2016, 67, 2063–2079. [Google Scholar] [CrossRef]
- Andrási, N.; Rigó, G.; Zsigmond, L.; Pérez-Salamó, I.; Papdi, C.; Klement, E.; Pettkó-Szandtner, A.; Baba, A.I.; Ayaydin, F.; Dasari, R.; et al. The mitogen-activated protein kinase 4-phosphorylated heat shock factor A4A regulates responses to combined salt and heat stresses. J. Exp. Bot. 2021, 72, 5781. [Google Scholar] [CrossRef]
- Gao, S.; Gao, J.; Zhu, X.Y.; Song, Y.; Li, Z.P.; Ren, G.D.; Zhou, X.; Kuai, B.K. ABF2, ABF3, and ABF4 Promote ABA-Mediated Chlorophyll Degradation and Leaf Senescence by Transcriptional Activation of Chlorophyll Catabolic Genes and Senescence-Associated Genes in Arabidopsis. Mol. Plant 2016, 9, 1272–1285. [Google Scholar] [CrossRef]
- Teng, Z.N.; Lyu, J.; Chen, Y.K.; Zhang, J.H.; Ye, N.H. Effects of stress-induced ABA on root architecture development: Positive and actions. Crop J. 2023, 11, 1072–1079. [Google Scholar] [CrossRef]
- Rawat, R.; Schwartz, J.; Jones, M.A.; Sairanen, I.; Cheng, Y.F.; Andersson, C.R.; Zhao, Y.D.; Ljung, K.; Harmer, S.L. REVEILLE1, a Myb-like transcription factor, integrates the circadian clock and auxin pathways. Proc. Natl. Acad. Sci. USA 2009, 106, 16883–16888. [Google Scholar] [CrossRef]
- Ohta, M.; Sato, A.; Renhu, N.; Yamamoto, T.; Oka, N.; Zhu, J.K.; Tada, Y.; Suzaki, T.; Miura, K. MYC-type transcription factors, MYC67 and MYC70, interact with ICE1 and negatively regulate cold tolerance in Arabidopsis. Sci. Rep. 2018, 8, 11622. [Google Scholar] [CrossRef] [PubMed]
- Michaletz, S.T.; Weiser, M.D.; Zhou, J.Z.; Kaspari, M.; Helliker, B.R.; Enquist, B.J. Plant Thermoregulation: Energetics, Trait-Environment Interactions, and Carbon Economics. Trends Ecol. Evol. 2015, 30, 714–724. [Google Scholar] [CrossRef]
- Calleja-Cabrera, J.; Boter, M.; Oñate-Sánchez, L.; Pernas, M. Root Growth Adaptation to Climate Change in Crops. Front. Plant Sci. 2020, 11, 544. [Google Scholar] [CrossRef]
- Liu, H.T.; Liu, Y.P.; Huang, W.D. Root-fed salicylic acid in grape involves the response caused by aboveground high temperature. J. Integr. Plant Biol. 2008, 50, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Talanova, V.V.; Akimova, T.V.; Titov, A.F. Effect of whole plant and local heating on the ABA content in cucumber seedling leaves and roots and on their heat tolerance. Russ. J. Plant Physiol. 2003, 50, 90–94. [Google Scholar] [CrossRef]
- Li, J.G.; Fan, M.; Hua, W.B.; Tian, Y.H.; Chen, L.G.; Sun, Y.; Bai, M.Y. Brassinosteroid and Hydrogen Peroxide Interdependently Induce Stomatal Opening by Promoting Guard Cell Starch Degradation. Plant Cell 2020, 32, 984–999. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- McLeay, R.C.; Bailey, T.L. Motif Enrichment Analysis: A unified framework and an evaluation on ChIP data. BMC Bioinform. 2010, 11, 165. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Tag | Regulation | Regulation | Official Full Name | Description |
|---|---|---|---|---|
| (42W-S)/FC | (42W-R)/FC | |||
| AT5G18670 | Up/3 | Down/−3.7 | BAM9 | b-Amylase BAM9 regulates starch breakdown, and its gene expression is responsive to several environmental changes [24]. |
| AT4G32480 | Up/6 | Down/−2.5 | - | Phosphorus (P) stress-inducible DUF506 gene family member [25]. |
| AT5G23350 | Up/4.3 | Down/−2.3 | ABA-responsive-like protein | GRAM domain protein/ABA-responsive-like protein |
| AT3G16560 | Down/−2.4 | Up/2.25 | Protein phosphatase 2C family protein | This gene belongs to PP2C subfamily C and is induced by cold and heat [26]. |
| AT5G01712 | Down/−4 | Up/12 | - | Unknown |
| AT5G62920 | Down/−3 | Up/4.4 | Type-A response regulator 6 | A Type-A response regulator that is responsive to cytokinin treatment and negative regulator of cytokinin [27]. |
| AT3G21560 | Down/−3.1 | Up/2 | UDP-GLUCOSYL TRANSFERASE 84A2 | brt1 showed smaller stomatal apertures than wild type under normal light conditions and UV-B irradiation [28]. |
| Locus Tag | Official Full Name | Localization | Description |
|---|---|---|---|
| 42S-S | |||
| Stress response | |||
| AT5G16110 | Hypothetical protein | Nucleus | Related to abiotic stress [33]. |
| AT1G13600 | bZIP-58 | Nucleus | Downstream target gene of HY5 [34]. |
| AT1G56300 | Chaperone DnaJ-domain protein | Nucleus | DJC53 may play a negative regulatory role in response to HS [35]. |
| AT5G64400 | Twin cysteine proteins | Mitochondrion | The loss of both At12cys-1 and At12cys-2 leads to enhanced tolerance to drought and light stress and increased antioxidant capacity [36]. |
| AT1G50740 | Transmembrane proteins 14C | Extracellular | Cellular response to hypoxia [37]. |
| AT5G42220 | Ubiquitin-like protein | Nucleus | Misfolded protein binding and polyubiquitin modification-dependent protein binding [38]. |
| AT1G29260 | PEX7 | Peroxisome | Under certain stressful conditions, such as in the presence of high amounts of H2O2 produced within peroxisomes, PEX7 may be damaged and thus accumulate on the peroxisomal membrane [39]. |
| AT3G62740 | Beta glucosidase 7 | Extracellular | The expression of AtBGLU7 was significantly up-regulated when treated with cold for 24 h [40]. |
| Other and unknown | |||
| AT4G30200 | VEL1 protein | Nucleus | VEL1 is a part of a polycomb repressive complex (PRC2) that is involved in epigenetic silencing of the FLC flowering locus [41]. |
| AT1G78150 | N-lysine methyltransferase | Chloroplast | Unknown function |
| AT4G19430 | Hypothetical protein | Mitochondrion | Unknown function |
| AT3G57810 | Cysteine proteinases protein | Mitochondrion | Unknown function |
| AT2G43630 | Nucleus envelope protein | Plastid | Unknown function |
| AT1G30190 | Cotton fiber protein | Nucleus | Unknown function |
| AT1G14970 | O-fucosyltransferase protein | Nucleus | Unknown function |
| AT1G21680 | DPP6 N-terminal domain-like protein | Extracellular | Unknown function |
| 42S-R | |||
| Stress response | |||
| AT3G25230 | FK506 binding protein 62 | Cytosol | Modulates thermotolerance by interacting with HSP90.1 and affecting the accumulation of HSFA2-regulated HSPs [42]. |
| AT3G04710 | Tetratricopeptide repeat 10 | Cytosol | Encodes 1 of the 36 carboxylate clamp (CC)-tetratricopeptide repeat (TPR) proteins with the potential to interact with HSP90/HSP70 as co-chaperones [43]. |
| AT3G13470 | Chloroplasts chaperonins | Plastid | Encodes a subunit of chloroplast chaperonins (CHAPERONIN-60BETA2, CPNB2). |
| Locus Tag | Official Full Name | Localization | Description |
|---|---|---|---|
| Plant hormone signal transduction | |||
| AT1G56010 | Transcription factor NAC1 | Nucleus | Encodes a transcription factor involved in auxin-mediated lateral root formation, auxin-activated signaling pathways, and lateral root development [44]. |
| Light signaling pathway | |||
| AT2G34430 | Photosystem II type I chlorophyll a/b-binding protein | Plastid | Light harvesting in photosystem II [45]. |
| Unknown | |||
| AT1G49030 | PLAC8 family protein | Plasma membrane | Unknown function |
| AT1G55430 | Cysteine/histidine-rich C1 domain protein | Nucleus | Unknown function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Liu, X.; Wang, S.; Liang, S.; Li, S.; Wang, J.; Liu, S.; Guo, Y.; Li, R. Comparative Transcriptome Analysis of Arabidopsis Seedlings Under Heat Stress on Whole Plants, Shoots, and Roots Reveals New HS-Regulated Genes, Organ-Specific Responses, and Shoots-Roots Communication. Int. J. Mol. Sci. 2025, 26, 2478. https://doi.org/10.3390/ijms26062478
Liu Z, Liu X, Wang S, Liang S, Li S, Wang J, Liu S, Guo Y, Li R. Comparative Transcriptome Analysis of Arabidopsis Seedlings Under Heat Stress on Whole Plants, Shoots, and Roots Reveals New HS-Regulated Genes, Organ-Specific Responses, and Shoots-Roots Communication. International Journal of Molecular Sciences. 2025; 26(6):2478. https://doi.org/10.3390/ijms26062478
Chicago/Turabian StyleLiu, Zhaojiao, Xinye Liu, Shuailei Wang, Shuang Liang, Saimei Li, Juntao Wang, Sitong Liu, Yi Guo, and Rui Li. 2025. "Comparative Transcriptome Analysis of Arabidopsis Seedlings Under Heat Stress on Whole Plants, Shoots, and Roots Reveals New HS-Regulated Genes, Organ-Specific Responses, and Shoots-Roots Communication" International Journal of Molecular Sciences 26, no. 6: 2478. https://doi.org/10.3390/ijms26062478
APA StyleLiu, Z., Liu, X., Wang, S., Liang, S., Li, S., Wang, J., Liu, S., Guo, Y., & Li, R. (2025). Comparative Transcriptome Analysis of Arabidopsis Seedlings Under Heat Stress on Whole Plants, Shoots, and Roots Reveals New HS-Regulated Genes, Organ-Specific Responses, and Shoots-Roots Communication. International Journal of Molecular Sciences, 26(6), 2478. https://doi.org/10.3390/ijms26062478