1. Introduction
Neurodegeneration is exemplified through a heterogeneous family of chronic, progressively worsening disorders characterized by gradual neuronal dysfunction and death, effectively compromising motor and cognitive function. The most common neurodegenerative diseases include Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and Huntington’s disease [
1,
2]. As the global human population ages, the number of cases of neurodegenerative diseases is projected to grow, from 13.5 million in 2000 to 21.2 million in 2025 and 36.7 million in 2050 [
3]. There are various factors leading to the pathogenesis of these diseases; however, mounting evidence suggests that ROS (reactive oxygen species) play a critical role in the onset and progression of the respective pathological conditions [
4]. Moderate ROS concentration can act as a regulator of physiological cellular processes, such as redox signaling transduction, gene expression, and receptor activation, thus leading to tissue turnover and cell proliferation [
5]. On the other hand, when indigenous antioxidant systems cannot modulate and control ROS accumulation due to an increase in the production of free radicals, oxidative stress sets in [
6]. Oxidative stress can influence all biomacromolecules, causing lipid peroxidation, protein misfolding and aggregation, RNA mistranscription, and DNA damage. It seems that ROS toxicity is mediated by post-translational modifications of signaling molecules and/or hallmark proteins of neurodegeneration, while at the same time, free radicals pass freely through the plasma membrane, resulting in cell leakage and death [
7]. In the face of such adversity, the brain is particularly vulnerable to oxidative damage because it contains a high concentration of radical-susceptible polyunsaturated fatty acids, while at the same time, high levels of redox-active iron can act as a prooxidant, thereby contributing to the induction of autophagic cell death. Furthermore, lipid peroxidation of unsaturated fatty acids leads to the production of highly toxic compounds, including aldehydes or dienals, which can cause neuronal apoptosis. Aside from the aforementioned, the brain is a major metabolizer of oxygen (with a consumption of nearly 20% of the consumption of the entire body), while at the same time, the protective antioxidant mechanism is relatively inefficient [
8]. In fact, the brain contains low levels of glutathione, almost no catalase, and compared to the liver, reduced concentrations of glutathione peroxidase and vitamin E [
9].
Natural products are currently a subject of great interest in the pharmaceutical, health, food, and cosmetic industry owing to their diverse biological activities. The plant kingdom remains an insufficiently known source of bioactive natural compounds with various beneficial activities, such as anti-cancer, immunostimulant, anti-inflammatory, antioxidant, neuroprotective, and hepatoprotective [
10]. The most studied forms of natural products are extracts. The advantage of an extract, compared to isolated molecules, is the possible synergistic effect that the plethora of molecules in the extract might have, thus augmenting its collective beneficial effects. However, through this approach, due to inadequate fractionation processes, it is challenging to identify individual molecules responsible for the observed phenotype and macroscopic activity of the extracts [
11].
Cornus mas L., also known as dogwood, is a plant of the Cornaceae family that grows in Eastern Europe and the Middle East [
12]. The plant is widely used in folk medicine against various diseases, such as diarrhea, hemorrhoids, diabetes, and sore throat. Many authors over the years have attributed these properties to the high concentration of anthocyanins due to their physiological role in plants. Apart from anthocyanins, the chemical composition of the specific fruit can be divided into four main structural groups, i.e., iridoids, phenolic acids, flavonoids, and tannins. The quantity of these compounds is greatly dependent on the plant genotype, cultivation conditions, and ripeness of the fruit [
13,
14]. These components have been linked to many health-promoting effects, either as isolated compounds or synergistic combinations thereof. The main properties of these compounds are (a) antioxidant, emanating from the present polyphenols; (b) anti-inflammatory, anti-osteoporotic, and antiglaucomic due to the presence of loganic acid; (c) hepatoprotective from the anthocyanins and iridoids; as well as (d) anti-atherosclerotic and antidiabetic from whole fruit extracts [
15].
Concurrently, metal ions are a necessary part of human physiology, the deficiencies or paucity of which can result in pathological aberrations. Zinc, in particular, is a known antioxidant in the immune system, with zinc supplementation also blocking NF-κB activity, a known mediator of pro-inflammatory gene induction. On the other hand, vanadium, due to its pluripotent coordination chemistry, the various physiologically relevant oxidation states of the metal ion, and its flexibility in promoting complexation chemistries, can act as a malleable therapeutic agent, depending on the desired medicinal characteristics [
16,
17,
18,
19]. Along the same lines, citric acid is an α-hydroxytricarboxylic acid found in the metabolism of all aerobic organisms, as a key energy intermediate in the citric acid cycle (Krebs cycle), widely used for its antioxidant capacity in pharmaceutical, food, and beverage industries. At the same time, citric acid can act as a very soluble and effective metal ion chelator, thus ending up being a very good option as a ligand for bioessential metal ions [
20,
21]. On the other hand, the supplementation of bioavailable metals, especially in the case of zinc ions, might provide certain challenges since there is well-established evidence detailing the dynamic interplay between zinc and calcium ions in cell physiology that could result in irregular protein aggregation [
22,
23] and bear down on the integrity and survival of cells. At the same time, the introduction of metal ions in physiological fluids could have an influence on the ionic strength of the medium, thereby influencing the type of aggregation of subcellular targets related to neurodegenerative diseases [
24].
On the basis of the aforementioned grounds and specified factors, research has been launched in our lab to seek, identify, study, delineate, and formulate hybrid formulations of natural product materials poised to contribute to the amelioration of human neuropathological conditions and/or contribute to the proactive aversion of the onset or retardation of oxidative stress-induced neurodegenerative events. Cognizant of the physicochemical power of the above-described natural agents of inorganic or organic nature, carefully designed and appropriately configured abiotic and biological forms of such agents were employed. To that end, an attempt was made in the herein-described work to delineate the effectiveness of Cornus mas L. extracts against oxidative stress as a major contributor to neurodegeneration, with comprehensive studies poised to discover, define, and test in vitro well-configured hybrid formulations of the extracts supplemented by bioinorganic complex forms of zinc and vanadium (both biologically relevant and essential metal ions in biological systems in nature), collectively presenting a comprehensive neuroprotective profile. The generated profile(s) of neuronal cell (a)toxicity, antioxidant activity, anti-inflammatory activity, and cellular mechanism of action, as well as the ability of the hybrid bioinorganic materials to enhance such actions, provide a platform of natural product derivatives with merit as nutraceutical supplements, acting against oxidative stress and potentially serving as neuroprotectants.
4. Materials and Methods
4.1. Reagents
For the synthesis of the hybrid materials, ZnCl2 was purchased from Merck (Darmstadt, Germany), anhydrous citric acid from ChemLab (Zedelgem, Belgium), ammonia from VWR Chemicals (Darmstadt, Germany), VCl3 from Aldrich (Waltham, MA, USA), and sodium hydroxide from Sigma-Aldrich (St. Louis, MO, USA).
For the photometric evaluation of the total phenolic contents (TPC) and Ferric Reduction Antioxidant Power (FRAP) of the extracts, the Folin–Ciocalteau (F–C) reagent was purchased from Supelco-Sigma (Steinheim, Germany), sodium carbonate from P. Bacacos (Thessaloniki, Greece), gallic acid from Sigma (Steinheim, Germany), ascorbic acid from BDH Chemicals (Darmstadt, Germany), acetic acid from Aldrich (Waltham, MA, USA), sodium acetate from Johnson Mathey (London, UK), 2,4,6-Tri(2-pyridyl)-1,3,5-triazine (TPTZ) from Alfa Aesar (Ward Hill, MA, USA), hydrochloric acid was obtained from Ferak (Berlin, Germany), iron(III) chloride was purchased from Fluka (Gillingham, UK), and ultrapure water was produced by our laboratory.
For the in vitro studies, High Glucose Dulbecco’s Modified Eagle’s Medium (DMEM) with stable Glutamine and sodium pyruvate (L-103), Dulbecco’s Modified Eagle’s Medium supplemented with Ham’s F-12 Nutrient Mixture in a volumetric ratio of 1:1 (DMEM-F12) with stable Glutamine and 15 mM Hepes (L0092), Fetal Bovine Serum (FBS) South America origin (S1810), penicillin–streptomycin (P/S) solution 100× (L0022), and trypsin-EDTA 1X solution in Phosphate Buffer Saline (PBS) w/o calcium, w/o magnesium and w/o phenol red (L0940) were purchased from Biowest (Nuaillé, France), with trypan blue purchased from Sigma (Steinheim, Germany). PBS (1× pH 7.4) was prepared by dissolving sodium chloride (Panreac AppliChem, Barcelona, Spain), disodium phosphate (Merck, Darmstadt, Germany), potassium dihydrogen phosphate (Panreac AppliChem, Spain) and potassium chloride (Riedel-de Haën, Hannover, Germany) under continuous stirring in sterile ultrapure water in our laboratory. The resulting solution was buffered using HCl (Ferak, Berlin, Germany) 1 M solution to pH 7.4 and then additional sterile ultrapure water was added. Subsequently, the solution was autoclaved and filtered prior to use. The XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) cell viability kit was purchased from Cell Signaling Technologies (Danvers, MA, USA), Triton X-100 for Molecular Biology from Sigma (Steinheim, Germany), hydrogen peroxide (H2O2) from ChemLab (Zedelgem, Belgium), 2′,7′-dichlorofluorescin diacetate (DCF-DA) from Sigma (Steinheim, Germany), paraformaldehyde (PFA) from BDH Chemicals (Darmstadt, Germany), water for molecular biology and Tritidy G were purchased from Panreac AppliChem (Spain), chloroform was purchased from VWR Chemicals (Darmstadt, Germany), ethanol from J.T. Baker (Phillipsburg, NJ, USA), isopropanol from Sigma (Steinheim, Germany), nitric acid from ChemLab (Zedelgem, Belgium), EDTA (Ethylenediaminetetraacetic acid) from Panreac (AppliChem, Spain), and RNase-free water from Qiagen (Hilden, Germany). All of the primers were pre-designed LNA (Locked Nucleic Acid) primers for each gene purchased from Qiagen (Hilden, Germany), with the cDNA synthesis kit (iScript gDNA Clear cDNA Synthesis Kit, 1725035) and PCR master mix (iTaq Universal SYBR Green Supermix, 1725120) having been purchased from Bio-Rad (Hercules, CA, USA).
4.2. Synthesis of Metal–Organic Hybrid Complex Materials
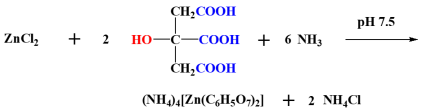
4.2.1. Synthesis of Zinc Citrate (Zn(II)-Cit)
Zinc citrate (Zn(II)-Cit) was synthesized as described in the literature, with slight modifications [
42]. Briefly, zinc chloride (ZnCl
2) and citric acid, in a molar ratio of 1:10, were dissolved in water, and their pH was adjusted to ~7.5, using aqueous ammonia. Ammonia acts both as a base to increase the pH of the solution while it concurrently provides the counter ions to the emerging anionic complex in (NH
4)
4[Zn(C
6H
5O
7)
2] (Zn(II)-Cit). The crystalline hybrid material was characterized through elemental analysis, spectroscopically, using FT-IR (Nicolet IR200 Infrared Spectrometer, Thermo-Fischer Scientific, Madison, WI, USA), and crystallographically, using a Bruker Kappa APEX II (Bruker, Bremen, Germany) diffractometer equipped with a triumph monochromator and Mo
Kα radiation.
4.2.2. Synthesis of Vanadium Citrate (V(IV)-Cit)
Vanadium citrate Na
4[V
2O
2(C
6H
4O
7)
2]•12H
2O (V(IV)-Cit) was produced and isolated as crystalline material using the methodology described in the literature, with slight modifications [
43]. In brief, vanadium(III) chloride and citric acid were mixed in water in a molar ratio of 1:1, the solution pH was adjusted to 8, using aqueous sodium hydroxide solution, and the reaction mixture was left for overnight stirring. On the following day, the blue solution was dried in vacuo, the residue was redissolved in water, and crystalline material was isolated a few days later using isopropanol as a precipitating agent. The sodium hydroxide solution, apart from the increase in pH, acts also as the source of counter ions to the anionic complex [V
2O
2(C
6H
4O
7)
2]
4−. The hybrid material was characterized through elemental analysis, spectroscopically, using the FT-IR technique (Nicolet IR-200 Infrared Spectrometer, Thermo-Fischer Scientific, Madison, WI, USA), and crystallographically, using Bruker Kappa APEX II (Bruker, Germany) diffractometer equipped with a triumph monochromator and Mo
Kα radiation.
4.2.3. Extraction and Isolation of Cornus mas L. Extracts
The lyophilized extracts were produced and optimized as described in the literature [
26]. Briefly, after washing and destoning a batch of fresh Cornelian cherries, their juice was removed, and the produced pomace was freeze-dried until further use. The extraction of the bioactive compounds was optimized using ultrasound-assisted extraction with independent variables: liquid-to-solid ratio (L/S), β-cyclodextrin concentration (Cβ-cd), in the aqueous solution, and sonication amplitude and duration, with the dependent responses being the total phenolic content (TPC), total anthocyanin content (TMA), DPPH radical scavenging activity, and loganic acid content. The optimized conditions were L/S of 50 mL/g, C
β-cd was 4.5 mg/mL, sonication amplitude 90%, and sonication time 22 min. The following studies were performed using optimized extracts, which were lyophilized and kept at −20 °C until further use. The desired solutions for the study of the extracts were freshly prepared prior to the experiments.
4.3. Determination of Total Phenolic Content (TPC) and Ferric Reduction Antioxidant Power (FRAP)
The photometric antioxidant capacity of aqueous
Cornus extracts was investigated using the total phenolic content (TPC) [
44] and Ferric Reduction Antioxidant Power (FRAP) [
45] assays, as reported in the literature, with slight modifications. Specifically, for TPC determination, 2.8 mL of ultrapure water was mixed with 0.1 mL aqueous extract solution and 0.1 mL F–C reagent. The emerging yellow mixture was vortexed for 1 min. To that reaction mixture, 2 mL of aqueous sodium carbonate solution 7.5%
w/v was added, and the blue solution was kept in the dark for 1 h. The absorbance of the final mixture was measured at 750 nm on a Hitachi U-1900 spectrophotometer (Hitachi, Ibaraki, Japan), and the results were expressed as mg GAE/g dry extract, using a gallic acid (68 mg/L–510 mg/L) calibration curve (y = 300.437x + 0.0034, r
2 = 0.998).
For determination of the ability of the extracts to reduce ferric ions (FRAP assay), the following solutions were prepared: 300 mM acetate buffer (CH3COOH/CH3COONa, with the pH adjusted to 3.6 using HCl (1 M) or NaOH (1 M)); 10 mM TPTZ prepared in 40 mM HCl; 20 mM FeCl3 6H2O solution in ultrapure water. The FRAP reagent was prepared by appropriate mixing of the aforementioned solution acetate buffer:TPTZ:iron chloride in a ratio 10:1:1. A volume of 80 μL of the desired aqueous Cornus extracts was mixed with 4 mL of the FRAP reagent, and the mixture was left for 15 min at 37 °C in the dark. Subsequently, the absorbance of the navy-blue solution was measured at 596 nm on a Hitachi U-1900 spectrophotometer (Hitachi, Ibaraki, Japan). The results are expressed in mg AAE/g dry extract, using an ascorbic acid (124 mg/L–1000 mg/L) standard calibration curve (y = 988.219x + 0.029, r2 = 0.999). Five replicates of each assay were run and the results are expressed as mean ± standard deviations (SD).
4.4. Cell Lines
For the in vitro evaluation of the potency of the extracts, two sensitive neuroblastoma cell lines were used, i.e., N2a58 (murine) and SH-SY5Y (human). The N2a58 cells were cultured in DMEM media, whereas the SH-SY5Y cells were cultured in DMEM-F12 media. Both media were supplemented with a 1% v/v penicillin-streptomycin solution and 10% v/v FBS to generate the complete media, while both cell lines were cultured in a humidified incubator at 37 °C and 5% CO2. When appropriate confluency was achieved, the medium of the cells was removed, and the attached cells were rinsed with PBS. To detach the cells, trypsin was added, and the plate was incubated at 37 °C. Subsequently, a complete medium was added to inactivate trypsin and the resulting mixture was centrifuged. The cell pellet was resuspended in a complete medium, and the concentration of viable cells was evaluated using the trypan blue exclusion assay.
4.5. Cytotoxicity Evaluation
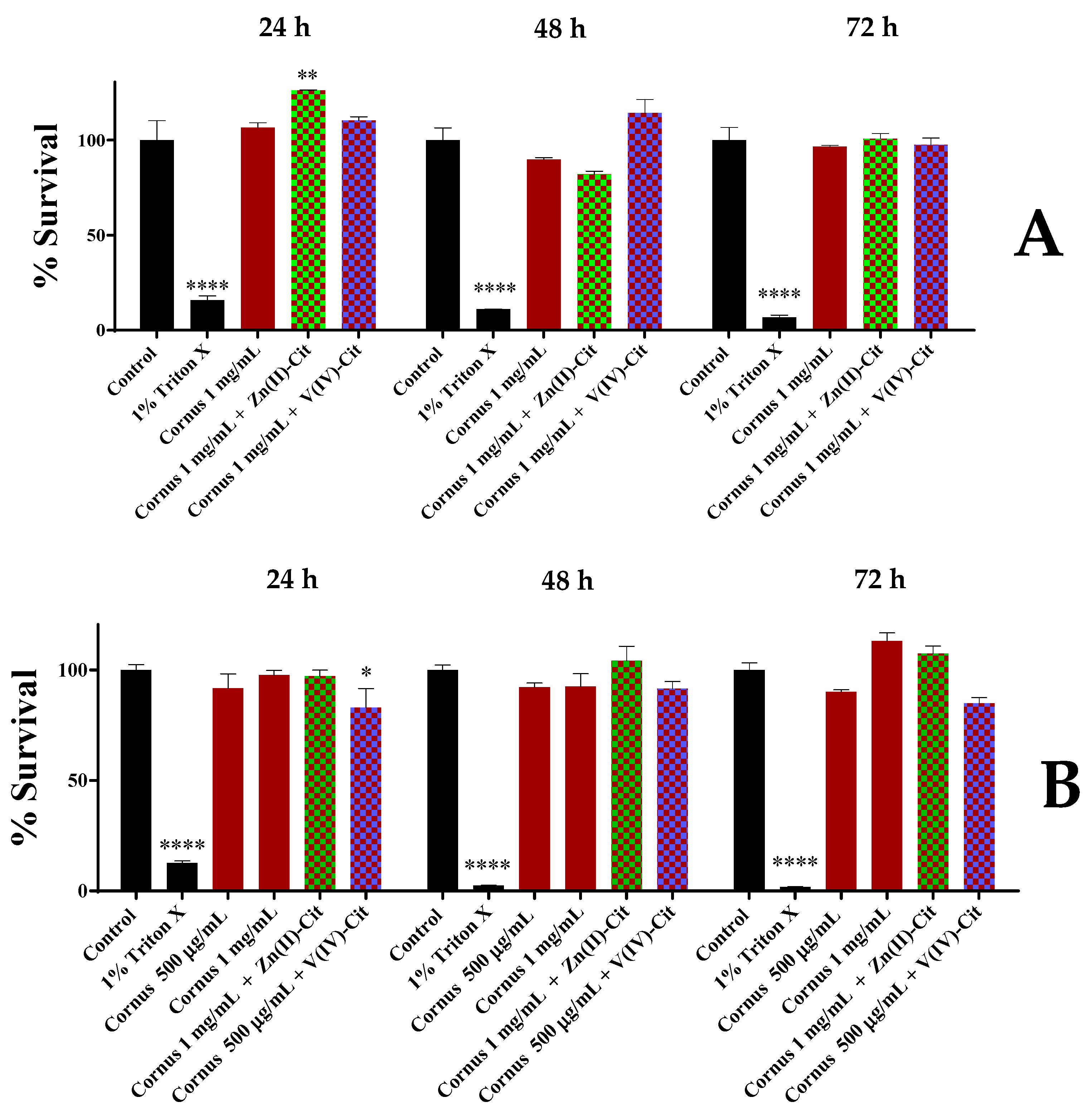
To evaluate the cytotoxicity profile of the extracts, the supplementary hybrid compounds Zn(II)-Cit and V(IV)-Cit, as well as combinations thereof, apart from cell viability, we studied the morphology and chemotacticity of the cells in each case, in a concentration-, time-, and cell line-dependent fashion.
4.5.1. Viability Evaluation
For the viability evaluation of the extracts and the hybrid compounds Zn(II)-Cit and V(IV)-Cit, as well as combinations thereof, the XTT cell viability assay was used according to manufacturer instructions. To pursue that, 5000 cells in 100 μL of the proper medium were seeded per well of a tissue culture 96-well plate, and the plate was left overnight in the incubator for the cells to attach to the plate surface. On the following day, the medium was replaced by 100 μL of medium, containing the title compound concentration, and the plate was incubated for the designated time. At the end of the timeframe, 50 μL of XTT detection solution (containing 49 μL of XTT reagent and 1 μL of electron coupling solution) was added in each well and the plate was incubated for 4 h. Subsequently, the absorbance A of each well was measured at 450 nm, using a BioTek Synergy H1 Multimode Reader (Agilent Technologies, Santa Clara, CA, USA). The survival percentage was calculated by subtracting the absorbance of the culture medium (CM) without cells from the absorbance of each experimental condition (EC), and subsequently dividing this difference with the difference in absorbance of the control medium with cells minus the absorbance of the control medium without cells, and multiplying with 100, as shown below. All of the experiments were performed in triplicates.
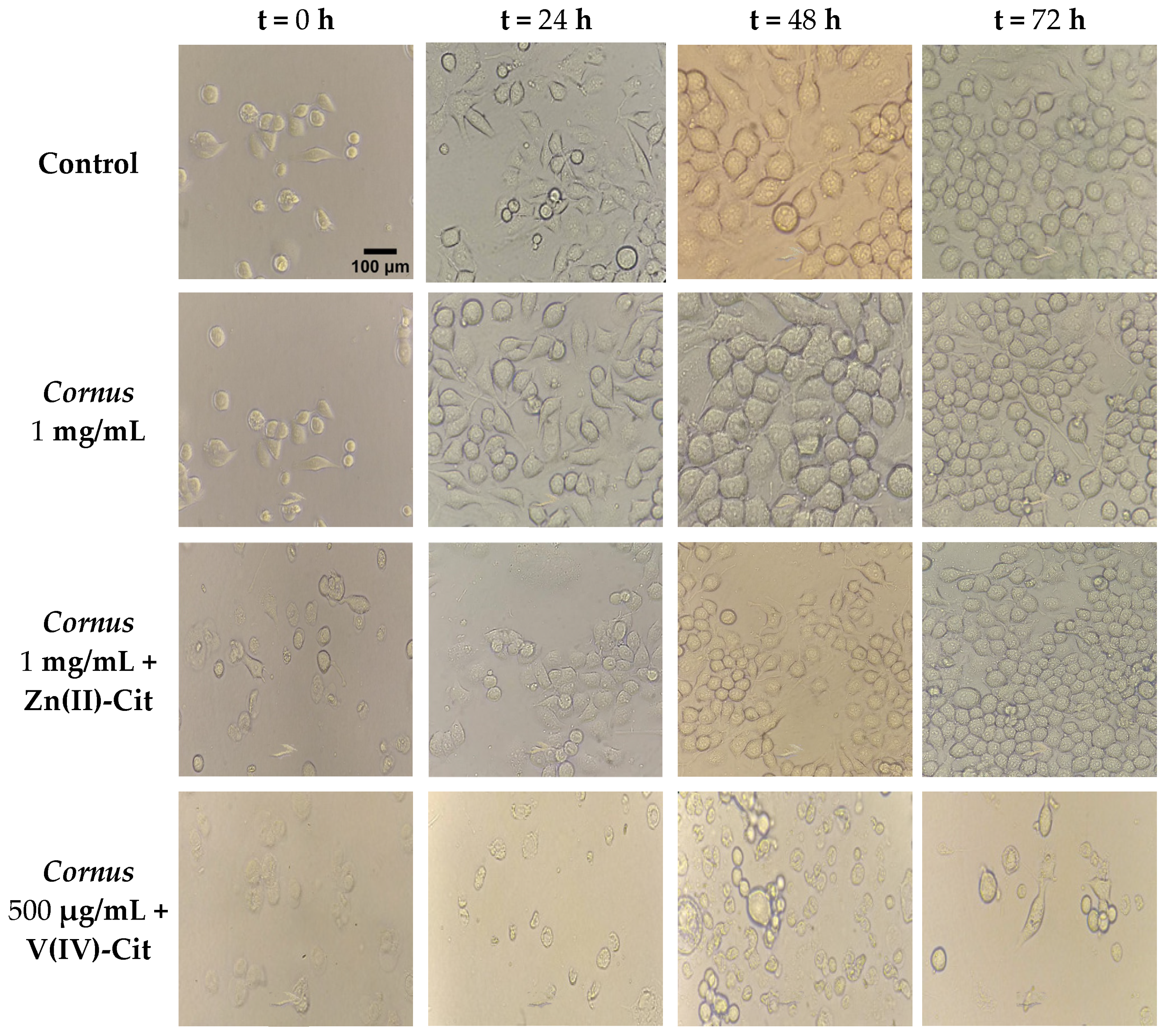
4.5.2. Morphology Studies
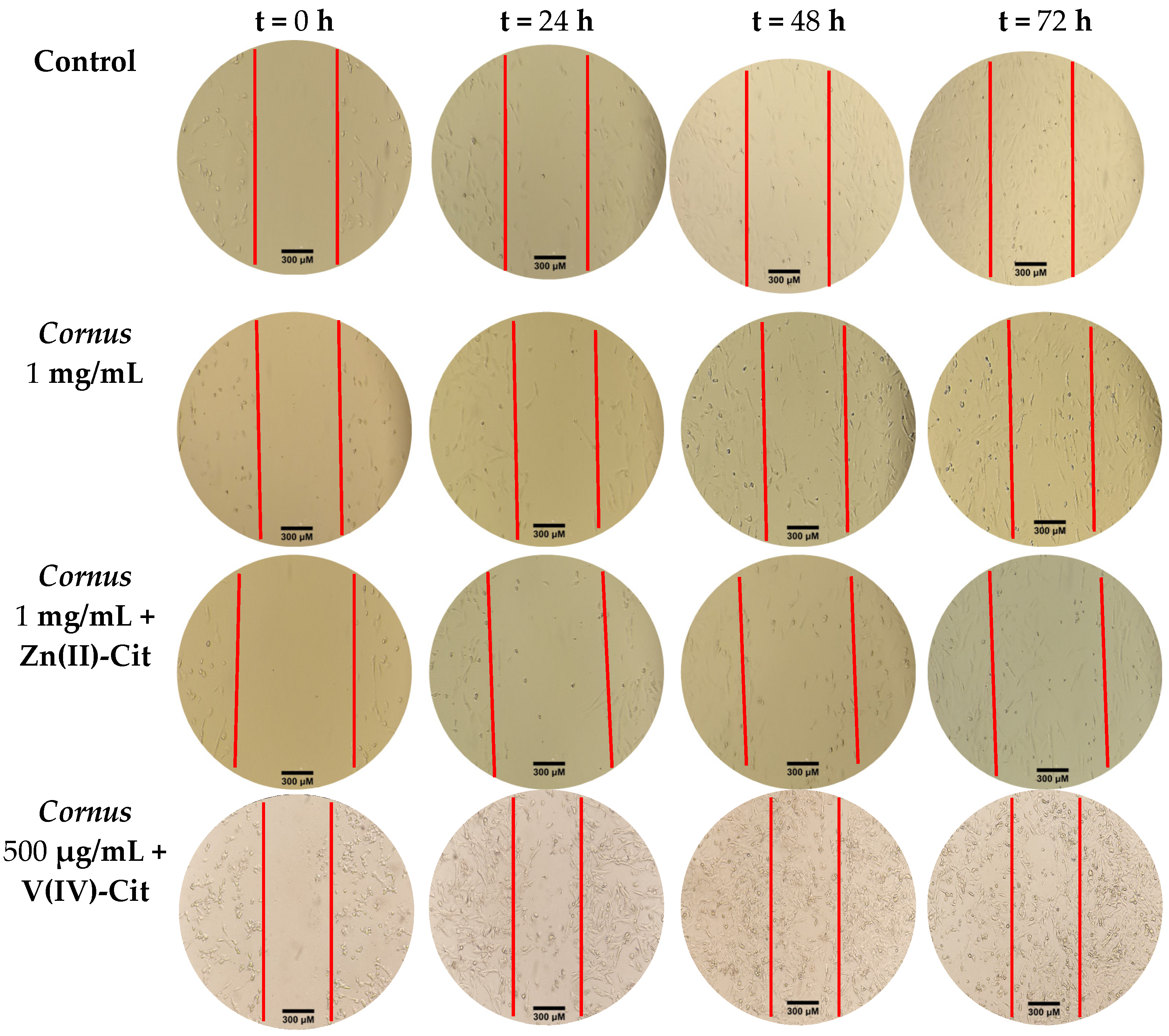
To better evaluate the cytotoxicity profile of each of the experimental conditions, morphology studies were pursued. To that end, 200,000 cells in 2 mL of proper medium were seeded per well of a tissue culture six-well plate, and the plate was incubated overnight. On the following day, the medium was replaced by 2 mL of medium containing the title concentration in each case. At specific time points (0, 24, 48, and 72 h), the plate was photographed via an Oxion Inverso biological microscope (Euromex, Duiven, The Netherlands) using both 10× and 40× lenses.
4.5.3. Chemotacticity
The chemotacticity of the cells under the specific experimental conditions was evaluated using the wound-healing (scratch) assay. Briefly, 200,000 cells in 2 mL of proper medium were seeded per well of a tissue culture six-well plate, and the plate was incubated until confluency had reached 80–90%. Subsequently, with a sterile yellow 200 μL tip, a vertical line was drawn across the well, thus disassociating the attached cells from the plate. Then, the medium of the well was replaced by 2 mL of medium containing the title compound concentration, and pictures were taken at specific time points (0, 24, 48, and 72 h) (vide supra) using the 10× lens. The pictures were imported in ImageJ software (version 1.53t, NIH, Bethesda, MD, USA), and the scratch distance was measured at each time point.
4.6. Antioxidant Activity
To evaluate the antioxidant potency of the extracts and the effectiveness of the supplementation using the hybrid compounds Zn(II)-Cit and V(IV)-Cit, as well as combinations thereof, H2O2, a well-known oxidant, was used to induce oxidative stress. For more information regarding the antioxidant properties, apart from the evaluation of the viability under oxidative stress, reactive oxygen species (ROS) were visualized using DCF-DA staining, and the cellular mechanism was studied using RT-PCR (Reverse Transcriptase Polymerase Chain Reaction).
4.6.1. Viability Under Oxidative Stress
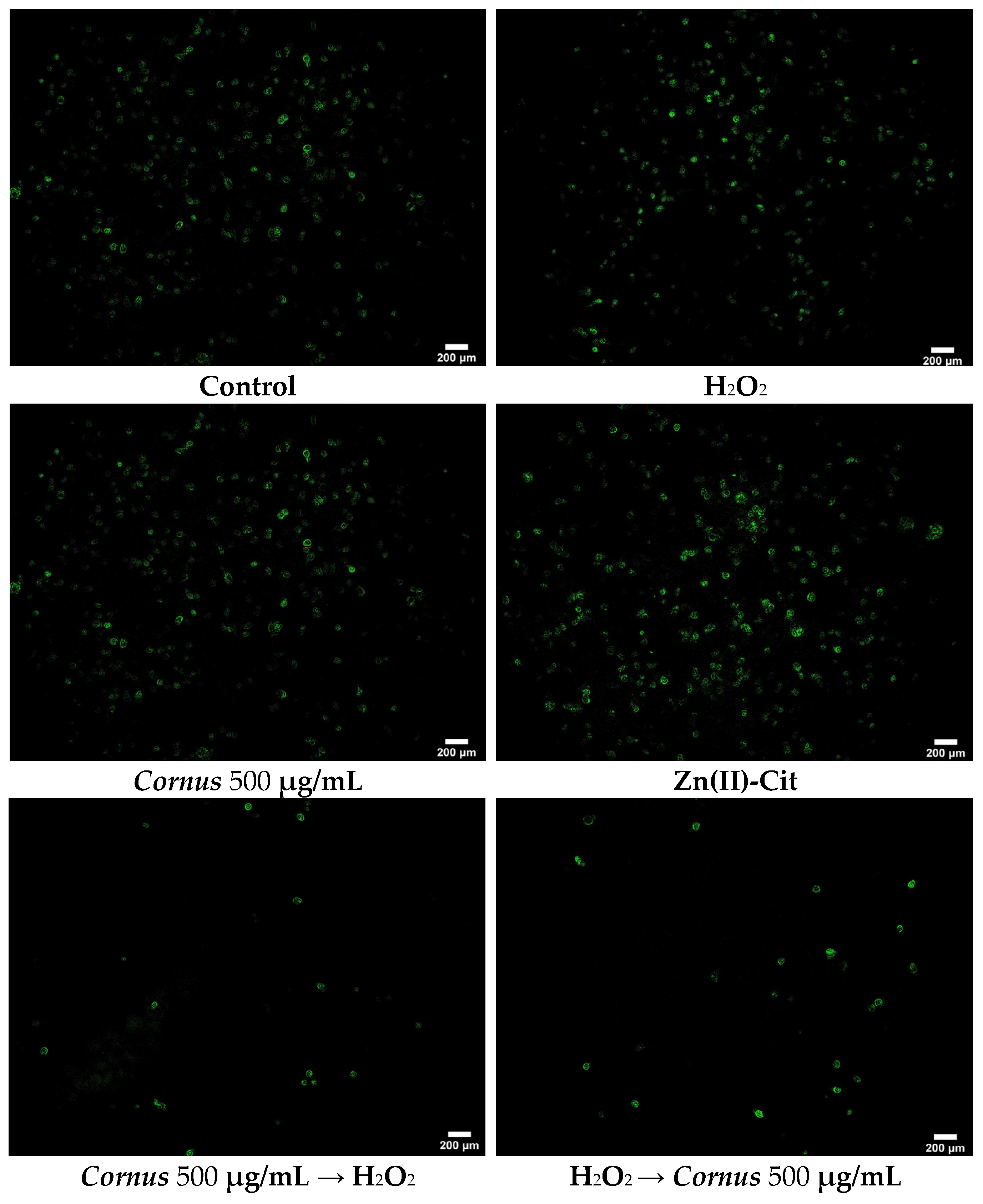
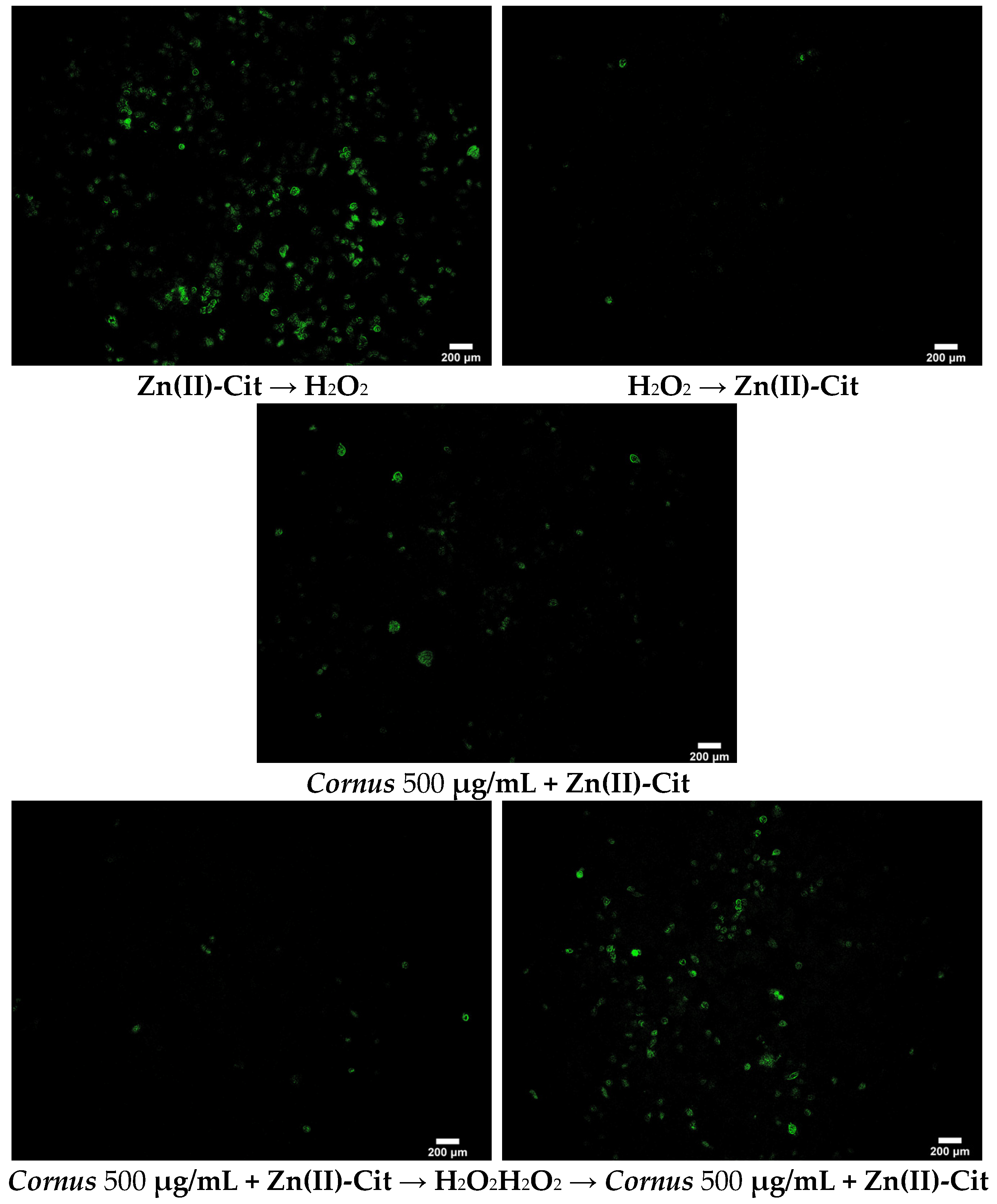
To evaluate the viability of the cell cultures under oxidative stress and the potency of the extracts and Zn(II)-Cit to increase viability, the XTT viability assay was used. The protocol was similar to the one mentioned above, but after overnight incubation, the medium was replaced by one containing H2O2. After 2 h of incubation with the oxidizing agent, the antioxidant agent (extracts, Zn(II)-Cit, combination thereof) was added, and the plate was incubated for an additional 24 h. Subsequently, the XTT assay was performed as mentioned above. Moreover, experiments were performed where the antioxidant was added prior to the oxidizing agent (2 h antioxidant pre-treatment). All of the experiments were performed in triplicates.
4.6.2. DCF-DA (2′,7′-Dichlorofluorescein Diacetate) Staining
To visualize the reduction of ROS in the presence of our potential antioxidant agent(s), the DCF-DA staining protocol was performed. Briefly, 200,000 N2a58 cells in 2 mL were seeded in small tissue culture dishes (35 × 10 mm) and incubated overnight. The medium was replaced by 2 mL of medium containing the title compound similar to the XTT assay (antioxidant treatment both prior to and after induction of the oxidative stress). After the end of the chosen timeframe, the treatment solution was removed, and the cells were rinsed twice with 2 mL PBS. After that, 1.5 mL of 4% PFA in PBS was added and the dishes were incubated for 30 min at room temperature. Subsequently, the PFA solution was discarded, cells were rinsed twice with 2 mL PBS, and 1.5 mL of 1 μM DCF-DA dye in PBS was added. The dishes were incubated for 30 min at 37 °C, and then the dye solution was discarded. Finally, after a rinse with PBS, 2 mL of PBS was added and the dishes were visualized using fluorescence microscope Axio Observer Z1 (Carl Zeiss, GmbH Jena, Jena, Germany). Images were captured on an AxioCam Hc camera (Carl Zeiss, GmbH Jena, Germany) and processed using ImageJ software (version 1.53t, NIH, Bethesda, MD, USA).
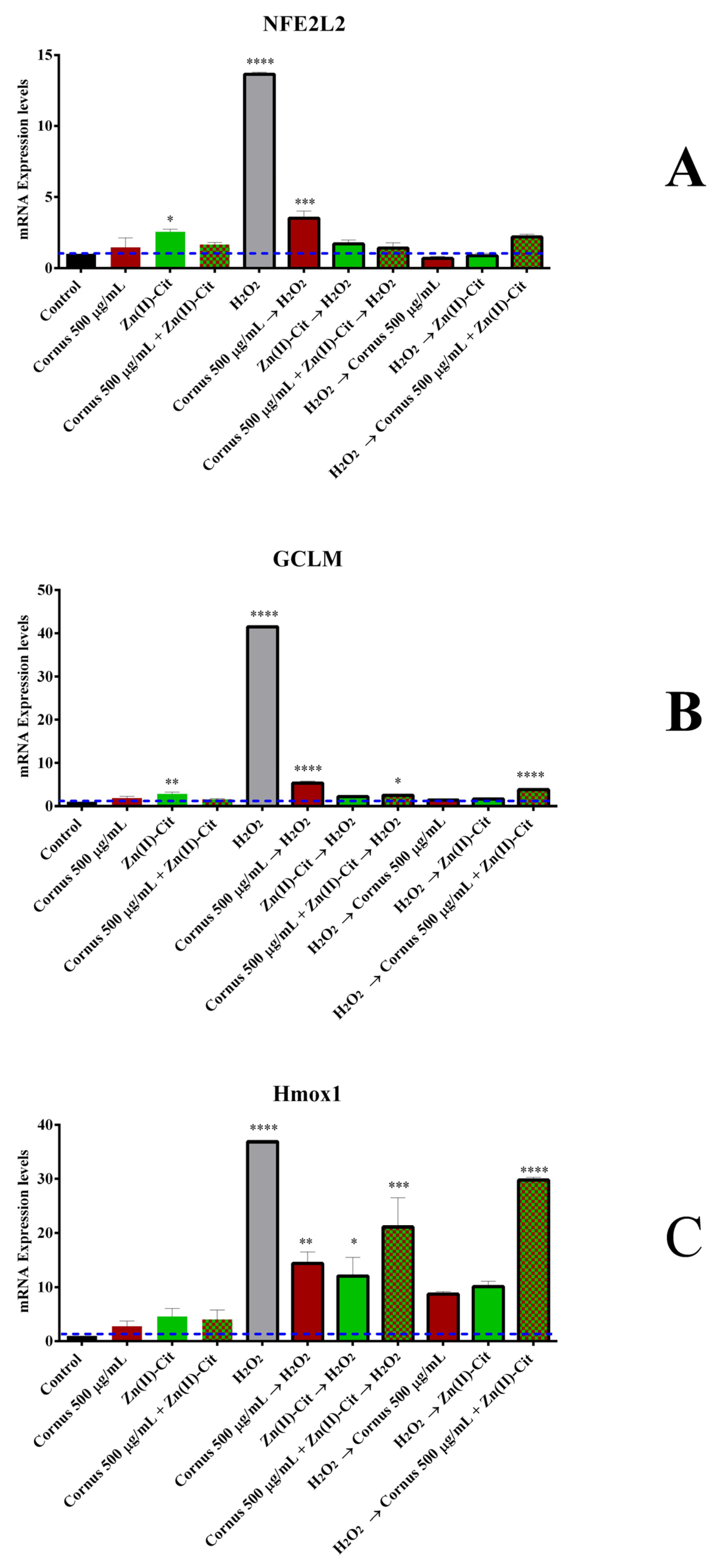
4.6.3. RT-PCR (Reverse Transcriptase Polymerase Chain Reaction) Assay
To evaluate the mechanism of action triggered by the presence of oxidizing and antioxidant agents, RT-PCR was used. Briefly, 200,000 N2a58 cells were seeded per well of a tissue culture six-well plate and incubated overnight. After the desired treatment with the oxidizing and/or the antioxidant agents (vide supra), treatment solutions were discarded, and cells were lysed using 1 mL of Tritidy G per well. The plate was subsequently incubated at room temperature for 20 min in the dark. The lysate was transferred to an Eppendorf tube, 200 μL of chloroform was added, and after shaking, the reaction mixture was incubated at room temperature for 15 min. After centrifugation at 12,000× g for 15 min at 4 °C, the upper clear phase (containing the RNA) was isolated, and 0.5 mL of ice-cold isopropanol was added. The resulting mixture was incubated at −20 °C for 45 min. The Eppendorf tube was centrifuged (vide supra), and the pellet was isolated and washed consecutively with 1 mL of 75% ethanol in sterile ultrapure water and then with 1 mL of pure ethanol. After the last wash, the supernatant was discarded, and the pellet was air-dried prior to resuspension of the RNA pellet in RNase-free water. The purity and concentration of the extracted RNA were evaluated using Nanodrop 2000 (Thermo Scientific, Waltham, MA, USA). Subsequently, 1 μg of RNA was used to produce cDNA, using the cDNA synthesis kit (iScript gDNA Clear cDNA Synthesis Kit) (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. The cDNA concentration was determined using Nanodrop 2000 (Thermo Scientific, Waltham, MA, USA). PCR amplification was performed in a Rotor-Gene Q (Qiagen, Hilden, Germany) thermocycler, using mouse LNA primers for the genes: GAPDH (MM_GAPDH_2176592), NFE2L2 (MM_NFE2L2_2096657), GCLM (MM_GCLM_2188527), and Hmox1 (MM_HMOX1_2180547), with the following protocol: 95 °C 10 min, 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s, followed by a hold at 72 °C for 5 min. The fluorescent signal was detected in the channel for the SYBR Green dye, and the results were quantified using delta delta Ct (2−ΔΔCt) analysis compared to the untreated condition, with GAPDH serving as the housekeeping gene.
4.7. Anti-Inflammatory Activity
To evaluate the anti-inflammatory activity of the extracts and the effect(s) of Zn(II)-Cit supplementation, RT-PCR was used. The treatment of the cells, the RNA extraction, and the cDNA synthesis were similar to the aforementioned protocol. For the primers, mouse LNA primers were used for the genes GAPDH (vide supra), TNF-a (MM_TNF_2032539), and IL-6 (MM_IL6_1986720), with the following protocol: 95 °C 2 min and 50 cycles of 95 °C for 5 s,60 °C for 10 s. The fluorescent signal was detected in the channel for the SYBR Green dye, and the results were quantified using delta delta Ct (2−ΔΔCt) analysis, compared to the untreated condition, with GAPDH being the housekeeping gene.
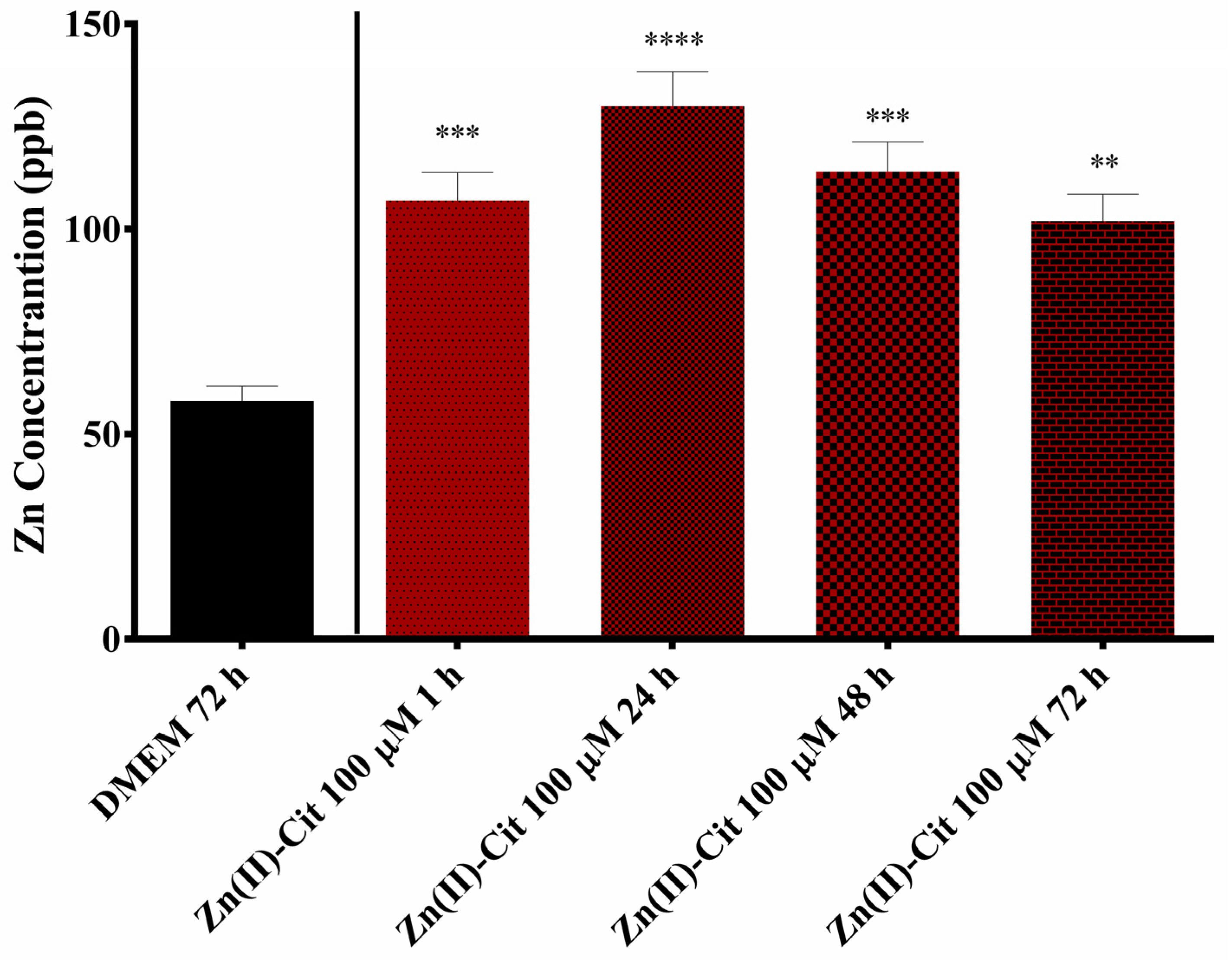
4.8. Metal Uptake Studies
To further delineate the effects of hybrid Zn(II)-Cit material in the cells, zinc uptake studies were performed in N2a58 cell cultures. Briefly, after the seeding of 200,000 N2a58 cells and their overnight incubation at 37 °C and 5% CO
2, cells were treated with 100 μM of Zn(II)-Cit for 1, 24, 48, and 72 h. All of the tips and the Eppendorf tubes used in this study were incubated overnight in 5% nitric acid solution and then rinsed with ultrapure water. Following the treatment of the cells, the solution was removed and kept in an Eppendorf tube; the cell monolayer was rinsed with 1 mL of 1 mM EDTA solution in PBS, a well-known and very potent zinc chelator [
46], to remove any loosely bound zinc residues from the cells. Subsequently, the cells were rinsed with PBS. To disassociate the cell monolayer from the plate, 1.5 mL of trypsin solution was added, and incubation took place at 37 °C for 5 min. To make sure that all of the cells had been disassociated from the plate, following trypsin incubation, the solution was pipetted several times and then stored in an Eppendorf tube. The samples were frozen immediately at −20 °C until the time of the measurement. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) analysis was carried out following established protocols described in the literature [
47], utilizing an Agilent 7500CE ICP-MS instrument (Agilent Technologies, Santa Clara, CA, USA), a high sensitivity and precise multielement detection instrumentation. Sample preparation was conducted using a CEM MARS 6 microwave digestion system (CEM Corporation, Matthews, NC, USA) to efficiently break down complex matrices for trace elemental analysis. The digestion procedure involves treating the samples with an acid mixture composed of concentrated nitric acid (HNO
3) and hydrogen peroxide (H
2O
2) in a volumetric ratio of 4:1. This mixture facilitates oxidative degradation of organic material and solubilization of metals. The digestion was performed under controlled conditions of elevated pressure, maintained at 200 psi, with a rise to pressure ramp followed by a 10-minute hold time to ensure complete sample dissolution and consistent results. These stringent parameters enhance the complete digestion of the samples investigated. Following the digestion, the samples are introduced into the Inductively Coupled Plasma Mass Spectrometry (ICP-MS) instrument (Agilent Technologies, Santa Clara, CA, USA), where a high-temperature plasma (approximately 10,000 K) ionizes the elements within the sample. The resulting ions are separated based on their mass-to-charge ratios using a quadrupole mass analyzer, enabling accurate quantification of trace elements. The intracellular concentrations were expressed as ppb (parts per billion). In the case of V(IV)-Cit, similar studies were not performed, mainly due to the lack of antioxidant activity (vide supra) and the low concentration of the treatment solution.
4.9. Statistical Analysis
The obtained experimental data are presented as average ± standard error mean (SEM) values of multiple sets of three independent measurements for the in vitro results and as average ± standard deviation (SD) values of multiple sets of independent measurements for the analytical studies. Mean cell survival rates and SEMs were calculated for each individual group. p-values were calculated using either one- or two-way analysis of variance, followed by Dunnet’s method, using GraphPad Prism v.6. Significance levels were assessed as follows: * p < 0.05 (significant), ** p < 0.01 (highly significant), *** p < 0.001 (extremely significant) and **** p ≤ 0.0001 (extremely significant) or non-significant (p > 0.05).
5. Conclusions
The pursuit of potent nutraceutical supplements counteracting the effects of oxidative stress linked to neurodegeneration has led to the utilization of bioactive materials derived from natural products to ensure their low cytotoxicity. Therefore, the employed approach targeted the use of natural extracts from Cornus mas L. and well-defined hybrid meta-organic complex species comprised of physiologically relevant constituents exemplified in Zn(II)-Cit and V(IV)-Cit, individually and collectively investigating potency, activity and potential synergism between the emerging binary bioactive combinations. To that end, the study focused and evaluated the bioactive profile of optimized aqueous extracts of Cornus mas L. photometrically, using FRAP and TPC methods, and determined in vitro their multifaceted activity, using two sensitive brain tissue cell lines; one human (SH-SY5Y) and one mouse (N2a58). To that end, the TPC and FRAP assays emphasized the significant antioxidant capacity of the aqueous extracts, thus justifying further work toward in vitro evaluation. The in vitro evaluation of the extracts provided the cytotoxicity profile in a dose-, time- and cell tissue-dependent fashion, evaluating not only the viability of cells but also the morphology and chemotacticity in each case, always factoring in the contribution of the zinc and vanadium complex species. The complete profile of cytotoxicity, evaluating the viability, morphological, and chemotactic data, indicates that the extracts are not toxic up to very high concentrations, while at the same time, supplementation of the extracts with the maximum atoxic concentrations of the hybrid materials revealed no additional cytotoxicity. Moreover, the extracts exhibit significant antioxidant activity, which was assessed through viability, ROS staining, and PCR studies, further enhanced by Zn(II)-Cit supplementation against H2O2-induced oxidative stress. Concurrently, the downregulation of pro-inflammatory genes (IL-6 and TNF-α) indicated the anti-inflammatory activity of the extracts, enhanced by Zn(II)-Cit supplementation. Finally, bioavailability studies on Zn(II)-Cit show that there are new potentially competitive metal–organic compounds of metal salt sources of Zn(II), exhibiting (a) solubility and biochemical reactivity characteristics commensurate with the nature and role of the organic acid in energy produced through cellular physiology, and (b) significant antioxidant activity commensurate with the nature of the organic acid substrate and the hybrid nature of a promising neuroprotective agent. These results provide solid evidence that zinc supplementation under the employed experimental conditions has a significant neuroprotective effect, with no ostensible undesired effects subverting neuronal physiology and integrity. The collectively formulated bioactivity profile of the extracts and the employed metal–organic species provide solid evidence of the tissue cell-specific beneficial nutraceutical properties of the Cornus mas L. extracts, further enhanced by a well-defined soluble and bioavailable hybrid form of zinc, thus setting the groundwork for further assessment of such composite natural products in averting oxidative stress-induced neurodegenerative events in sensitive human tissue loci. In pursuit of such a goal, the in vivo bioactivity profile of the above-mentioned materials needs to be investigated, in order to (a) validate the effectiveness of the extracts with and without the supplements, and (b) delineate the role of each participant in the formulation of new neutraceuticals acting proactively to protect, avert, and/or retard neurodegeneration.




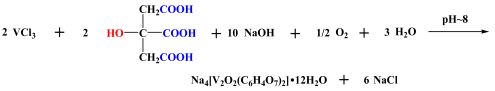




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}