
Reinforcing Nrf2 Signaling: Help in the Alzheimer’s Disease Context

 , ,
, ,  and
and
Abstract
:
1. Introduction
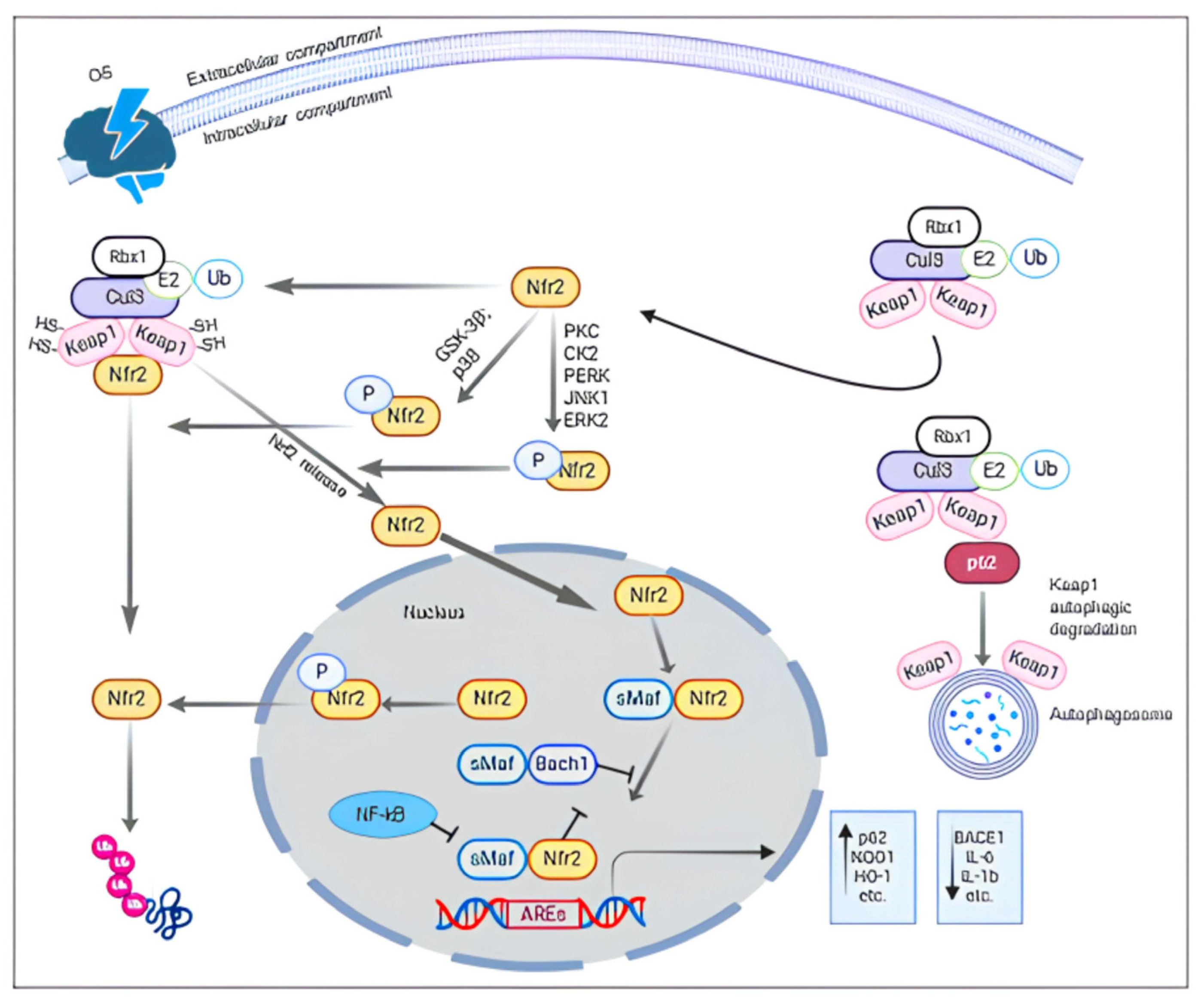
2. Nrf2 Structure and Function
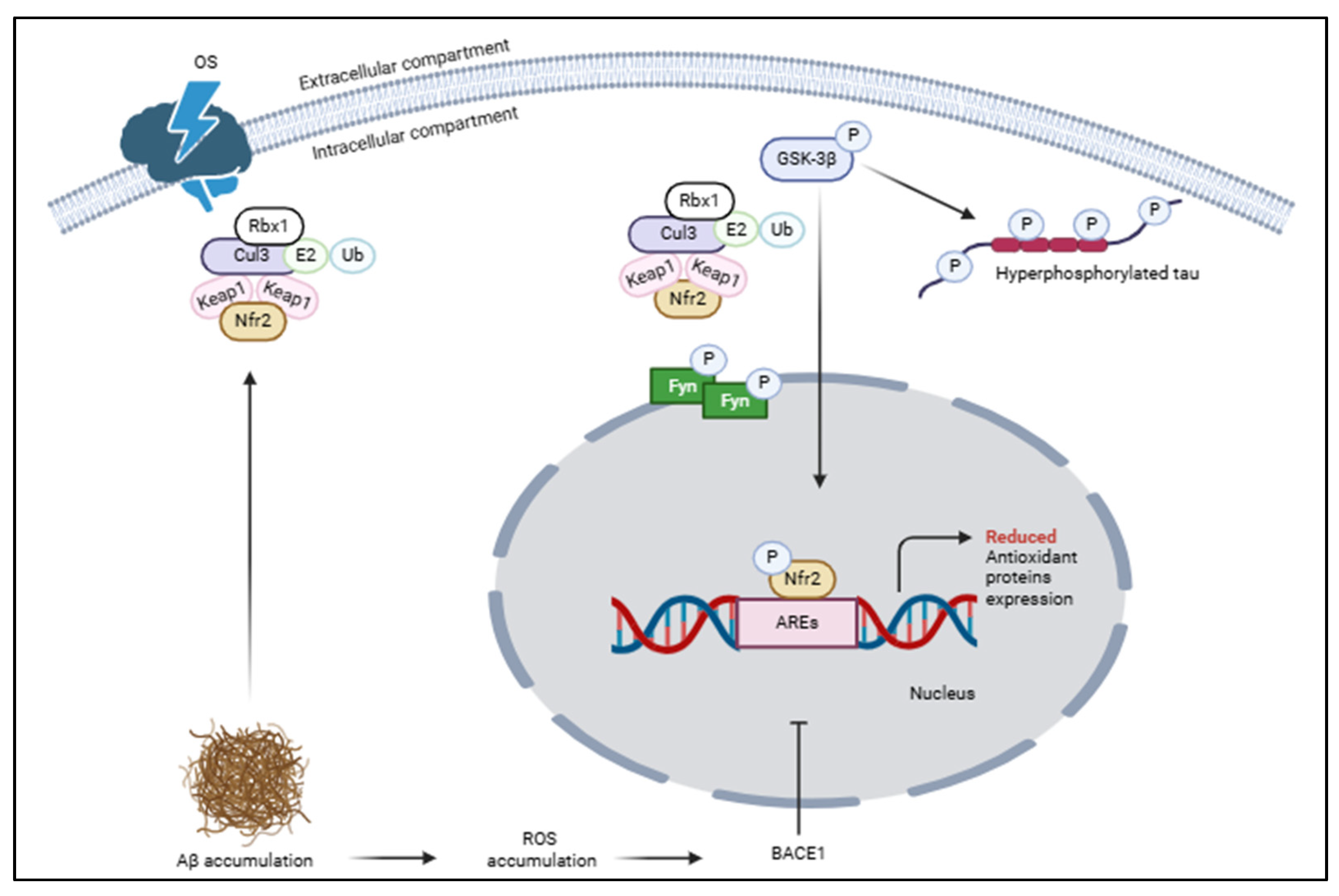
3. Nrf2 in the Brain: Physiological and Pathophysiological Conditions
4. FDA-Approved Treatments for Alzheimer’s
5. The Employment of Nrf2-Activating Natural and Synthetic Compounds for AD: The State of the Art
6. Inside the Molecular Nrf2-Activating Mechanisms by the Action of Natural Compounds
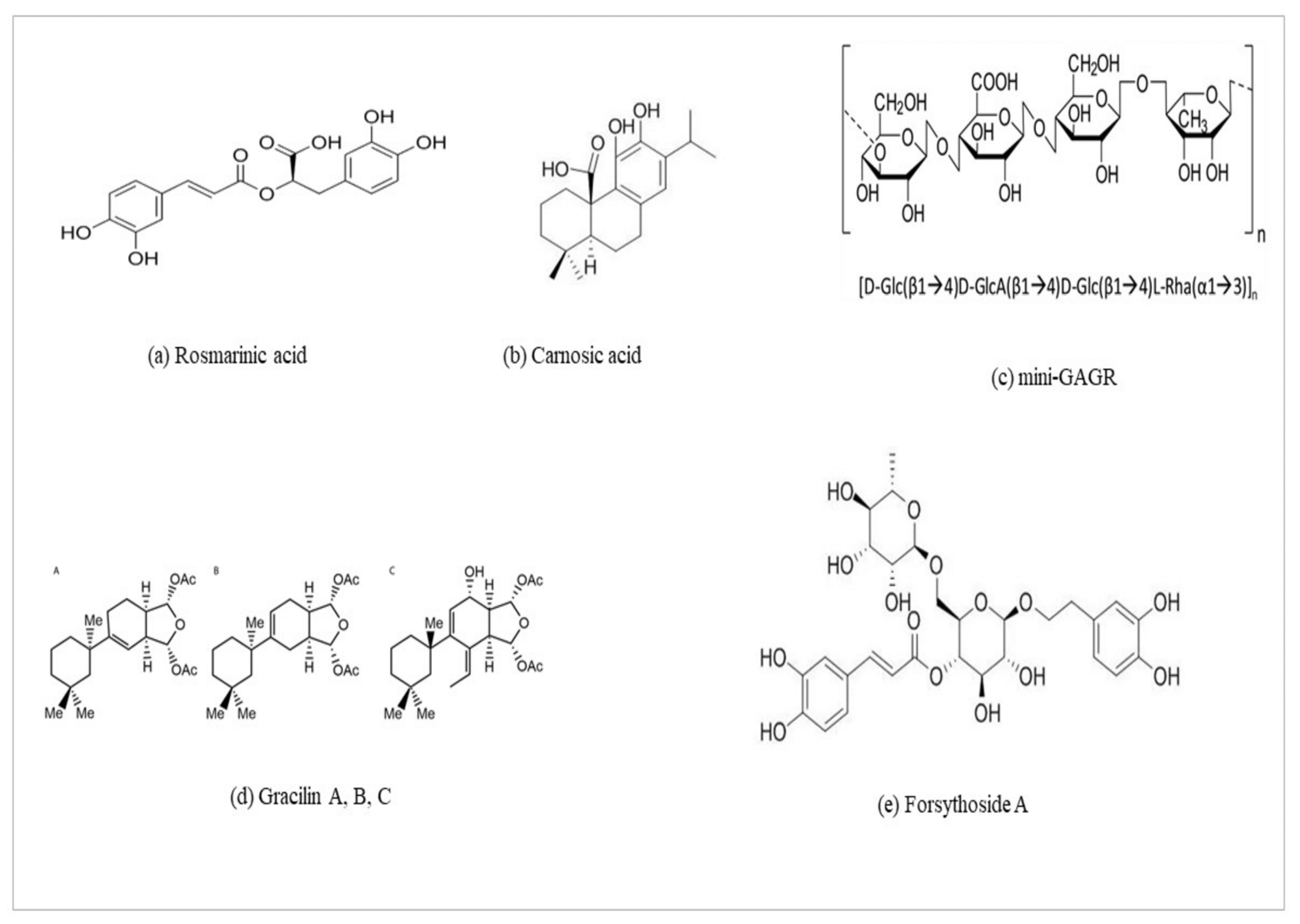
6.1. Rosmarinic Acid (ROSA)
6.2. Carnosic Acid (CA)
6.3. Mini-GAGR and Gracilins
6.4. Forsythoside A
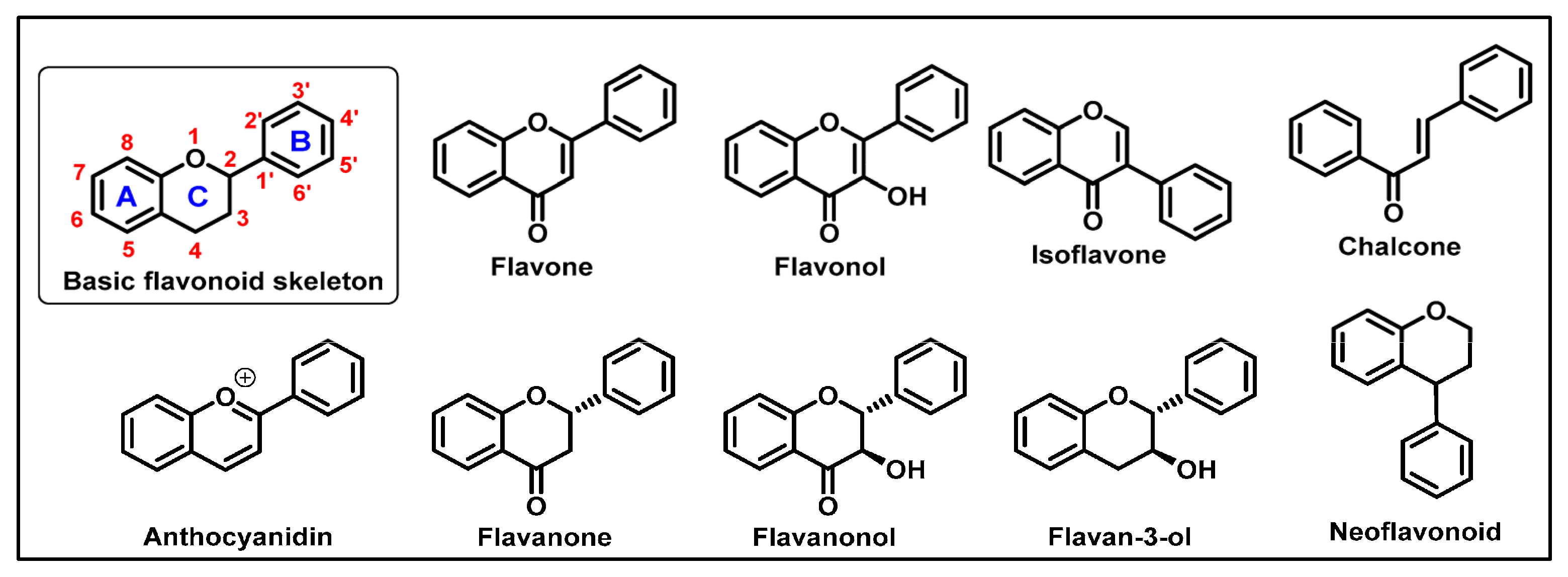
6.5. Flavonoids
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Kumar, A.; Sidhu, J.; Lui, F.; Tsao, J.W. Alzheimer Disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Soldner, F.; Jaenisch, R. Dissecting risk haplotypes in sporadic Alzheimer’s disease. Cell Stem Cell 2015, 16, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Statista. Leading Causes of Death Worldwide in 2019 (In Millions). Available online: https://www.statista.com/statistics/288839/leading-causes-of-death-worldwide/ (accessed on 4 April 2024).
- Alzheimer’s Disease International. World Alzheimer Report 2023. Available online: https://www.alzint.org/u/World-Alzheimer-Report-2023.pdf (accessed on 18 November 2024).
- la Torre, A.; Lo Vecchio, F.; Greco, A. Epigenetic Mechanisms of Aging and Aging-Associated Diseases. Cells 2023, 12, 1163. [Google Scholar] [CrossRef] [PubMed]
- Amelimojarad, M.; Amelimojarad, M.; Cui, X. The emerging role of brain neuroinflammatory responses in Alzheimer’s disease. Front. Aging Neurosci. 2024, 16, 1391517. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxidative Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [CrossRef]
- Irshad, M.; Chaudhuri, P.S. Oxidant-antioxidant system: Role and significance in human body. Indian J. Exp. Biol. 2002, 40, 1233–1239. [Google Scholar]
- Murphy, K.; Llewellyn, K.; Wakser, S.; Pontasch, J.; Samanich, N.; Flemer, M.; Hensley, K.; Kim, D.S.; Park, J. Mini-GAGR, an intranasally applied poly-saccharide, activates the neuronal Nrf2-mediated antioxidant defense system. J. Biol. Chem. 2018, 293, 18242–18269. [Google Scholar] [CrossRef]
- Kim, T.S.; Pae, C.U.; Yoon, S.J.; Jang, W.Y.; Lee, N.J.; Kim, J.J.; Lee, S.J.; Lee, C.; Paik, I.H.; Lee, C.U. Decreased plasma antioxidants in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2006, 21, 344–348. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Carapeto, A.P.; Marcuello, C.; Faísca, P.F.N.; Rodrigues, M.S. Morphological and Biophysical Study of S100A9 Protein Fibrils by Atomic Force Microscopy Imaging and Nanomechanical Analysis. Biomolecules 2024, 14, 1091. [Google Scholar] [CrossRef]
- Sedaghat, F.; Notopoulos, A. S100 protein family and its application in clinical practice. Hippokratia 2008, 12, 198–204. [Google Scholar]
- Gruden, M.A.; Davydova, T.V.; Wang, C.; Naekevich, V.B.; Fomina, V.G.; Kudrin, V.S.; Morozova-Roche, L.A.; Sewell, R.D.E. The misfolded pro-inflammatory protein S100A9 disrupts memory via neurochemical remodelling instigating an Alzheimer’s disease-like cognitive deficit. Behav. Brain Res. 2016, 306, 106–116. [Google Scholar] [CrossRef]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Wärmländer, S.K.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef]
- Eren-Koçak, E.; Dalkara, T. Ion Channel Dysfunction and Neuroinflammation in Migraine and Depression. Front. Pharmacol. 2021, 12, 777607. [Google Scholar] [CrossRef]
- Guan, P.P.; Cao, L.L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef]
- Kim, N.; Lee, H.J. Redox-Active Metal Ions and Amyloid-Degrading Enzymes in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7697. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Sternke-Hoffmann, R.; Zheng, W.; Mörman, C.; Luo, J. Multivariate effects of pH, salt, and Zn2+ ions on Aβ40 fibrillation. Commun. Chem. 2022, 5, 171. [Google Scholar] [CrossRef]
- Zidar, J.; Merzel, F. Probing amyloid-beta fibril stability by increasing ionic strengths. J. Phys. Chem. B 2011, 115, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Ziaunys, M.; Sakalauskas, A.; Mikalauskaite, K.; Smirnovas, V. Polymorphism of Alpha-Synuclein Amyloid Fibrils Depends on Ionic Strength and Protein Concentration. Int. J. Mol. Sci. 2021, 22, 12382. [Google Scholar] [CrossRef] [PubMed]
- Moubarak, M.M.; Pagano Zottola, A.C.; Larrieu, C.M.; Cuvellier, S.; Daubon, T.; Martin, O.C.B. Exploring the multifaceted role of NRF2 in brain physiology and cancer: A comprehensive review. Neuro-Oncol. Adv. 2023, 6, vdad160. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- De Plano, L.M.; Calabrese, G.; Rizzo, M.G.; Oddo, S.; Caccamo, A. The Role of the Transcription Factor Nrf2 in Alzheimer’s Disease: Therapeutic Opportunities. Biomolecules 2023, 13, 549. [Google Scholar] [CrossRef]
- Gureev, A.P.; Khorolskaya, V.G.; Sadovnikova, I.S.; Shaforostova, E.A.; Cherednichenko, V.R.; Burakova, I.Y.; Plotnikov, E.Y.; Popov, V.N. Age-Related Decline in Nrf2/ARE Signaling Is Associated with the Mitochondrial DNA Damage and Cognitive Impairments. Int. J. Mol. Sci. 2022, 23, 15197. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Bahn, G.; Park, J.S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef]
- Kanninen, K.; Malm, T.M.; Jyrkkänen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Ylä-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef]
- Karunatilleke, N.C.; Fast, C.S.; Ngo, V.; Brickenden, A.; Duennwald, M.L.; Konermann, L.; Choy, W.Y. Nrf2, the Major Regulator of the Cellular Oxidative Stress Response, is Partially Disordered. Int. J. Mol. Sci. 2021, 22, 7434. [Google Scholar] [CrossRef]
- Nguyen, T.; Sherratt, P.J.; Huang, H.C.; Yang, C.S.; Pickett, C.B. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003, 278, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Hushpulian, D.M.; Kaidery, N.A.; Dutta, D.; Sharma, S.M.; Gazaryan, I.; Thomas, B. Emerging small molecule inhibitors of Bach1 as therapeutic agents: Rationale, recent advances, and future perspectives. BioEssays 2024, 46, e2300176. [Google Scholar] [CrossRef]
- Ahuja, M.; Kaidery, N.A.; Dutta, D.; Attucks, O.C.; Kazakov, E.H.; Gazaryan, I.; Matsumoto, M.; Igarashi, K.; Sharma, S.M.; Thomas, B. Harnessing the Therapeutic Potential of the Nrf2/Bach1 Signaling Pathway in Parkinson’s Disease. Antioxidants 2022, 11, 1780. [Google Scholar] [CrossRef]
- Yu, C.; Xiao, J.H. The Keap1-Nrf2 System: A Mediator between Oxidative Stress and Aging. Oxidative Med. Cell. Longev. 2021, 2021, 6635460. [Google Scholar] [CrossRef]
- Fakhri, S.; Pesce, M.; Patruno, A.; Moradi, S.Z.; Iranpanah, A.; Farzaei, M.H.; Sobarzo-Sánchez, E. Attenuation of Nrf2/Keap1/ARE in Alzheimer’s Disease by Plant Secondary Metabolites: A Mechanistic Review. Molecules 2020, 25, 4926. [Google Scholar] [CrossRef]
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.N. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–32850. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Velichkova, M.; Hasson, T. KEAP1 regulates the oxidation-sensitive shuttling of NRF2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol. Cell. Biol. 2005, 25, 4501–4513. [Google Scholar] [CrossRef]
- Shaw, M.; Cohen, P. Role of protein kinase B and the MAP kinase cascade in mediating the EGF-dependent inhibition of glycogen synthase kinase 3 in Swiss 3T3 cells. FEBS Lett. 1999, 461, 120–124. [Google Scholar] [CrossRef]
- Thornton, T.M.; Pedraza-Alva, G.; Deng, B.; Wood, C.D.; Aronshtam, A.; Clements, J.L.; Sabio, G.; Davis, R.J.; Matthews, D.E.; Doble, B.; et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK-3β inactivation. Science 2008, 320, 667–670. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Hyötyläinen, I.; Trontti, K.; Rolova, T.; Wojciechowski, S.; Koskuvi, M.; Viitanen, M.; Levonen, A.-L.; Hovatta, I.; Roybon, L.; et al. NF-E2-related factor 2 activation boosts antioxidant defenses and ameliorates inflammatory and amyloid properties in human Presenilin-1 mutated Alzheimer’s disease astrocytes. Glia 2020, 68, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fahl, W.E. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem. Biophys. Res. Commun. 2001, 289, 212–219. [Google Scholar] [CrossRef]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Shi, Z.M.; Han, Y.W.; Han, X.H.; Zhang, K.; Chang, Y.N.; Hu, Z.M.; Qi, H.X.; Ting, C.; Zhen, Z.; Hong, W. Upstream regulators and downstream effectors of NF-κB in Alzheimer’s disease. J. Neurol. Sci. 2016, 366, 127–134. [Google Scholar] [CrossRef]
- Ren, P.; Chen, J.; Li, B.; Zhang, M.; Yang, B.; Guo, X.; Chen, Z.; Cheng, H.; Wang, P.; Wang, S.; et al. Nrf2 Ablation Promotes Alzheimer’s Disease-Like Pathology in APP/PS1 Transgenic Mice: The Role of Neuroinflammation and Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 3050971. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Profumo, E.; Tucci, P.; Saso, L. A Perspective on Nrf2 Signaling Pathway for Neuroinflammation: A Potential Therapeutic Target in Alzheimer’s and Parkinson’s Diseases. Front. Cell. Neurosci. 2022, 15, 787258. [Google Scholar] [CrossRef]
- Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells 2021, 10, 1884. [Google Scholar] [CrossRef]
- Yu, L.; Wang, S.; Chen, X.; Yang, H.; Li, X.; Xu, Y.; Zhu, X. Orientin alleviates cognitive deficits and oxidative stress in Abeta1-42-induced mouse model of Alzheimer’s disease. Life Sci. 2015, 121, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.; Tan, C.Y.; Tian, H.Z.; Liu, L.H.; Yang, M.W.; Hong, F.F.; Yang, S.L. Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci. Ther. 2020, 26, 155–166. [Google Scholar] [CrossRef]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Nilsson, P.; Saido, T.C. Dual roles for autophagy: Degradation and secretion of Alzheimer’s disease Aβ peptide. BioEssays 2014, 36, 570–578. [Google Scholar] [CrossRef]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef]
- Darios, F.; Stevanin, G. Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [CrossRef]
- Kim, S.; Viswanath, A.N.I.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. Nrf2 activator via interference of Nrf2-Keap1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989. [Google Scholar] [CrossRef]
- Steele, J.W.; Fan, E.; Kelahmetoglu, Y.; Tian, Y.; Bustos, V. Modulation of Autophagy as a Therapeutic Target for Alzheimer’s Disease. Postdoc J. 2013, 1, 21–34. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Cheng, J.; Hou, L. Drug Development for Alzheimer’s Disease: Microglia Induced Neuroinflammation as a Target? Int. J. Mol. Sci. 2019, 20, 558. [Google Scholar] [CrossRef] [PubMed]
- Vodičková, A.; Koren, S.A.; Wojtovich, A.P. Site-specific mitochondrial dysfunction in neurodegeneration. Mitochondrion 2022, 64, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Han, C.; Xia, Y.; Wan, F.; Yin, S.; Li, Y.; Kou, L.; Chi, X.; Hu, J.; Sun, Y.; et al. Efficacy and safety of 3-n-butylphthalide for the treatment of cognitive impairment: A systematic review and meta-analysis. CNS Neurosci. Ther. 2022, 28, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Feldman, H.H.; Zhang, S.; Flowers, S.A.; Luchsinger, J.A. Pharmacological thiamine levels as a therapeutic approach in Alzheimer’s disease. Front. Med. 2022, 9, 1033272. [Google Scholar] [CrossRef]
- Gibson, G.E.; Luchsinger, J.A.; Cirio, R.; Chen, H.; Franchino-Elder, J.; Hirsch, J.A.; Bettendorff, L.; Chen, Z.; Flowers, S.A.; Gerber, L.M.; et al. Benfotiamine and Cognitive Decline in Alzheimer’s Disease: Results of a Randomized Placebo-Controlled Phase IIa Clinical Trial. J. Alzheimer’s Dis. 2020, 78, 989–1010. [Google Scholar] [CrossRef]
- Baum, L.; Lam, C.W.; Cheung, S.K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef]
- Maiti, P.; Dunbar, G.L. Use of Curcumin, a Natural Polyphenol for Targeting Molecular Pathways in Treating Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1637. [Google Scholar] [CrossRef]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial Dysfunction in Alzheimer’s Disease: Opportunities for Drug Development. Curr. Neuropharmacol. 2022, 20, 675–692. [Google Scholar] [CrossRef]
- Effects of Sulforaphane in Patients With Prodromal to Mild Alzheimer’s Disease. Available online: https://cdek.pharmacy.purdue.edu/trial/NCT04213391/ (accessed on 20 November 2024).
- Vu, G.H.; Nguyen, H.D. Molecular mechanisms of sulforaphane in Alzheimer’s disease: Insights from an in-silico study. Silico Pharmacol. 2024, 12, 96. [Google Scholar] [CrossRef]
- Oregon Health and Science Universit. Lipoic Acid and Omega-3 Fatty Acids for Alzheimer’s Disease. Available online: https://clinicaltrials.gov/study/NCT01058941?tab=results (accessed on 20 November 2024).
- Center for Research Innovation in Biotechnology (CRIB). Telmisartan vs. Perindopril in Mild-Moderate Alzheimer’s Disease Patients. Available online: https://cdek.pharmacy.purdue.edu/trial/NCT02085265/ (accessed on 20 November 2024).
- Hamilton Health Sciences Corporation. Effects of Doxycycline and Rifampicin on Biomarkers of Alzheimer’s Disease in the Cerebrospinal Fluid. Available online: https://clinicaltrials.gov/study/NCT00439166?tab=results (accessed on 20 November 2024).
- Gonzales, M.M.; Garbarino, V.R.; Kautz, T.F.; Palavicini, J.P.; Lopez-Cruzan, M.; Dehkordi, S.K.; Mathews, J.J.; Zare, H.; Xu, P.; Zhang, B.; et al. Senolytic therapy in mild Alzheimer’s disease: A phase 1 feasibility trial. Nat. Med. 2023, 29, 2481–2488. [Google Scholar] [CrossRef]
- Viña, J.; Escudero, J.; Baquero, M.; Cebrián, M.; Carbonell-Asíns, J.A.; Muñoz, J.E.; Satorres, E.; Meléndez, J.C.; Ferrer-Rebolleda, J.; Cózar-Santiago, M.D.P.; et al. Genistein effect on cognition in prodromal Alzheimer’s disease patients. The GENIAL clinical trial. Alzheimer’s Res. Ther. 2022, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Plutacorresponding, R.; Ułamek-Kozioł, M. Chapter 4 Tau Protein-Targeted Therapies in Alzheimer’s Disease: Current State and Future Perspectives. In Alzheimer’s Disease: Drug Discovery [Internet]; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- National Institute on Aging (NIA). VITAL-VITamins to Slow ALzheimer’s Disease (Homocysteine Study). Available online: https://clinicaltrials.gov/study/NCT00056225 (accessed on 21 November 2024).
- Teng, E.; Manser, P.T.; Pickthorn, K.; Brunstein, F.; Blendstrup, M.; Sanabria Bohorquez, S.; Wildsmith, K.R.; Toth, B.; Dolton, M.; Ramakrishnan, V.; et al. Safety and Efficacy of Semorinemab in Individuals With Prodromal to Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2022, 79, 758–767. [Google Scholar] [CrossRef] [PubMed]
- El Kantar, S.; Yassin, A.; Nehmeh, B.; Labaki, L.; Mitri, S.; Naser Aldine, F.; Hirko, A.; Caballero, S.; Monck, E.; Garcia-Maruniak, A.; et al. Deciphering the therapeutical potentials of rosmarinic acid. Sci. Rep. 2022, 12, 15489. [Google Scholar] [CrossRef]
- Rong, H.; Liang, Y.; Niu, Y. Rosmarinic acid attenuates β-amyloid-induced OS via Akt/GSK-3β/Fyn-mediated Nrf2 activation in PC12 cells. Free. Radic. Biol. Med. 2018, 120, 114–123. [Google Scholar] [CrossRef]
- Zhou, M.W.; Jiang, R.H.; Kim, K.D.; Lee, J.H.; Kim, C.D.; Yin, W.T.; Lee, J.H. Rosmarinic acid inhibits poly(I:C)-induced inflammatory reaction of epidermal keratinocytes. Life Sci. 2016, 155, 189–194. [Google Scholar] [CrossRef]
- Osakabe, N.; Takano, H.; Sanbongi, C.; Yasuda, A.; Yanagisawa, R.; Inoue, K.; Yoshikawa, T. Anti-inflammatory and anti-allergic effect of rosmarinic acid (RA); inhibition of seasonal allergic rhinoconjunctivitis (SAR) and its mechanism. BioFactors 2004, 21, 127–131. [Google Scholar] [CrossRef]
- Cui, H.Y.; Zhang, X.J.; Yang, Y.; Zhang, C.; Zhu, C.H.; Miao, J.Y.; Chen, R. Rosmarinic acid elicits neuroprotection in ischemic stroke via Nrf2 and heme oxygenase 1 signaling. Neural Regen. Res. 2018, 13, 2119–2128. [Google Scholar]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, Z.; Yue, Z.; Ding, L.; Zhou, Y.; Huang, Z.; Huang, H. Rosmarinic Acid Ameliorates H2O2-Induced OS in L02 Cells Through MAPK and Nrf2 Pathways. Rejuvenation Res. 2019, 22, 289–298. [Google Scholar] [CrossRef]
- Mirza, F.J.; Zahid, S.; Holsinger, R.M.D. Neuroprotective Effects of Carnosic Acid: Insight into Its Mechanisms of Action. Molecules 2023, 28, 2306. [Google Scholar] [CrossRef]
- Tamaki, Y.; Tabuchi, T.; Takahashi, T.; Kosaka, K.; Satoh, T. Activated glutathione metabolism participates in protective effects of carnosic acid against oxidative stress in neuronal HT22 cells. Planta Medica 2010, 76, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Rezaie, T.; Nutter, A.; Lopez, K.M.; Parker, J.; Kosaka, K.; Satoh, T.; McKercher, S.R.; Masliah, E.; Nakanishi, N. Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 2016, 7, e2499. [Google Scholar] [CrossRef]
- Wu, C.R.; Tsai, C.W.; Chang, S.W.; Lin, C.Y.; Huang, L.C.; Tsai, C.W. Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: Involvement of antioxidative enzymes induction. Chem.-Biol. Interact. 2015, 225, 40–46. [Google Scholar] [CrossRef]
- Kosaka, K.; Yokoi, T. Carnosic acid, a component of rosemary (Rosmarinus officinalis L.), promotes synthesis of nerve growth factor in T98G human glioblastoma cells. Biol. Pharm. Bull. 2003, 26, 1620–1622. [Google Scholar] [CrossRef]
- Mimura, J.; Kosaka, K.; Maruyama, A.; Satoh, T.; Harada, N.; Yoshida, H.; Satoh, K.; Yamamoto, M.; Itoh, K. Nrf2 regulates NGF mRNA induction by carnosic acid in T98G glioblastoma cells and normal human astrocytes. J. Biochem. 2011, 150, 209–217. [Google Scholar] [CrossRef]
- Chen, J.H.; Ou, H.P.; Lin, C.Y.; Lin, F.J.; Wu, C.R.; Chang, S.W.; Tsai, C.W. Carnosic acid prevents 6-hydroxydopamine-induced cell death in SH-SY5Y cells via mediation of glutathione synthesis. Chem. Res. Toxicol. 2012, 25, 1893–1901. [Google Scholar] [CrossRef]
- Miller, D.M.; Singh, I.N.; Wang, J.A.; Hall, E.D. Administration of the Nrf2-ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free. Radic. Biol. Med. 2013, 57, 1–9. [Google Scholar] [CrossRef]
- Teng, L.; Fan, L.; Peng, Y.; He, X.; Chen, H.; Duan, H.; Yang, F.; Lin, D.; Lin, Z.; Li, H.; et al. Carnosic Acid Mitigates Early Brain Injury After Subarachnoid Hemorrhage: Possible Involvement of the SIRT1/p66shc Signaling Pathway. Front. Neurosci. 2019, 13, 26. [Google Scholar] [CrossRef]
- Leirós, M.; Alonso, E.; Rateb, M.E.; Houssen, W.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. Gracilins: Spongionella-derived promising compounds for Alzheimer disease. Neuropharmacology 2015, 93, 285–293. [Google Scholar] [CrossRef]
- McGrath, L.T.; McGleenon, B.M.; Brennan, S.; McColl, D.; McILroy, S.; Passmore, A.P. Increased oxidative stress in Alzheimer’s disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM 2001, 94, 485–490. [Google Scholar] [CrossRef]
- Ayton, S.; Portbury, S.; Kalinowski, P.; Agarwal, P.; Diouf, I.; Schneider, J.A.; Morris, M.C.; Bush, A.I. Regional brain iron associated with deterioration in Alzheimer’s disease: A large cohort study and theoretical significance. Alzheimer’s Dement. 2021, 17, 1244–1256. [Google Scholar] [CrossRef]
- Ayton, S.; Janelidze, S.; Kalinowski, P.; Palmqvist, S.; Belaidi, A.A.; Stomrud, E.; Roberts, A.; Roberts, B.; Hansson, O.; Bush, A.I. CSF ferritin in the clinicopathological progression of Alzheimer’s disease and associations with APOE and inflammation biomarkers. J. Neurol. Neurosurg. Psychiatry 2023, 94, 211–219. [Google Scholar] [CrossRef]
- Ayton, S.; Diouf, I.; Bush, A.I.; Alzheimer’s Disease Neuroimaging Initiative. Evidence that iron accelerates Alzheimer’s pathology: A CSF biomarker study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 456–460. [Google Scholar]
- Diouf, I.; Fazlollahi, A.; Bush, A.I.; Ayton, S.; Alzheimer’s Disease Neuroimaging Initiative. Cerebrospinal fluid ferritin levels predict brain hypometabolism in people with underlying β-amyloid pathology. Neurobiol. Dis. 2019, 124, 335–339. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Osama, A.; Zhang, J.; Yao, J.; Yao, X.; Fang, J. Nrf2: A dark horse in Alzheimer’s disease treatment. Ageing Res. Rev. 2020, 64, 101206. [Google Scholar] [CrossRef]
- Wang, C.; Chen, S.; Guo, H.; Jiang, H.; Liu, H.; Fu, H.; Wang, D. Forsythoside A Mitigates Alzheimer’s-like Pathology by Inhibiting Ferroptosis-mediated Neuroinflammation via Nrf2/GPX4 Axis Activation. Int. J. Biol. Sci. 2022, 18, 2075–2090. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Huang, H.; Zhang, H.; Lu, W.; Fu, G.; Zhu, Y. Forsythoside A inhibited S. aureus stimulated inflammatory response in primary bovine mammary epithelial cells. Microb. Pathog. 2018, 116, 158–163. [Google Scholar] [CrossRef]
- Rothschild, D.E.; McDaniel, D.K.; Ringel-Scaia, V.M.; Allen, I.C. Modulating inflammation through the negative regulation of NF-κB signaling. J. Leukoc. Biol. 2018, 103, 1131–1150. [Google Scholar] [CrossRef]
- Gao, W.; Guo, L.; Yang, Y.; Wang, Y.; Xia, S.; Gong, H.; Zhang, B.K.; Yan, M. Dissecting the Crosstalk Between Nrf2 and NF-κB Response Pathways in Drug-Induced Toxicity. Front. Cell Dev. Biol. 2022, 9, 809952. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Munir, S.; Badshah, S.L.; Khan, N.; Ghani, L.; Poulson, B.G.; Emwas, A.H.; Jaremko, M. Important Flavonoids and Their Role as a Therapeutic Agent. Molecules 2020, 25, 5243. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef]
- Suraweera, L.T.; Rupasinghe, H.P.V.; Dellaire, G.; Xu, Z. Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants 2020, 9, 973. [Google Scholar] [CrossRef]
- Li, Y.R.; Li, G.H.; Zhou, M.X.; Xiang, L.; Ren, D.M.; Lou, H.X.; Wang, X.N.; Shen, T. Discovery of natural flavonoids as activators of Nrf2-mediated defense system: Structure-activity relationship and inhibition of intracellular oxidative insults. Bioorg. Med. Chem. 2018, 26, 5140–5150. [Google Scholar] [CrossRef]
- Khan, H.; Tundis, R.; Ullah, H.; Aschner, M.; Belwal, T.; Mirzaei, H.; Akkol, E.K. Flavonoids targeting NRF2 in neurodegenerative disorders. Food Chem. Toxicol. 2020, 146, 111817. [Google Scholar] [CrossRef]
- Dong, F.; Wang, S.; Wang, Y.; Yang, X.; Jiang, J.; Wu, D.; Qu, X.; Fan, H.; Yao, R. Quercetin ameliorates learning and memory via the Nrf2-ARE signaling pathway in d-galactose-induced neurotoxicity in mice. Biochem. Biophys. Res. Commun. 2017, 491, 636–641. [Google Scholar] [CrossRef]
- Assi, A.A.; Farrag, M.M.Y.; Badary, D.M.; Allam, E.A.H.; Nicola, M.A. Protective effects of curcumin and Ginkgo biloba extract combination on a new model of Alzheimer’s disease. Inflammopharmacology 2023, 31, 1449–1464. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number Identifier | Study Type | Compound | Data/Status | Outcomes | Main Side Effects |
|---|---|---|---|---|---|
| NCT02711683 [74] | Observational | DL-3-n-butylphthalide | 2019-12 (completed) | Slower cognitive decline | Gastrointestinal disorders, including nausea, vomiting, and gastrointestinal discomfort Abnormal liver function Neurological disorders, including insomnia, dizziness, fatigue, and psychiatric symptoms [75] |
| NCT02292238 [76] | Interventional | Benfotiamine | 2022-06 (completed) | Reduced AD-like changes in tangles, and plaques. Diminished inflammation, neuron loss, and memory | Data not provided [77] |
| NCT015004854 | Resveratrol | 2014-03 (completed) | Modest improvement of cognitive function | Data not provided | |
| NCT00164749 [78] | Curcumin | 2006-07 (completed) | No differences in Aβ levels between treatments or MMSE scores | Gastrointestinal discomfort, chest tightness, skin rashes, and swollen skin, and allergic reactions, including dermatitis [79] | |
| NCT04213391 [80,81] | Sulforaphane | 2022-12 (completed) | Results awaited | Brain swelling, microbleeds, fatigue, nausea [82] | |
| NCT01058941 [83] | Alpha Lipoic Acid and Omega-3 Fatty Acid | 2014-12 (completed) [60] | Greater impairment in functional and cognitive ability | Cardiac disorders, including tachycardia and atrial fibrillations. Gastrointestinal disorders, including gastrointestinal obstruction, gastric ulcer, and hemorrhage. Pneumonia. Head injury. Pleura effusion, subdural hematoma evacuation. Fatigue, allergic conditions. Behavioral and psychiatric symptoms of dementia | |
| NCT02085265 [84] | Perindopril | 2023-09 (recruiting) [61] | Results not provided | Data not provided | |
| NCT00439166 [85] | Doxycycline | 2010-12 (completed) | Results not provided | Data not provided | |
| NCT04063124 [86] | Quercetin | 2023-01 (completed) | Safety, tolerability, and feasibility of the treatment | Diarrhea and emesis, urinary tract infection, hypoglycemia | |
| NCT01982578 [87] | Genistein | 2020-12 (completed) [62] | Lower Aβ deposition and slowdown of cognitive decline in prodromal AD patients | Mild diarrhea | |
| NCT00948259 [88] | Tideglusib | 2009-30 (completed) [63] | Safety of the drug. Positive, but not significant, trends in cognitive health | Data not provided | |
| NCT00056225 [89] | Pyridoxine | 2009-30 (completed) | Results not provided | Depression | |
| NCT03289143 [90] | Semorinemab | 2021-01 (completed) | Well-tolerated and acceptable safety profile No slowdown of AD progression | Nasopharyngitis, and infusion-related reaction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
la Torre, A.; Lo Vecchio, F.; Angelillis, V.S.; Gravina, C.; D’Onofrio, G.; Greco, A. Reinforcing Nrf2 Signaling: Help in the Alzheimer’s Disease Context. Int. J. Mol. Sci. 2025, 26, 1130. https://doi.org/10.3390/ijms26031130
la Torre A, Lo Vecchio F, Angelillis VS, Gravina C, D’Onofrio G, Greco A. Reinforcing Nrf2 Signaling: Help in the Alzheimer’s Disease Context. International Journal of Molecular Sciences. 2025; 26(3):1130. https://doi.org/10.3390/ijms26031130
Chicago/Turabian Stylela Torre, Annamaria, Filomena Lo Vecchio, Valentina Soccorsa Angelillis, Carolina Gravina, Grazia D’Onofrio, and Antonio Greco. 2025. "Reinforcing Nrf2 Signaling: Help in the Alzheimer’s Disease Context" International Journal of Molecular Sciences 26, no. 3: 1130. https://doi.org/10.3390/ijms26031130
APA Stylela Torre, A., Lo Vecchio, F., Angelillis, V. S., Gravina, C., D’Onofrio, G., & Greco, A. (2025). Reinforcing Nrf2 Signaling: Help in the Alzheimer’s Disease Context. International Journal of Molecular Sciences, 26(3), 1130. https://doi.org/10.3390/ijms26031130