
Hypertrophic Cardiomyopathy-Associated CRYABR123W Activates Calcineurin, Reduces Calcium Sequestration, and Alters the CRYAB Interactome and the Proteomic Response to Pathological Hypertrophy



Abstract
1. Introduction
2. Results
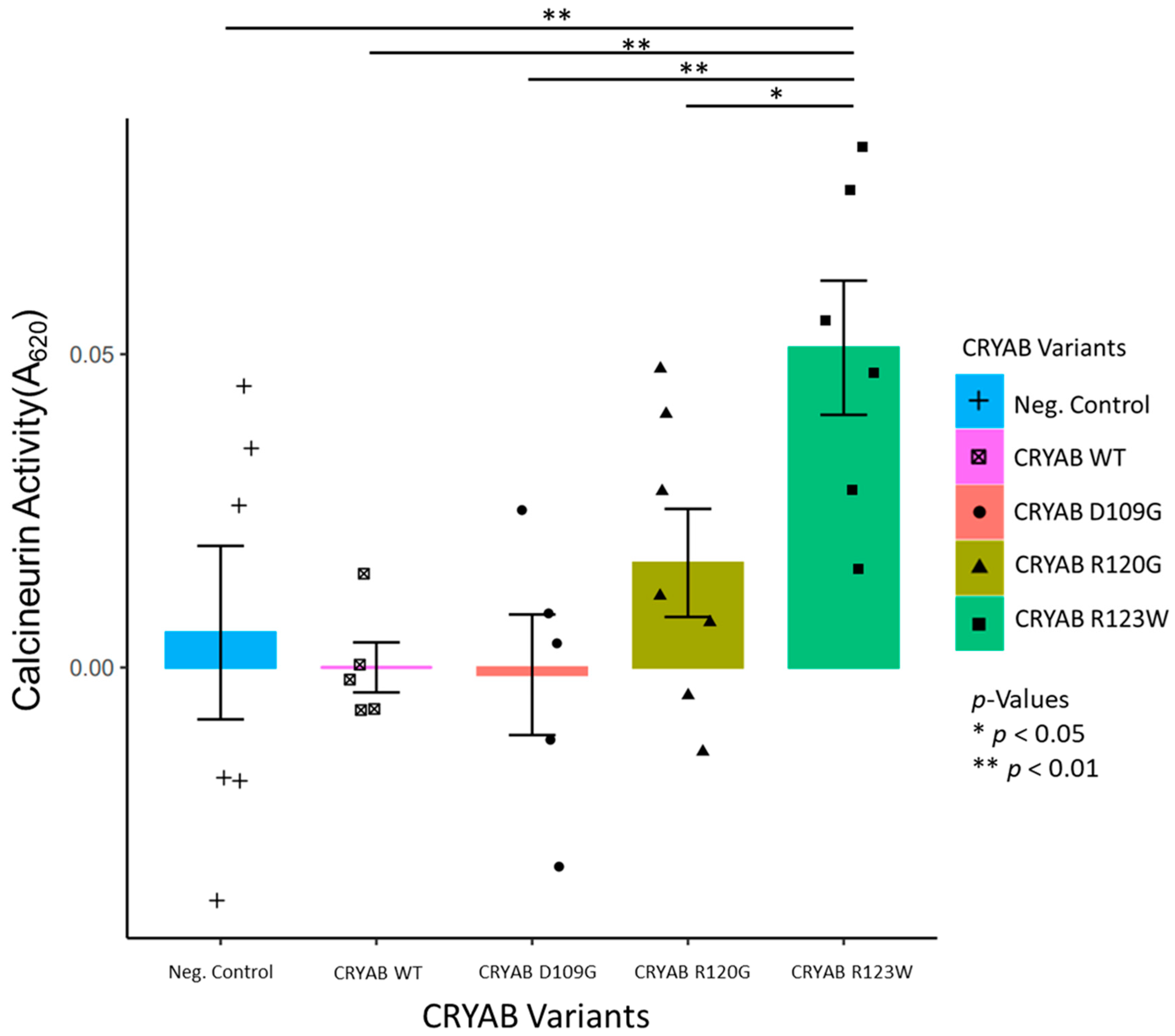
2.1. CRYABR123W Increases Calcineurin Activity
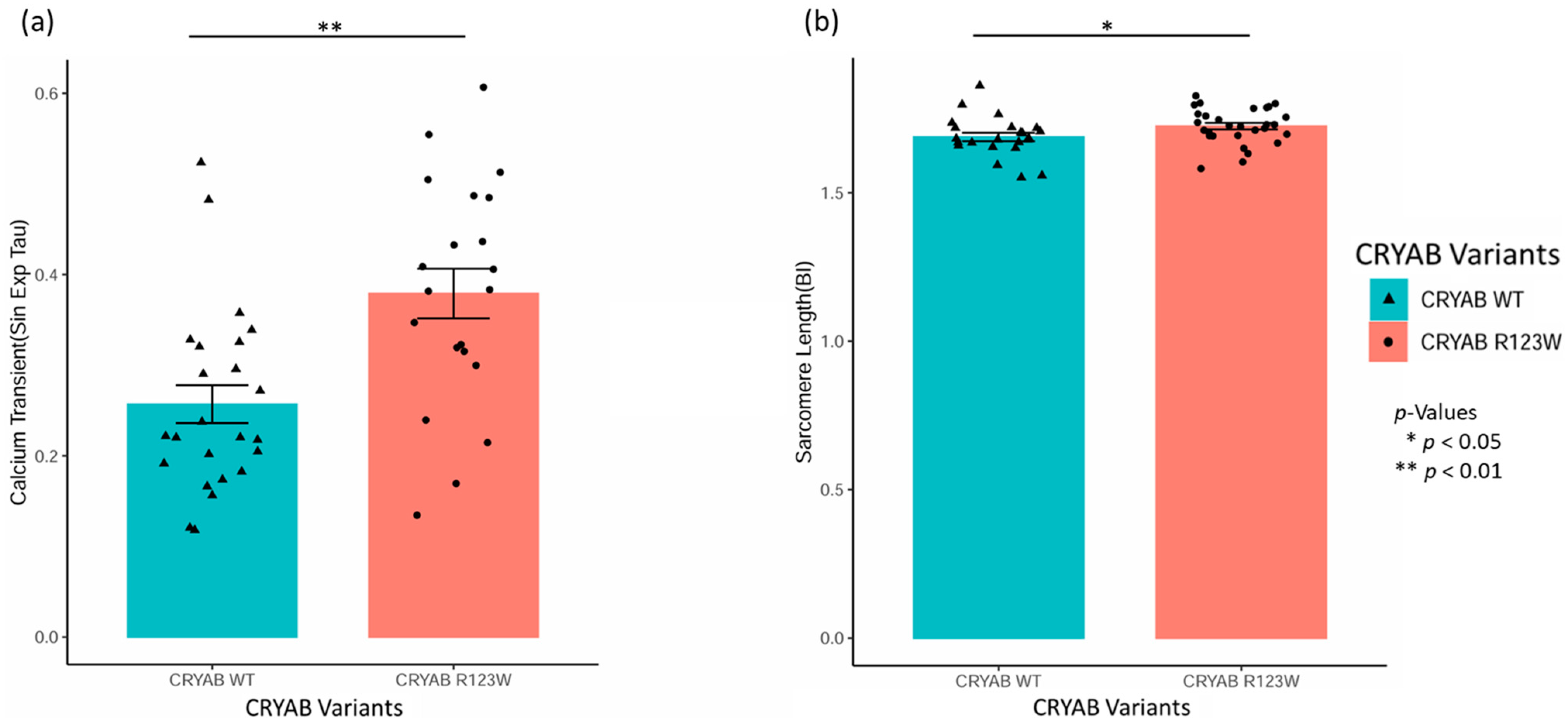
2.2. CRYABR123W Causes Impaired Calcium Reuptake and Myocyte Relaxation
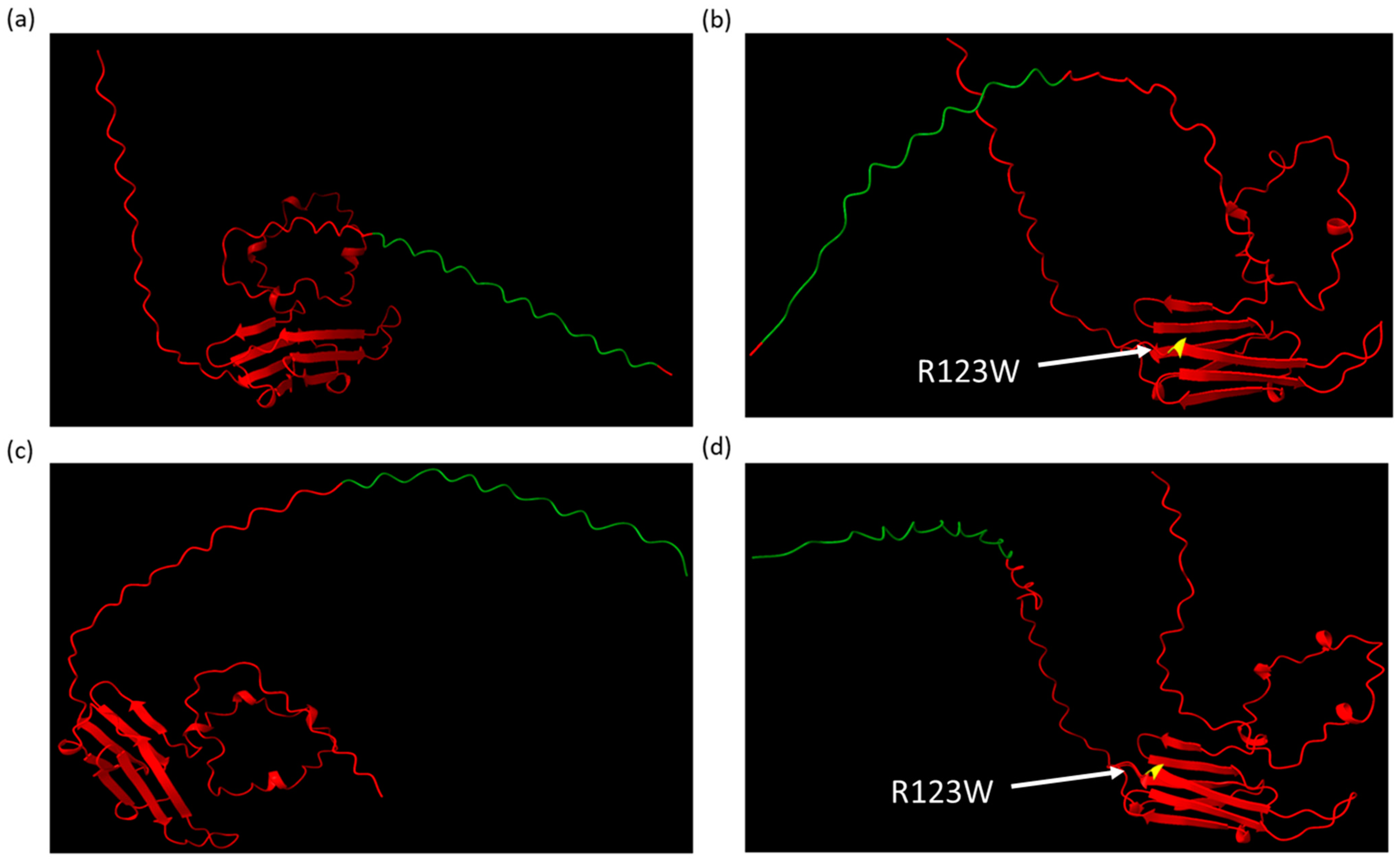
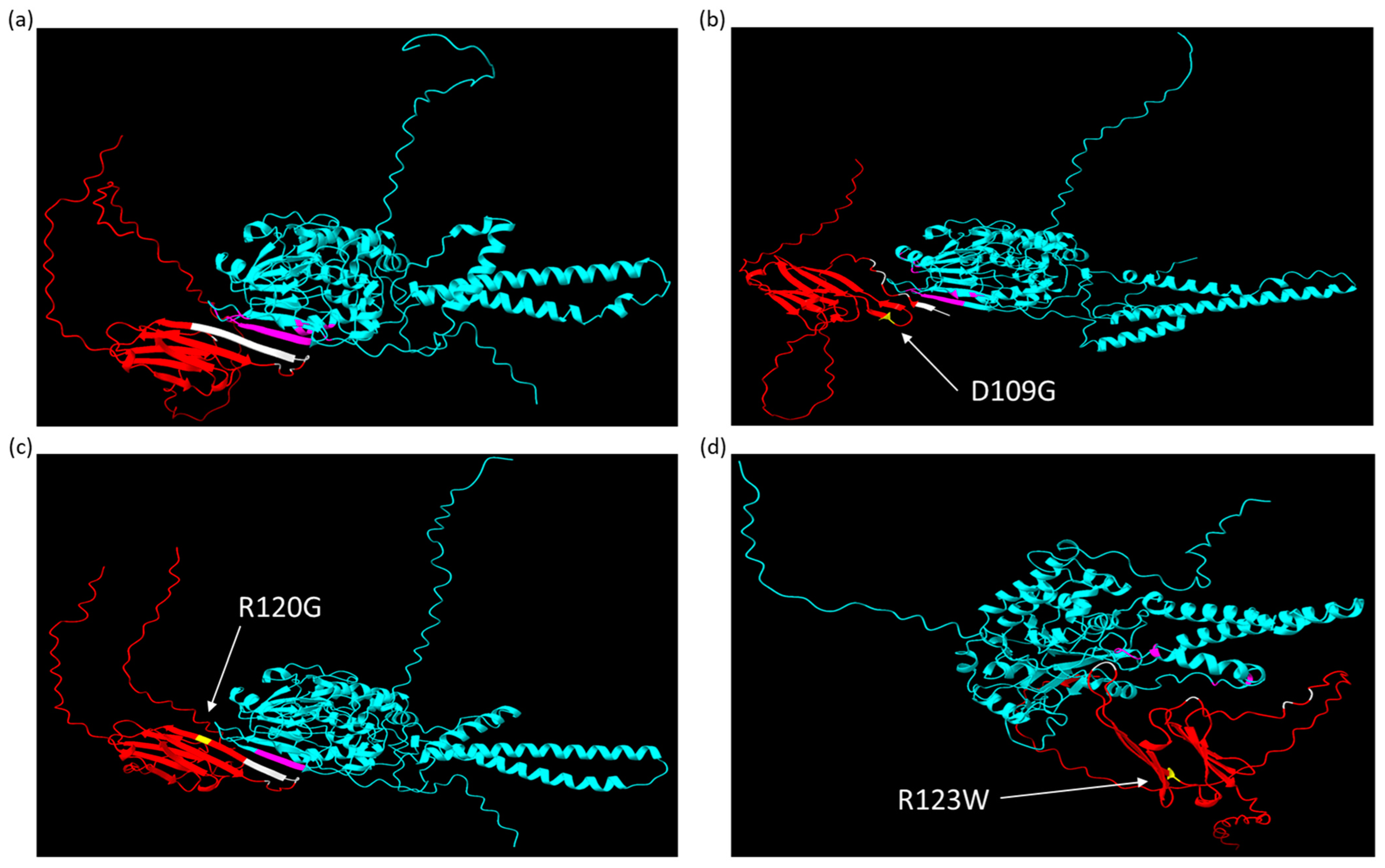
2.3. AlphaFold Predicts That CRYABR123W Binds to the Autoinhibitory Domain of Calcineurin
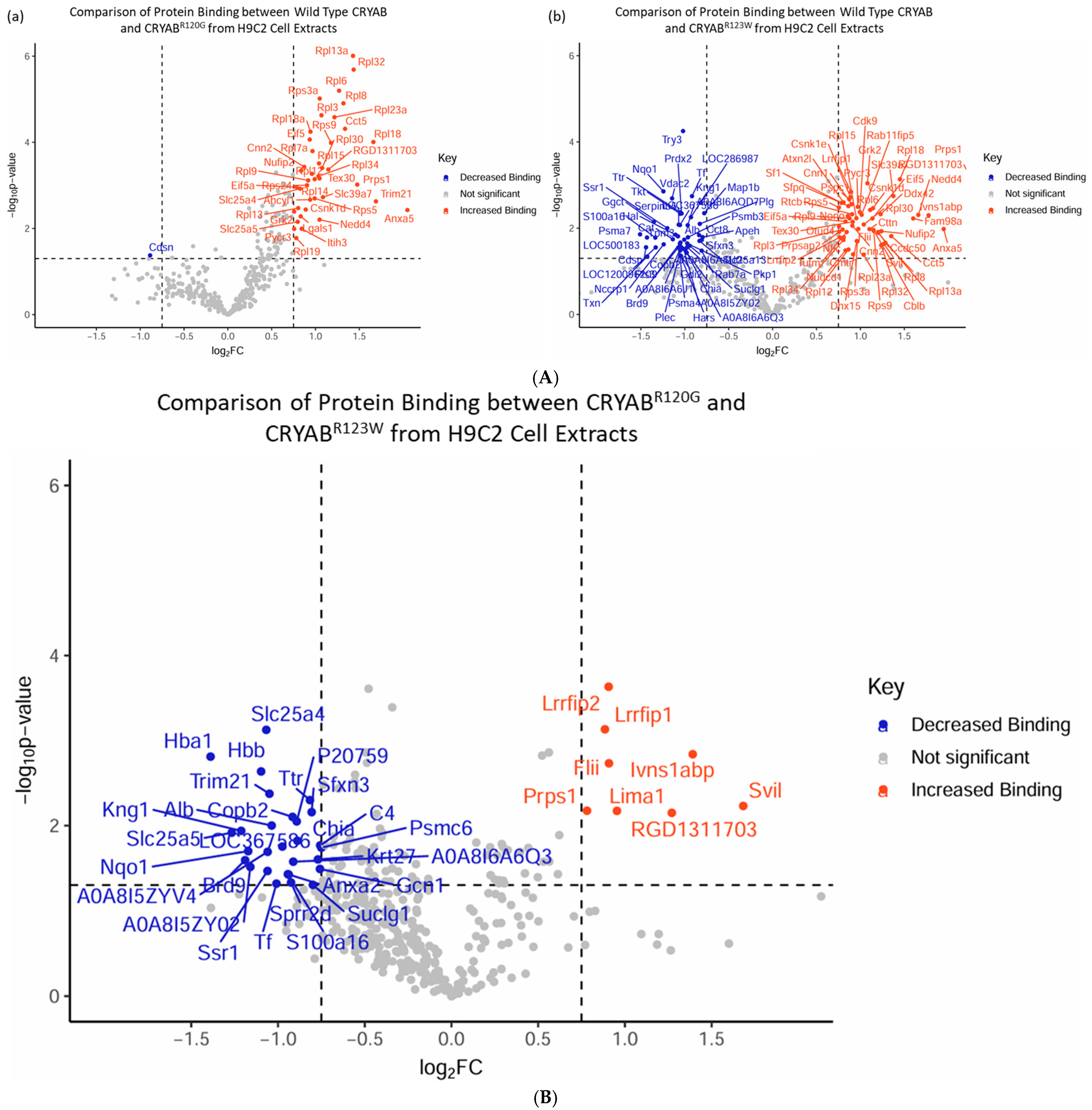
2.4. CRYABR123W Protein Interactome Diverges from CRYAB and CRYABR120G
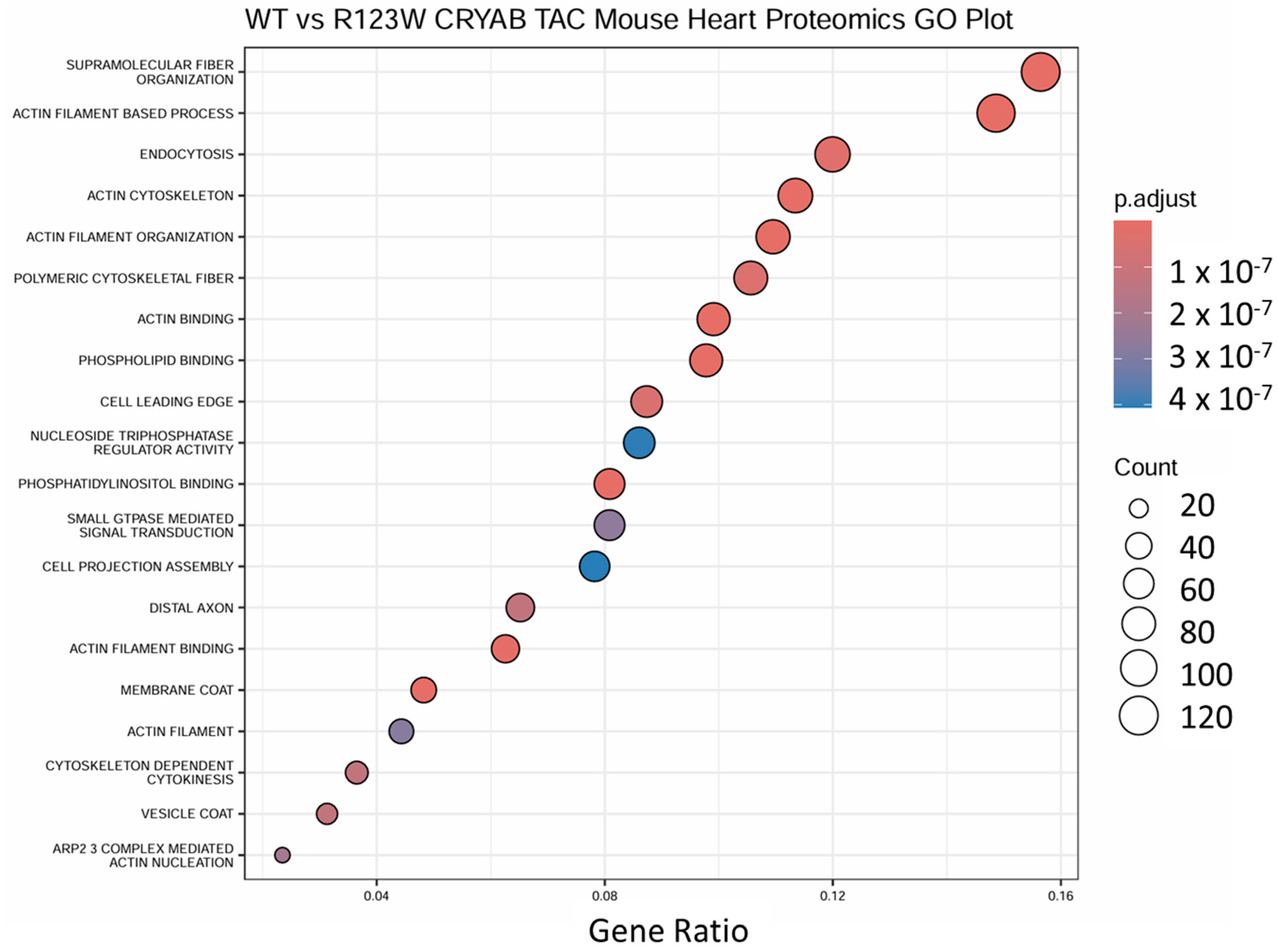
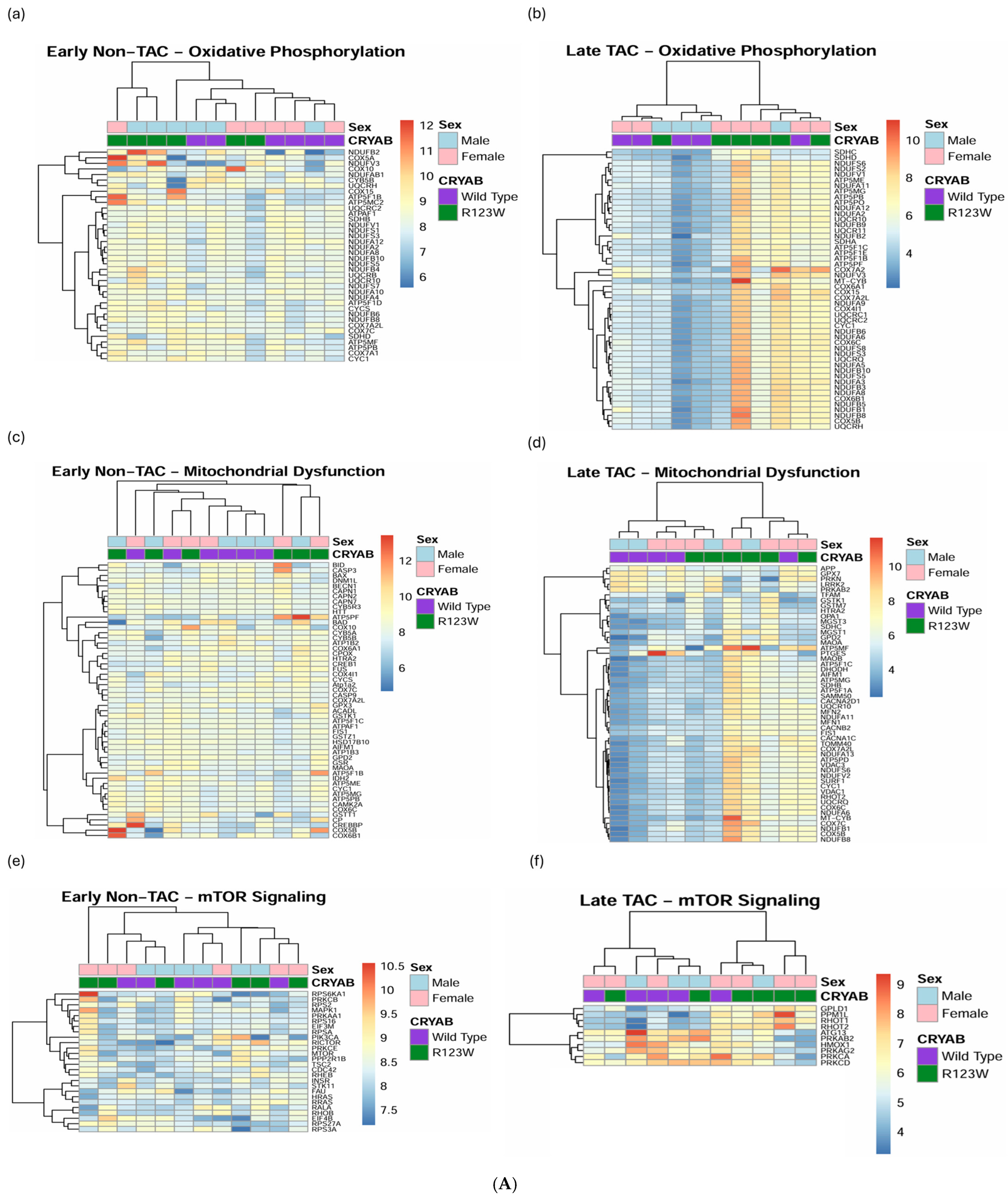
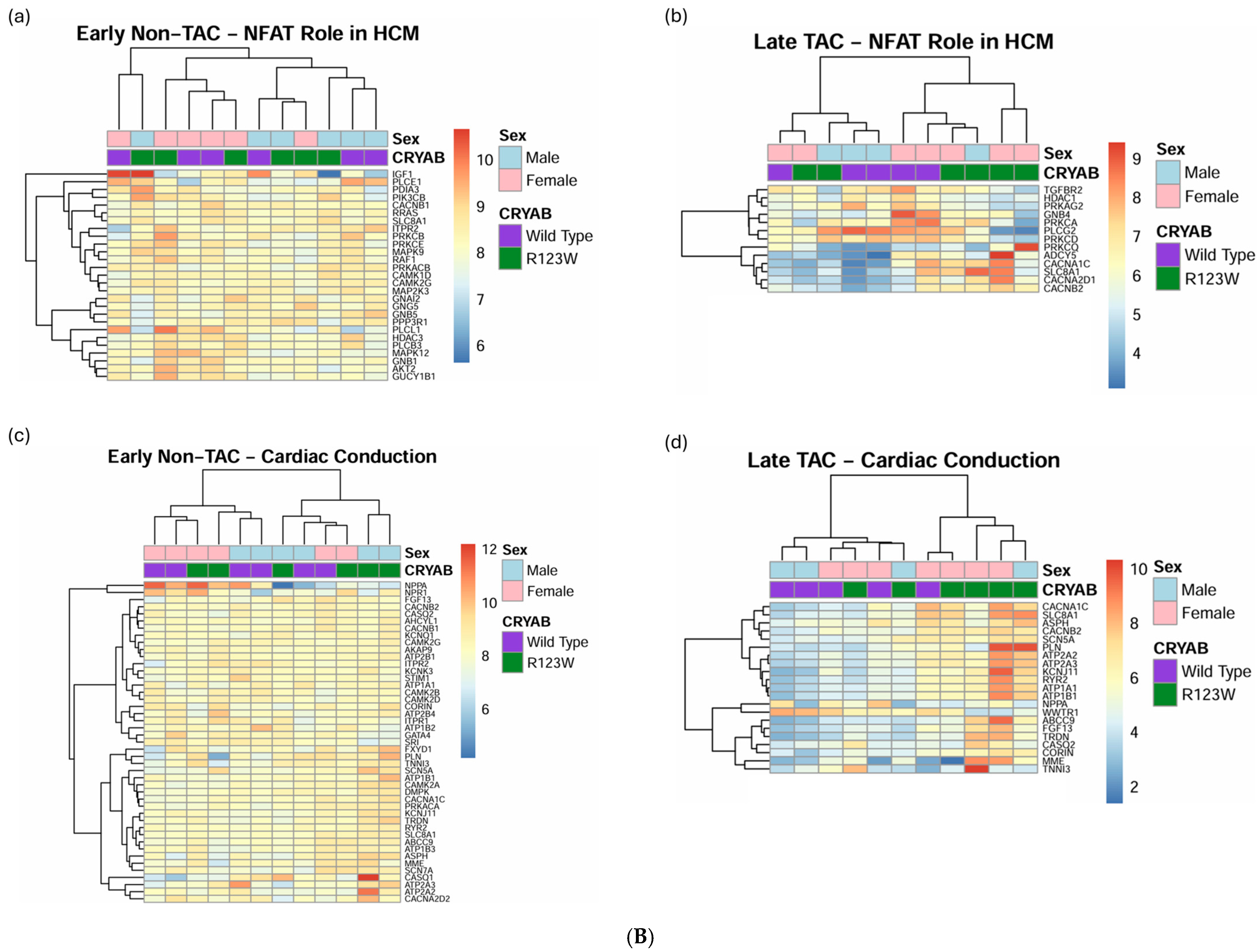
2.5. Global Proteomics and Phosphoproteomics Reveal Cytoskeletal, Metabolic, Immune, and Cardiac Dysfunction in CRYABR123W Mouse Hearts
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maron, B.J. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002, 287, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Roberts, R. The molecular genetic basis for hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2001, 33, 655–670. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marian, A.J. Modifier genes for hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2002, 17, 242–252. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Geisterfer-Lowrance, A.A.; Kass, S.; Tanigawa, G.; Vosberg, H.P.; McKenna, W.; Seidman, C.E.; Seidman, J.G. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990, 62, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Watkins, H.; MacRae, C.; Thierfelder, L.; Chou, Y.H.; Frenneaux, M.; McKenna, W.; Seidman, J.G.; Seidman, C.E. A disease locus for familial hypertrophic cardiomyopathy maps to chromosome 1q3. Nat. Genet. 1993, 3, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Thierfelder, L.; Watkins, H.; MacRae, C.; Lamas, R.; McKenna, W.; Vosberg, H.P.; Seidman, J.G.; Seidman, C.E. Alpha-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell 1994, 77, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Maron, M.S.; Maron, B.A.; Loscalzo, J. Moving Beyond the Sarcomere to Explain Heterogeneity in Hypertrophic Cardiomyopathy: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ingles, J.; Burns, C.; Bagnall, R.D.; Lam, L.; Yeates, L.; Sarina, T.; Puranik, R.; Briffa, T.; Atherton, J.J.; Driscoll, T.; et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ. Cardiovasc. Genet. 2017, 10, e001620. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Klaassen, S.; Brodehl, A. The genetic landscape of cardiomyopathies. Genet. Causes Card. Dis. 2019, 7, 45–91. [Google Scholar] [CrossRef]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prévost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tomé, F.; Dupret, J.M.; et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Kumarapeli, A.R.; Su, H.; Huang, W.; Tang, M.; Zheng, H.; Horak, K.M.; Li, M.; Wang, X. Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ. Res. 2008, 103, 1473–1482. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bennardini, F.; Wrzosek, A.; Chiesi, M. Alpha B-crystallin in cardiac tissue. Assoc. Actin Desmin. Filaments. Circ. Res. 1992, 71, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Nicholl, I.D.; Quinlan, R.A. Chaperone activity of alpha-crystallins modulates intermediate filament assembly. EMBO J. 1994, 13, 945–953. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, K.; Spector, A. alpha-crystallin stabilizes actin filaments and prevents cytochalasin-induced depolymerization in a phosphorylation-dependent manner. Eur. J. Biochem. 1996, 242, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Tannous, P.; Zhu, H.; Johnstone, J.L.; Shelton, J.M.; Rajasekaran, N.S.; Benjamin, I.J.; Nguyen, L.; Gerard, R.D.; Levine, B.; Rothermel, B.A.; et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 9745–9750. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bhuiyan, M.S.; Pattison, J.S.; Osinska, H.; James, J.; Gulick, J.; McLendon, P.M.; Hill, J.A.; Sadoshima, J.; Robbins, J. Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Investig. 2013, 123, 5284–5297. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitra, A.; Basak, T.; Datta, K.; Naskar, S.; Sengupta, S.; Sarkar, S. Role of α-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013, 4, e582. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chis, R.; Sharma, P.; Bousette, N.; Miyake, T.; Wilson, A.; Backx, P.H.; Gramolini, A.O. α-Crystallin B prevents apoptosis after H2O2 exposure in mouse neonatal cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H967–H978. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bagnéris, C.; Bateman, O.A.; Naylor, C.E.; Cronin, N.; Boelens, W.C.; Keep, N.H.; Slingsby, C. Crystal structures of alpha-crystallin domain dimers of alphaB-crystallin and Hsp20. J. Mol. Biol. 2009, 392, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Laganowsky, A.; Benesch, J.L.; Landau, M.; Ding, L.; Sawaya, M.R.; Cascio, D.; Huang, Q.; Robinson, C.V.; Horwitz, J.; Eisenberg, D. Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 2010, 19, 1031–1043. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mchaourab, H.S.; Godar, J.A.; Stewart, P.L. Structure and mechanism of protein stability sensors: Chaperone activity of small heat shock proteins. Biochemistry 2009, 48, 3828–3837. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Okamoto, K.; Nakayama, H.; Isobe, T.; Kato, K. Phosphorylation of alphaB-crystallin in response to various types of stress. J. Biol. Chem. 1997, 272, 29934–29941. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Féasson, L.; Antoine, J.C.; Pécheux, C.; Bernard, R.; Cobo, A.M.; Casarin, A.; Salviati, L.; Desnuelle, C.; Urtizberea, A. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul. Disord. 2012, 22, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Osinska, H.; Klevitsky, R.; Gerdes, A.M.; Nieman, M.; Lorenz, J.; Hewett, T.; Robbins, J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ. Res. 2001, 89, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Bova, M.P.; Yaron, O.; Huang, Q.; Ding, L.; Haley, D.A.; Stewart, P.L.; Horwitz, J. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc. Natl. Acad. Sci. USA 1999, 96, 6137–6142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maron, B.J.; Rowin, E.J.; Arkun, K.; Rastegar, H.; Larson, A.M.; Maron, M.S.; Chin, M.T. Adult Monozygotic Twins With Hypertrophic Cardiomyopathy and Identical Disease Expression and Clinical Course. Am. J. Cardiol. 2020, 127, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Martin, G.L.; Perera, G.; Awata, J.; Larson, A.; Blanton, R.; Chin, M.T. A novel αB-crystallin R123W variant drives hypertrophic cardiomyopathy by promoting maladaptive calcium-dependent signal transduction. Front. Cardiovasc. Med. 2023, 10, 1223244. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Somee, L.R.; Barati, A.; Shahsavani, M.B.; Hoshino, M.; Hong, J.; Kumar, A.; Moosavi-Movahedi, A.A.; Amanlou, M.; Yousefi, R. Understanding the structural and functional changes and biochemical pathomechanism of the cardiomyopathy-associated p.R123W mutation in human αB-crystallin. Biochim. Biophys Acta Gen. Subj. 2024, 1868, 130579. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thorkelsson, A.; Chin, M.T. Role of the Alpha-B-Crystallin Protein in Cardiomyopathic Disease. Int. J. Mol. Sci. 2024, 25, 2826. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pope, R.K.; Pestonjamasp, K.N.; Smith, K.P.; Wulfkuhle, J.D.; Strassel, C.P.; Lawrence, J.B.; Luna, E.J. Cloning, characterization, and chromosomal localization of human superillin (SVIL). Genomics 1998, 52, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Maul, R.S.; Song, Y.; Amann, K.J.; Gerbin, S.C.; Pollard, T.D.; Chang, D.D. EPLIN regulates actin dynamics by cross-linking and stabilizing filaments. J. Cell Biol. 2003, 160, 399–407. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oltion, K.; Carelli, J.D.; Yang, T.; See, S.K.; Wang, H.Y.; Kampmann, M.; Taunton, J. An E3 ligase network engages GCN1 to promote the degradation of translation factors on stalled ribosomes. Cell 2023, 186, 346–362.e17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, X.; Yang, L.; Chen, S.; Zheng, J.; Zhang, H.; Ren, L. Multiple Roles of TRIM21 in Virus Infection. Int. J. Mol. Sci. 2023, 24, 1683. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Burger, A.; Berendes, R.; Liemann, S.; Benz, J.; Hofmann, A.; Göttig, P.; Huber, R.; Gerke, V.; Thiel, C.; Römisch, J.; et al. The crystal structure and ion channel activity of human annexin II, a peripheral membrane protein. J. Mol. Biol. 1996, 257, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Barefield, D.; Kumar, M.; Gorham, J.; Seidman, J.G.; Seidman, C.E.; de Tombe, P.P.; Sadayappan, S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J. Mol. Cell. Cardiol. 2015, 79, 234–243. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vikstrom, K.L.; Factor, S.M.; Leinwand, L.A. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol. Med. 1996, 2, 556–567. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ertz-Berger, B.R.; He, H.; Dowell, C.; Factor, S.M.; Haim, T.E.; Nunez, S.; Schwartz, S.D.; Ingwall, J.S.; Tardiff, J.C. Changes in the chemical and dynamic properties of cardiac troponin T cause discrete cardiomyopathies in transgenic mice. Proc. Natl. Acad. Sci. USA 2005, 102, 18219–18224. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lowey, S.; Lesko, L.M.; Rovner, A.S.; Hodges, A.R.; White, S.L.; Low, R.B.; Rincon, M.; Gulick, J.; Robbins, J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J. Biol. Chem. 2008, 283, 20579–20589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vakrou, S.; Fukunaga, R.; Foster, D.B.; Sorensen, L.; Liu, Y.; Guan, Y.; Woldemichael, K.; Pineda-Reyes, R.; Liu, T.; Tardiff, J.C.; et al. Allele-specific differences in transcriptome, miRNome, and mitochondrial function in two hypertrophic cardiomyopathy mouse models. JCI Insight 2018, 3, e94493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Marstrand, P.; Han, L.; Day, S.M.; Olivotto, I.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Wittekind, S.G.; Helms, A.; Saberi, S.; et al. Hypertrophic Cardiomyopathy with Left Ventricular Systolic Dysfunction: Insights From the SHaRe Registry. Circulation 2020, 141, 1371–1383. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Beltrami, M.; Bartolini, S.; Pastore, M.C.; Milli, M.; Cameli, M. Relationship between measures of left ventricular systolic and diastolic dysfunction and clinical and biomarker status in patients with hypertrophic cardiomyopathy. Arch. Cardiovasc. Dis. 2022, 115, 598–609. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 1 December 2023).
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Perrino, B.A. Regulation of calcineurin phosphatase activity by its autoinhibitory domain. Arch. Biochem. Biophys. 1999, 372, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Chin, M.T. Pathogenic Mechanisms of Hypertrophic Cardiomyopathy beyond Sarcomere Dysfunction. Int. J. Mol. Sci. 2021, 22, 8933. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Golenhofen, N.; Ness, W.; Koob, R.; Htun, P.; Schaper, W.; Drenckhahn, D. Ischemia-induced phosphorylation and translocation of stress protein alpha B-crystallin to Z lines of myocardium. Am. J. Physiol. 1998, 274, H1457–H1464. [Google Scholar] [CrossRef] [PubMed]
- Maksimiuk, M.; Sobiborowicz, A.; Tuzimek, A.; Deptała, A.; Czerw, A.; Badowska-Kozakiewicz, A.M. αB-crystallin as a promising target in pathological conditions—A review. Ann. Agric. Environ. Med. 2020, 27, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Head, M.W.; Hurwitz, L.; Kegel, K.; Goldman, J.E. AlphaB-crystallin regulates intermediate filament organization in situ. Neuroreport 2000, 11, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Kurnellas, M.P.; Brownell, S.; Adams, C.M.; Su, L.; Axtell, R.C.; Chen, R.; Fathman, C.G.; Robinson, W.H.; Steinman, L. Therapeutic effects of systemic administration of chaperone αB-crystallin associated with binding proinflammatory plasma proteins. J. Biol. Chem. 2012, 287, 9708–9721. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, W.; Guo, Y.; Huang, Z.; Zhao, H.; Zhou, M.; Huang, Y.; Wen, D.; Song, J.; Zhu, Z.; Sun, M.; et al. Small heat shock protein CRYAB inhibits intestinal mucosal inflammatory responses and protects barrier integrity through suppressing IKKβ activity. Mucosal. Immunol. 2019, 12, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Arac, A.; Brownell, S.E.; Rothbard, J.B.; Chen, C.; Ko, R.M.; Pereira, M.P.; Albers, G.W.; Steinman, L.; Steinberg, G.K. Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 13287–13292. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Velotta, J.B.; Kimura, N.; Chang, S.H.; Chung, J.; Itoh, S.; Rothbard, J.; Yang, P.C.; Steinman, L.; Robbins, R.C.; Fischbein, M.P. αB-crystallin improves murine cardiac function and attenuates apoptosis in human endothelial cells exposed to ischemia-reperfusion. Ann. Thorac. Surg. 2011, 91, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Helms, A.S.; Alvarado, F.J.; Yob, J.; Tang, V.T.; Pagani, F.; Russell, M.W.; Valdivia, H.H.; Day, S.M. Genotype-Dependent and -Independent Calcium Signaling Dysregulation in Human Hypertrophic Cardiomyopathy. Circulation 2016, 134, 1738–1748. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Coppini, R.; Ferrantini, C.; Mugelli, A.; Poggesi, C.; Cerbai, E. Altered Ca2+ and Na+ Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for Arrhythmogenesis. Front. Physiol. 2018, 9, 1391. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wilkins, B.J.; Dai, Y.S.; Bueno, O.F.; Parsons, S.A.; Xu, J.; Plank, D.M.; Jones, F.; Kimball, T.R.; Molkentin, J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004, 94, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Golden, R.J.; Chen, B.; Li, T.; Braun, J.; Manjunath, H.; Chen, X.; Wu, J.; Schmid, V.; Chang, T.C.; Kopp, F.; et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 2017, 542, 197–202. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Froger, A.; Hall, J.E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 2007, 6, 253. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef]
- Richards, D.A.; Aronovitz, M.J.; Calamaras, T.D.; Tam, K.; Martin, G.L.; Liu, P.; Bowditch, H.K.; Zhang, P.; Huggins, G.S.; Blanton, R.M. Distinct Phenotypes Induced by Three Degrees of Transverse Aortic Constriction in Mice. Sci. Rep. 2019, 9, 5844. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef] [PubMed]
- Schweppe, D.K.; Prasad, S.; Belford, M.W.; Navarrete-Perea, J.; Bailey, D.J.; Huguet, R.; Jedrychowski, M.P.; Rad, R.; McAlister, G.; Abbatiello, S.E.; et al. Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Anal. Chem. 2019, 91, 4010–4016. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Li, J.; Mintseris, J.; O’Connell, J.; Gygi, S.P.; Schweppe, D.K. Improved Monoisotopic Mass Estimation for Deeper Proteome Coverage. J. Proteome Res. 2021, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Navarrete-Perea, J.; Yu, Q.; Gygi, S.P.; Paulo, J.A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17, 2226–2236. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, J.; Cai, Z.; Bomgarden, R.D.; Pike, I.; Kuhn, K.; Rogers, J.C.; Roberts, T.M.; Gygi, S.P.; Paulo, J.A. TMTpro-18plex: The Expanded and Complete Set of TMTpro Reagents for Sample Multiplexing. J. Proteome Res. 2021, 20, 2964–2972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schweppe, D.K.; Eng, J.K.; Yu, Q.; Bailey, D.; Rad, R.; Navarrete-Perea, J.; Huttlin, E.L.; Erickson, B.K.; Paulo, J.A.; Gygi, S.P. Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J. Proteome Res. 2020, 19, 2026–2034. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gassaway, B.M.; Li, J.; Rad, R.; Mintseris, J.; Mohler, K.; Levy, T.; Aguiar, M.; Beausoleil, S.A.; Paulo, J.A.; Rinehart, J.; et al. A multi-purpose, regenerable, proteome-scale, human phosphoserine resource for phosphoproteomics. Nat. Methods 2022, 19, 1371–1375. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sambrano, G.R.; Fraser, I.; Han, H.; Ni, Y.; O’Connell, T.; Yan, Z.; Stull, J.T. Navigating the signalling network in mouse cardiac myocytes. Nature 2002, 420, 712–714. [Google Scholar] [CrossRef]
- Xiang, F.; Sakata, Y.; Cui, L.; Youngblood, J.M.; Nakagami, H.; Liao, J.K.; Liao, R.; Chin, M.T. Transcription factor CHF1/Hey2 suppresses cardiac hypertrophy through an inhibitory interaction with GATA4. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1997–H2006. [Google Scholar] [CrossRef]
- Liu, Y.; Korte, F.S.; Moussavi-Harami, F.; Yu, M.; Razumova, M.; Regnier, M.; Chin, M.T. Transcription factor CHF1/Hey2 regulates EC coupling and heart failure in mice through regulation of FKBP12.6. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1860–H1870. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, T.D.; Rodrigo, M.C.; Simpson, P.C. Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 2007, 357, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Hadley, W. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wickham, H.; François, R.; Henry, L.; Müller, K.; Vaughan, D. Dplyr: A Grammar of Data Manipulation. R package Version 1.1.4. 2023. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 1 December 2023).
- Hadley, W. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar]
- Kassambara, A. Ggpubr: ‘Ggplot2’ Based Publication Ready Plots. R Package Version 0.6.0. 2023. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 1 December 2023).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Neuwirth, E. _RColorBrewer: ColorBrewer Palettes. R Package Version 1.1-3. 2022. Available online: https://CRAN.R-project.org/package=RColorBrewer (accessed on 1 December 2023).
- Slowikowski, K. Ggrepel: Automatically Position Non-Overlapping Text Labels with ‘ggplot2’. R Package Version 0.9.6. 2024. Available online: https://CRAN.R-project.org/package=ggrepel (accessed on 1 December 2023).
- Xu, S.; Hu, E.; Cai, Y.; Xie, Z.; Luo, X.; Zhan, L.; Tang, W.; Wang, Q.; Liu, B.; Wang, R.; et al. Using clusterProfiler to characterize multiomics data. Nat. Protoc. 2024, 19, 3292–3320. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Yan, G.R.; He, Q.Y. DOSE: An R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015, 31, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M. org.Mm.eg.db: Genome Wide Annotation for Mouse. R Package Version 3.19.1. 2024. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html (accessed on 1 December 2023).
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.24.4. 2024. Available online: https://yulab-smu.top/biomedical-knowledge-mining-book/ (accessed on 1 December 2023).
- Ahlmann-Eltze, C. Ggupset: Combination Matrix Axis for ‘ggplot2’ to Create ‘UpSet’ Plots. R Package Version 0.4.0. 2024. Available online: https://CRAN.R-project.org/package=ggupset (accessed on 1 December 2023).
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 1 December 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Kinases | G Protein-Coupled Receptors | Phosphatases | Ion Channels |
|---|---|---|---|---|
| DDX3X DHX36 | EEF2K EGFR | ADORA2A ADRB1 | PPP3CA PTP4A1 | KCNJ2 MCOLN1 |
| FAAH | EIF2AK4 | AGTR2 | SCN1B | |
| HRAS | FLT1 | CX3CR1 | ||
| KRAS | INSR | GPR174 | ||
| POLG | IPMK | MC5R | ||
| MAP2K6 | ||||
| MAP3K12 | ||||
| MAPK14 | ||||
| MKNK1 | ||||
| MOS | ||||
| PRKAG3 | ||||
| PRKCZ | ||||
| PTK2 | ||||
| RAF1 | ||||
| RET | ||||
| ROCK1 | ||||
| ROCK2 TGFBR2 TK1 TTN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thorkelsson, A.; Chou, C.; Tripp, A.; Ali, S.A.; Galper, J.; Chin, M.T. Hypertrophic Cardiomyopathy-Associated CRYABR123W Activates Calcineurin, Reduces Calcium Sequestration, and Alters the CRYAB Interactome and the Proteomic Response to Pathological Hypertrophy. Int. J. Mol. Sci. 2025, 26, 2383. https://doi.org/10.3390/ijms26062383
Thorkelsson A, Chou C, Tripp A, Ali SA, Galper J, Chin MT. Hypertrophic Cardiomyopathy-Associated CRYABR123W Activates Calcineurin, Reduces Calcium Sequestration, and Alters the CRYAB Interactome and the Proteomic Response to Pathological Hypertrophy. International Journal of Molecular Sciences. 2025; 26(6):2383. https://doi.org/10.3390/ijms26062383
Chicago/Turabian StyleThorkelsson, Andres, Chun Chou, Audrey Tripp, Samia A. Ali, Jonas Galper, and Michael T. Chin. 2025. "Hypertrophic Cardiomyopathy-Associated CRYABR123W Activates Calcineurin, Reduces Calcium Sequestration, and Alters the CRYAB Interactome and the Proteomic Response to Pathological Hypertrophy" International Journal of Molecular Sciences 26, no. 6: 2383. https://doi.org/10.3390/ijms26062383
APA StyleThorkelsson, A., Chou, C., Tripp, A., Ali, S. A., Galper, J., & Chin, M. T. (2025). Hypertrophic Cardiomyopathy-Associated CRYABR123W Activates Calcineurin, Reduces Calcium Sequestration, and Alters the CRYAB Interactome and the Proteomic Response to Pathological Hypertrophy. International Journal of Molecular Sciences, 26(6), 2383. https://doi.org/10.3390/ijms26062383