

CX3CR1–Fractalkine Dysregulation Affects Retinal GFAP Expression, Inflammatory Gene Induction, and LPS Response in a Mouse Model of Hypoxic Retinopathy

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Results
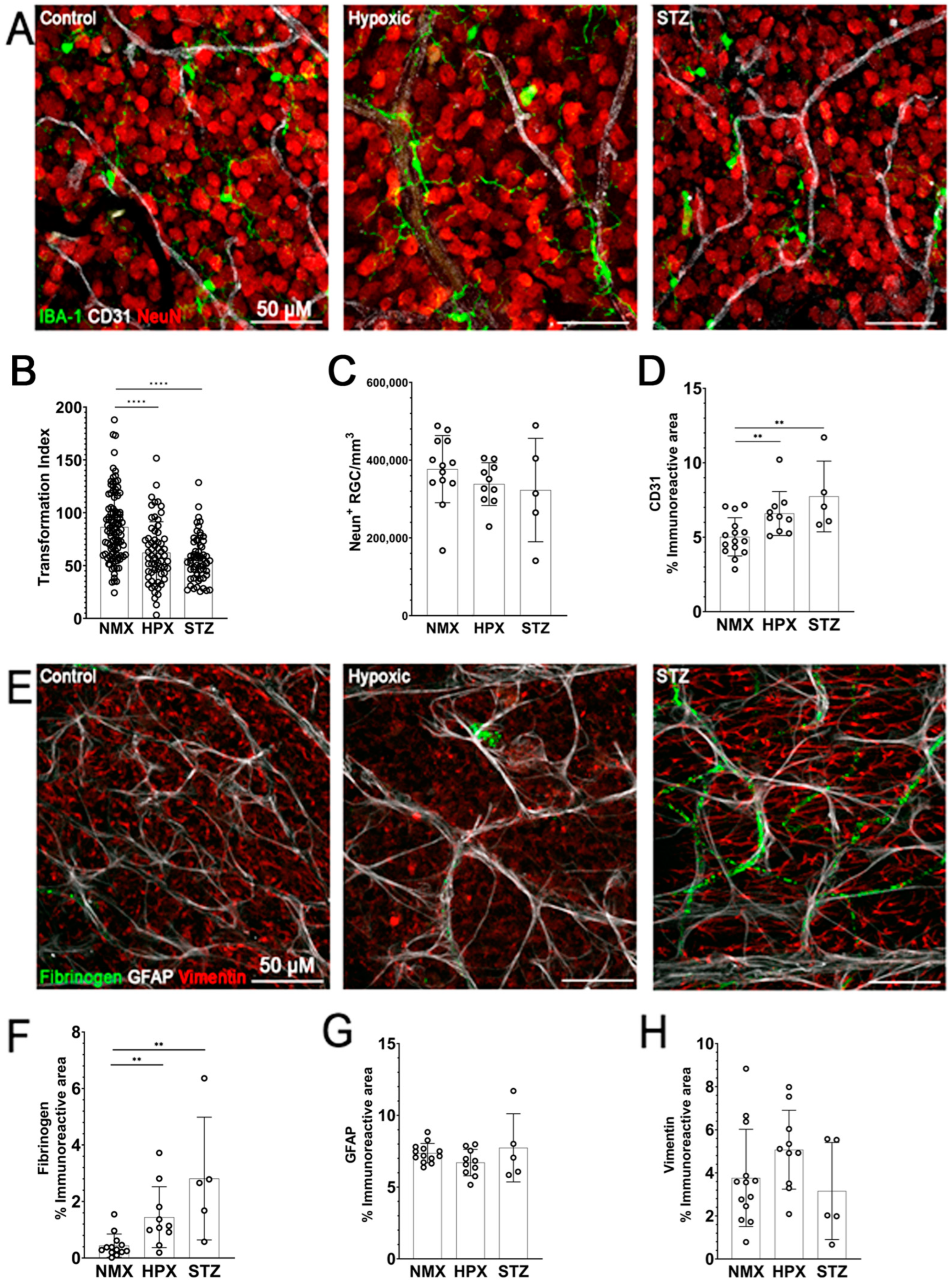
2.1. Microglia Reactivity, Reduced GFAP Expression, and Vascular Damage Are Observed in Both Models of Hypoxia- and STZ-Induced Retinopathy
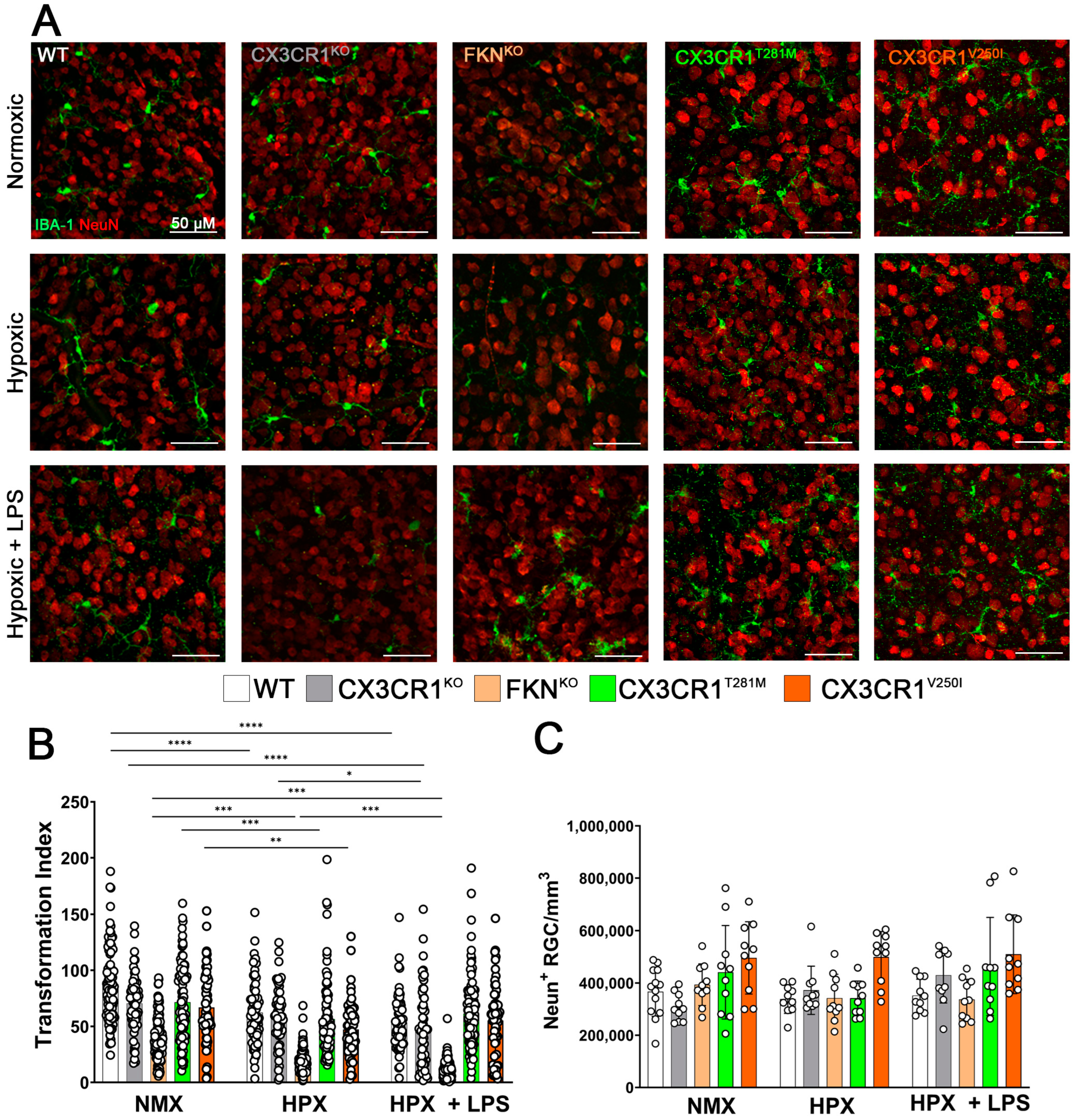
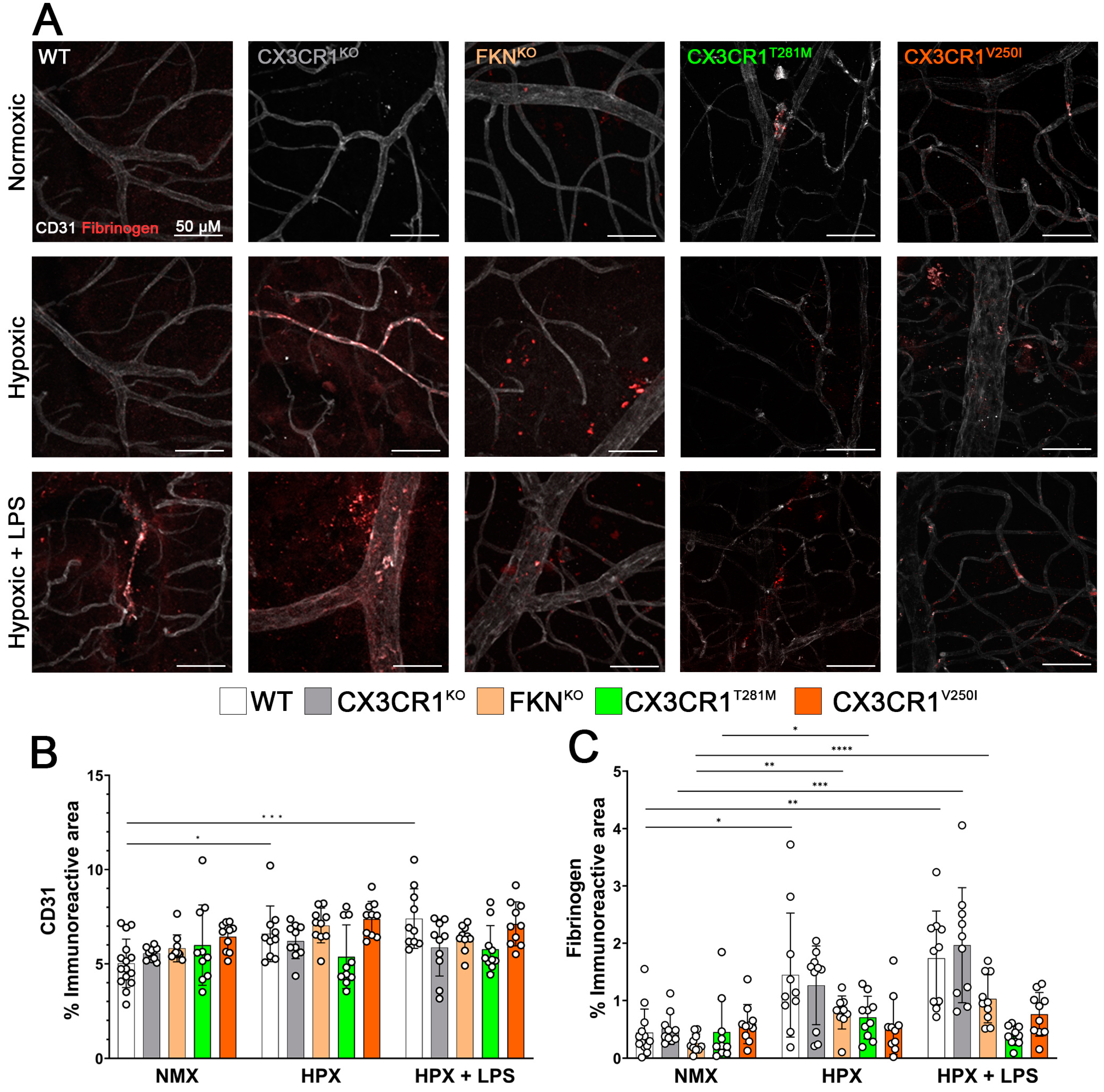
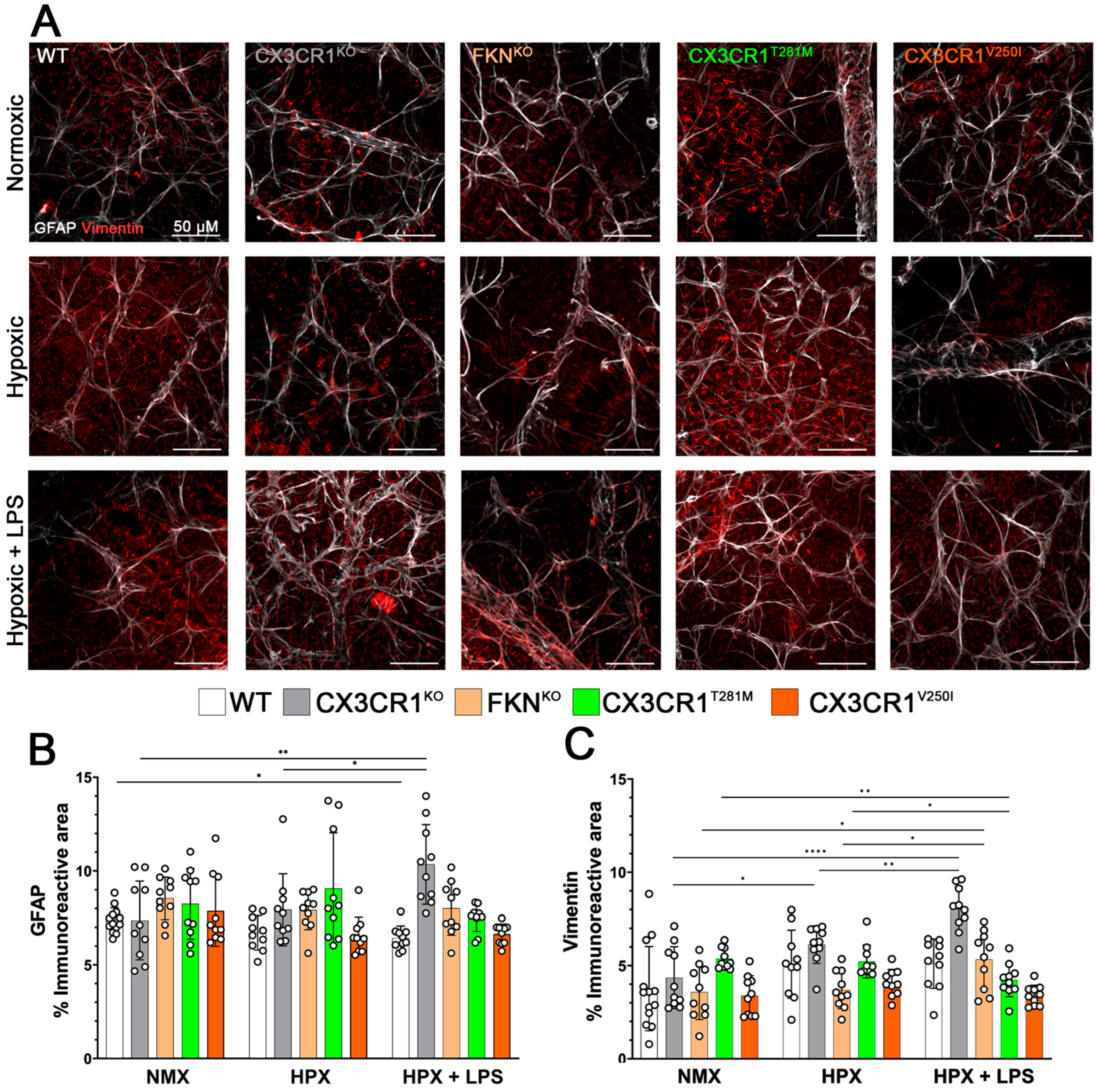
2.2. Hypoxia Induces Reactive Microgliosis, but Astrocyte and Müller Glial Responses to Hypoxia and Systemic Inflammation Are Dependent on Specific Defects in FKN-CX3CR1 Signaling
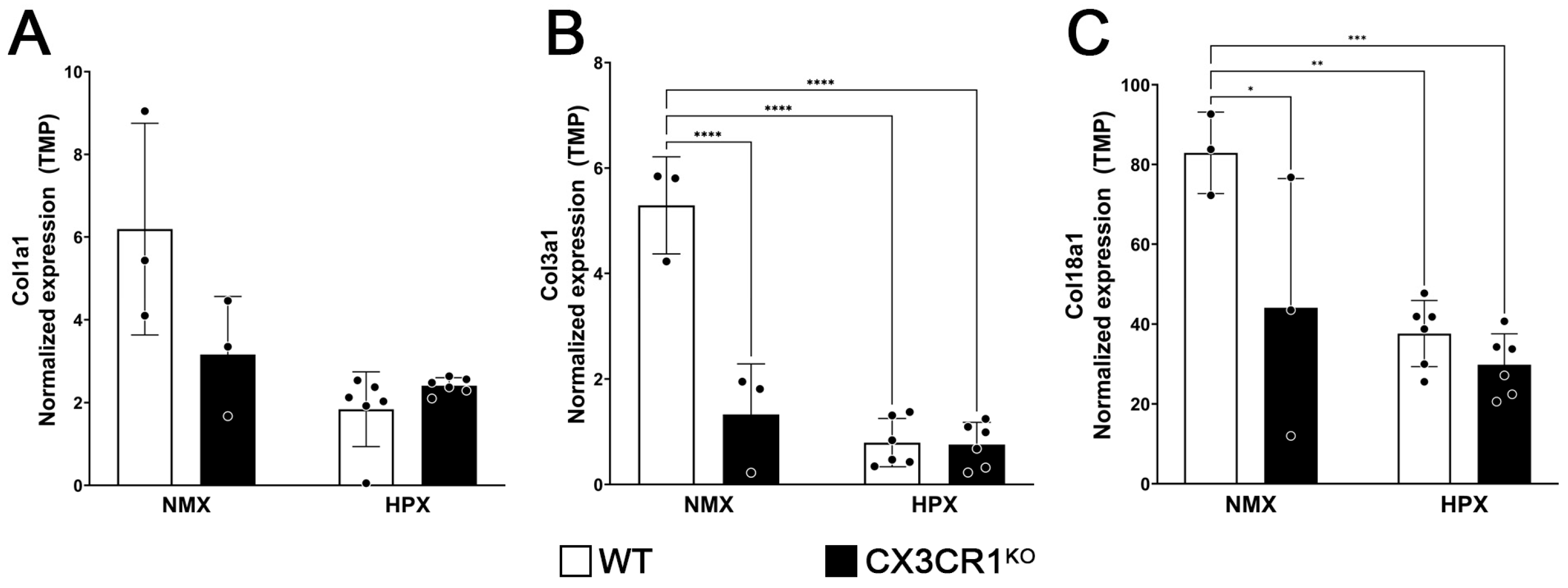
2.3. CX3CR1 Regulation Affects the Apoptosis, Adaptive Immune, and Extracellular Matrix Organization Responses to Hypoxia-Induced Retinopathy
2.4. Hypoxia Induces a Reduction in Endostatin Immunoreactivity
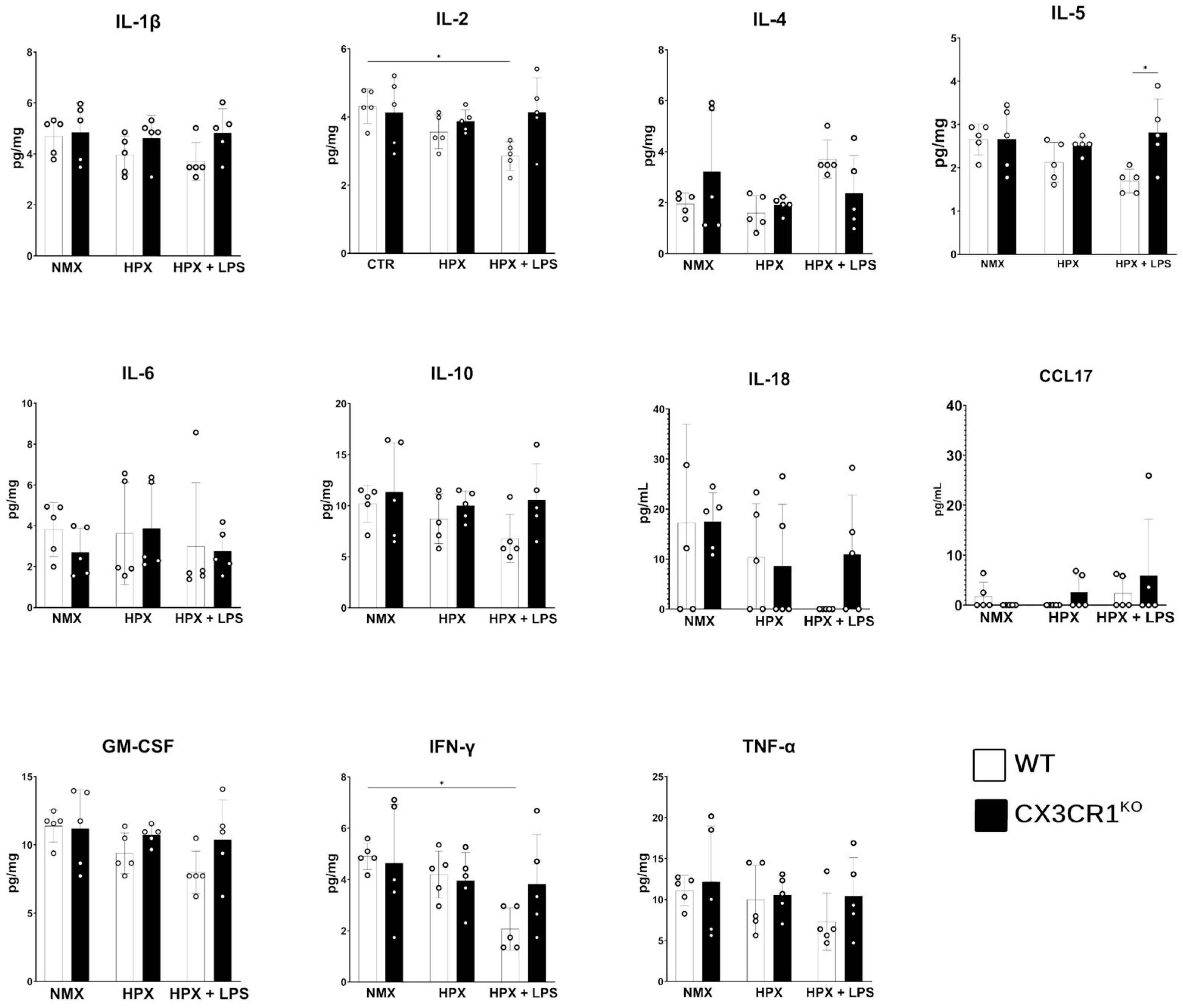
2.5. Hypoxia Ameliorates the LPS-Induced Inflammatory Cytokine Response
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Two-Hit Model of Hypoxia-Induced Retinopathy and Systemic Inflammation
4.3. Streptozotocin-Induced Hyperglycemia
4.4. Tissue Collection
4.5. Retinal RNA Collection and Processing
4.6. Immunofluorescent Staining
4.7. Tissue Imaging and Quantification
4.8. Quantification of Endostatin in Human Retinal Tissue
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez, D.; Church, K.A.; Pietramale, A.N.; Cardona, S.M.; Vanegas, D.; Rorex, C.; Leary, M.C.; Muzzio, I.A.; Nash, K.R.; Cardona, A.E. Fractalkine isoforms differentially regulate microglia-mediated inflammation and enhance visual function in the diabetic retina. J. Neuroinflammation 2024, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.A.; Jobling, A.I.; Dixon, M.A.; Bui, B.V.; Vessey, K.A.; Phipps, J.A.; Greferath, U.; Venables, G.; Wong, V.H.Y.; Wong, C.H.Y.; et al. Fractalkine-induced microglial vasoregulation occurs within the retina and is altered early in diabetic retinopathy. Proc. Natl. Acad. Sci. USA 2021, 118, e2112561118. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.; Church, K.A.; Smith, C.T.; Vanegas, D.; Cardona, S.M.; Muzzio, I.A.; Nash, K.R.; Cardona, A.E. Therapeutic Delivery of Soluble Fractalkine Ameliorates Vascular Dysfunction in the Diabetic Retina. Int. J. Mol. Sci. 2024, 25, 1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Mattagajasingh, S.; Su, S.; Chen, G.; Cai, Z.; Fox-Talbot, K.; Irani, K.; Becker, L.C. Fractalkine Upregulates Intercellular Adhesion Molecule-1 in Endothelial Cells Through CX3CR1 and the Jak–Stat5 Pathway. Circ. Res. 2007, 101, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Cardona, S.M.; Mendiola, A.S.; Yang, Y.-C.; Adkins, S.L.; Torres, V.; Cardona, A.E. Disruption of Fractalkine Signaling Leads to Microglial Activation and Neuronal Damage in the Diabetic Retina. ASN Neuro. 2015, 7, 1759091415608204. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.S.; Garza, R.; Cardona, S.M.; Mythen, S.A.; Lira, S.A.; Akassoglou, K.; Cardona, A.E. Fractalkine Signaling Attenuates Perivascular Clustering of Microglia and Fibrinogen Leakage during Systemic Inflammation in Mouse Models of Diabetic Retinopathy. Front. Cell. Neurosci. 2017, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef]
- de Hoz, R.; Rojas, B.; Ramírez, A.I.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Retinal Macroglial Responses in Health and Disease. BioMed Res. Int. 2016, 2016, 2954721. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef] [PubMed]
- Sueishi, K.; Hata, Y.; Murata, T.; Nakagawa, K.; Ishibashi, T.; Inomata, H. Endothelial and glial cell interaction in diabetic retinopathy via the function of vascular endothelial growth factor (VEGF). Pol. J. Pharmacol. 1996, 48, 307–316. [Google Scholar]
- Fehér, J.; Taurone, S.; Spoletini, M.; Biró, Z.; Varsányi, B.; Scuderi, G.; Orlando, M.P.; Turchetta, R.; Micera, A.; Artico, M. Ultrastructure of neurovascular changes in human diabetic retinopathy. Int. J. Immunopathol. Pharmacol. 2017, 31, 0394632017748841. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-Q.; Ma, C.-G.; Ding, Z.-B.; Song, L.-J.; Wang, Q.; Kumar, G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Sareddy, G.R.; Wang, J.; Zhang, Q.; Tang, F.-L.; Pratap, U.P.; Tekmal, R.R.; Vadlamudi, R.K.; Brann, D.W. Neuron-Derived Estrogen Is Critical for Astrocyte Activation and Neuroprotection of the Ischemic Brain. J. Neurosci. 2020, 40, 7355–7374. [Google Scholar] [CrossRef]
- Neal, M.; Luo, J.; Harischandra, D.S.; Gordon, R.; Sarkar, S.; Jin, H.; Anantharam, V.; Désaubry, L.; Kanthasamy, A.; Kanthasamy, A. Prokineticin-2 promotes chemotaxis and alternative A2 reactivity of astrocytes. Glia 2018, 66, 2137–2157. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Krohne, T.U.; Aguilar, E.; Kurihara, T.; Takeda, N.; Dorrell, M.I.; Simon, M.C.; Haase, V.H.; Friedlander, M.; Johnson, R.S. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia 2010, 58, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Shortt, A.J.; Howell, K.; O’Brien, C.; McLoughlin, P. Chronic systemic hypoxia causes intra-retinal angiogenesis. J. Anat. 2004, 205, 349–356. [Google Scholar] [CrossRef]
- Touhami, S.; Béguier, F.; Yang, T.; Augustin, S.; Roubeix, C.; Blond, F.; Conart, J.B.; Sahel, J.A.; Bodaghi, B.; Delarasse, C.; et al. Hypoxia Inhibits Subretinal Inflammation Resolution Thrombospondin-1 Dependently. Int. J. Mol. Sci. 2022, 23, 681. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Halder, S.K.; Milner, R. Mild hypoxia triggers transient blood-brain barrier disruption: A fundamental protective role for microglia. Acta Neuropathol. Commun. 2020, 8, 175. [Google Scholar] [CrossRef]
- Li, L.; Welser, J.V.; Dore-Duffy, P.; del Zoppo, G.J.; Lamanna, J.C.; Milner, R. In the hypoxic central nervous system, endothelial cell proliferation is followed by astrocyte activation, proliferation, and increased expression of the alpha 6 beta 4 integrin and dystroglycan. Glia 2010, 58, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Onnis, B.; Fer, N.; Rapisarda, A.; Perez, V.S.; Melillo, G. Autocrine production of IL-11 mediates tumorigenicity in hypoxic cancer cells. J. Clin. Investig. 2013, 123, 1615–1629. [Google Scholar] [CrossRef]
- Lin, P.K.; Davis, G.E. Extracellular Matrix Remodeling in Vascular Disease: Defining Its Regulators and Pathological Influence. Arter. Thromb. Vasc. Biol. 2023, 43, 1599–1616. [Google Scholar] [CrossRef] [PubMed]
- Anakha, J.; Dobariya, P.; Sharma, S.S.; Pande, A.H. Recombinant human endostatin as a potential anti-angiogenic agent: Therapeutic perspective and current status. Med. Oncol. 2023, 41, 24. [Google Scholar] [CrossRef] [PubMed]
- Tejerina-Miranda, S.; Pedrero, M.; Blázquez-García, M.; Serafín, V.; Montero-Calle, A.; Garranzo-Asensio, M.; Reviejo, A.J.; Pingarrón, J.M.; Barderas, R.; Campuzano, S. Angiogenesis inhibitor or aggressiveness marker? The function of endostatin in cancer through electrochemical biosensing. Bioelectrochemistry 2023, 155, 108571. [Google Scholar] [CrossRef] [PubMed]
- Bonet, A.; Valença, A.; Mendes-Jorge, L.; Casellas, A.; Rodríguez-Baeza, A.; Nacher, V.; Ramos, D.; Pampalona, J.; Simó, R.; Ruberte, J. Decreased endostatin in db/db retinas is associated with optic disc intravitreal vascularization. Exp. Eye Res. 2021, 212, 108801. [Google Scholar] [CrossRef] [PubMed]
- Ebner, L.J.A.; Samardzija, M.; Storti, F.; Todorova, V.; Karademir, D.; Behr, J.; Simpson, F.; Thiersch, M.; Grimm, C. Transcriptomic analysis of the mouse retina after acute and chronic normobaric and hypobaric hypoxia. Sci. Rep. 2021, 11, 16666. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Akira, S.; Miyake, K.; Ueda, H. Retinal cell type-specific prevention of ischemia-induced damages by LPS-TLR4 signaling through microglia. J. Neurochem. 2013, 126, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak-Wiercioch, A.; Sałat, K. Lipopolysaccharide-Induced Model of Neuroinflammation: Mechanisms of Action, Research Application and Future Directions for Its Use. Molecules 2022, 27, 5481. [Google Scholar] [CrossRef] [PubMed]
- Terraneo, L.; Paroni, R.; Bianciardi, P.; Giallongo, T.; Carelli, S.; Gorio, A.; Samaja, M. Brain adaptation to hypoxia and hyperoxia in mice. Redox Biol. 2017, 11, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Depner, M.; Kormann, M.S.; Klopp, N.; Illig, T.; Vogelberg, C.; Weiland, S.K.; von Mutius, E.; Combadière, C.; Kabesch, M. CX3CR1 Polymorphisms Are Associated with Atopy but Not Asthma in German Children. Int. Arch. Allergy Immunol. 2007, 144, 91–94. [Google Scholar] [CrossRef]
- Li, D.; Peng, X.; Sun, H. Association of CX3CR1 (V249I and T280M) polymorphisms with age-related macular degeneration: A meta-analysis. Can. J. Ophthalmol. 2015, 50, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Lupiañez, C.B.; Martínez-Bueno, M.; Sánchez-Maldonado, J.M.; Badiola, J.; Cunha, C.; Springer, J.; Lackner, M.; Segura-Catena, J.; Canet, L.M.; Alcazar-Fuoli, L.; et al. Polymorphisms within the ARNT2 and CX3CR1 Genes Are Associated with the Risk of Developing Invasive Aspergillosis. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Dang, G.; Yang, S.; Duan, L.; Zhang, Y. CX3CR1 polymorphisms and the risk of age-related macular degeneration. Int. J. Clin. Exp. Pathol. 2015, 8, 9592–9596. [Google Scholar] [PubMed]
- Woźny, Ł.; Żywiec, J.; Gosek, K.; Kuźniewicz, R.; Górczyńska-Kosiorz, S.; Trautsolt, W.; Śnit, M.; Grzeszczak, W. Association of CX3CR1 Gene Polymorphisms with Fractalkine, Fractalkine Receptor, and C-Reactive Protein Levels in Patients with Kidney Failure. Int. J. Environ. Res. Public Health 2021, 18, 2202. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Jin, P.; Sherchan, P.; Liu, S.; Cui, Y.; Huang, L.; Zhang, J.H.; Gong, Y.; Tang, J. Recombinant CCL17-dependent CCR4 activation alleviates neuroinflammation and neuronal apoptosis through the PI3K/AKT/Foxo1 signaling pathway after ICH in mice. J. Neuroinflammation 2021, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.-C.; Xu, W.-D.; Liu, X.-Y.; Liu, X.-Y.; Huang, A.-F.; Su, L.-C. Biology of IL-36 Signaling and Its Role in Systemic Inflammatory Diseases. Front. Immunol. 2019, 10, 2532. [Google Scholar] [CrossRef] [PubMed]
- Rorex, C.; Cardona, S.M.; Church, K.A.; Rodriguez, D.; Vanegas, D.; Saldivar, R.; Faz, B.; Cardona, A.E. Astrogliosis in the GFAP-CreERT2:Rosa26iDTR Mouse Model Does Not Exacerbate Retinal Microglia Activation or Müller Cell Gliosis under Hypoxic Conditions. Biomolecules 2024, 14, 567. [Google Scholar] [CrossRef]
- Muñoz, E.; Zubiaga, A.M.; Huber, B.T. Interleukin-1 induces c-fos and c-jun gene expression in T helper type II cells through different signal transmission pathways. Eur. J. Immunol. 1992, 22, 2101–2106. [Google Scholar] [CrossRef]
- Skelly, D.T.; Hennessy, E.; Dansereau, M.-A.; Cunningham, C. A Systematic Analysis of the Peripheral and CNS Effects of Systemic LPS, IL-1Β, TNF-α and IL-6 Challenges in C57BL/6 Mice. PLoS ONE 2013, 8, e69123. [Google Scholar] [CrossRef]
- Varma, T.K.; Lin, C.Y.; Toliver-Kinsky, T.E.; Sherwood, E.R. Endotoxin-Induced Gamma Interferon Production: Contributing Cell Types and Key Regulatory Factors. Clin. Vaccine Immunol. 2002, 9, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, X.; Zhao, T.; Qiao, M.; Zhao, X.; Zhao, M.; Xu, L.; Zhao, Y.; Wu, L.; Wu, K.; et al. Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav. Immun. 2017, 64, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Jaskiewicz, M.; Moszynska, A.; Serocki, M.; Kroliczewski, J.; Bartoszewska, S.; Collawn, J.F.; Bartoszewski, R. Hypoxia-inducible factor (HIF)-3α2 serves as an endothelial cell fate executor during chronic hypoxia. EXCLI J. 2022, 21, 454–469. [Google Scholar] [CrossRef]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and Characterization of Hypoxia-Inducible Factor (HIF)-3α in Human Kidney: Suppression of HIF-Mediated Gene Expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-L.; Wu, C.; Xiong, Z.-F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.-W.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-α isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zou, H. The role of lipopolysaccharides in diabetic retinopathy. BMC Ophthalmol. 2022, 22, 86. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.M.G.; Costa, J.A.; Alfenas, R.C.G. Metabolic endotoxemia and diabetes mellitus: A systematic review. Metabolism 2017, 68, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zeng, T.; Roux, M.; Lazar, D.; Chen, L.; Tudzarova, S. The Role of HIF1α-PFKFB3 Pathway in Diabetic Retinopathy. J. Clin. Endocrinol. Metab. 2021, 106, 2505–2519. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, J.; Babapoor-Farrokhran, S.; Applewhite, B.; Deshpande, M.; Megarity, H.; Flores-Bellver, M.; Aparicio-Domingo, S.; Ma, T.; Rui, Y.; et al. PAI-1 is a vascular cell-specific HIF-2-dependent angiogenic factor that promotes retinal neovascularization in diabetic patients. Sci. Adv. 2022, 8, eabm1896. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, T.; Liu, J.; Wei, S.; Yang, Y.; Liu, J.; Zhang, B.; Li, W. Phloretin alleviates doxorubicin-induced cardiotoxicity through regulating Hif3a transcription via targeting transcription factor Fos. Phytomedicine 2023, 120, 155046. [Google Scholar] [CrossRef] [PubMed]
- Church, K.A.; Rodriguez, D.; Mendiola, A.S.; Vanegas, D.; Gutierrez, I.L.; Tamayo, I.; Amadu, A.; Velazquez, P.; Cardona, S.M.; Gyoneva, S.; et al. Pharmacological depletion of microglia alleviates neuronal and vascular damage in the diabetic CX3CR1-WT retina but not in CX3CR1-KO or hCX3CR1I249/M280-expressing retina. Front. Immunol. 2023, 14, 1130735. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Fujita, H.; Tanaka, J.; Toku, K.; Tateishi, N.; Suzuki, Y.; Matsuda, S.; Sakanaka, M.; Maeda, N. Effects of GM-CSF and ordinary supplements on the ramification of microglia in culture: A morphometrical study. Glia 1996, 18, 269–281. [Google Scholar] [CrossRef]
- Green, T.R.F.; Murphy, S.M.; Rowe, R.K. Comparisons of quantitative approaches for assessing microglial morphology reveal inconsistencies, ecological fallacy, and a need for standardization. Sci. Rep. 2022, 12, 18196. [Google Scholar] [CrossRef]
- Sarker, B.; Cardona, S.M.; Cardona, S.M.; Church, K.A.; Church, K.A.; Vanegas, D.; Vanegas, D.; Velazquez, P.; Velazquez, P. Defibrinogenation Ameliorates Retinal Microgliosis and Inflammation in A CX3CR1-Independent Manner. ASN Neuro. 2022, 14, 17590914221131446. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | ||||
| Target | Host 1 | RRID | Dilution | Cellular target |
| Anti-ionized calcium-binding adaptor molecule-1 (Iba1) | Rb | AB_839504 | 1:4000 | Microglia |
| Anti-neuronal nuclei (NeuN) | Ms | AB_2298772 | 1:4000 | Retinal ganglion cells |
| Anti-glial fibrillary acidic protein (GFAP) | Rt | AB_2532994 | 1:4000 | Astrocytes |
| Anti-platelet endothelial cell adhesion molecule (PECAM-1/CD31) | Rt | AB_393571 | 1:500 | Endothelial cells |
| Anti-fibrinogen | Rb | AB_578481 | 1:2000 | N/A |
| Anti-vimentin | Ck | AB_2216267 | 1:1000 | Müller glia |
| Anti-beta tubulin III (TUJ1) | Ms | AB_10063408 | 1:1000 | Retinal ganglion cell axons |
| Anti-endostatin | Rb | AB_2245014 | 1:200 | Endostatin |
| Secondary Antibodies | ||||
| Target | Host | RRID | Dilution | |
| Anti-rabbit 488 | Dk | AB_2313584 | 1:1000 | |
| Anti-rabbit Cy3 | Gt | AB_2338006 | 1:1000 | |
| Anti-rat Cy5 | Dk | AB_2340694 | 1:1000 | |
| Anti-rat Cy3 | Gt | AB_2338394 | 1:1000 | |
| Anti-mouse Cy3 | Gt | AB_2338709 | 1:1000 | |
| Anti-mouse Cy3 | Gt | AB_2338713 | 1:1000 | |
| Anti-chicken Biotin | Dk | AB_2340363 | 1:1000 | |
| SA Cy3 | N/A | AB_2337244 | 1:1000 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rorex, C.; Cardona, S.M.; Church, K.A.; Rodriguez, D.; Vanegas, D.; Saldivar, R.A.; El-Sheikh, A.; Wang, Y.; Gyoneva, S.; Cotleur, A.C.; et al. CX3CR1–Fractalkine Dysregulation Affects Retinal GFAP Expression, Inflammatory Gene Induction, and LPS Response in a Mouse Model of Hypoxic Retinopathy. Int. J. Mol. Sci. 2025, 26, 1131. https://doi.org/10.3390/ijms26031131
Rorex C, Cardona SM, Church KA, Rodriguez D, Vanegas D, Saldivar RA, El-Sheikh A, Wang Y, Gyoneva S, Cotleur AC, et al. CX3CR1–Fractalkine Dysregulation Affects Retinal GFAP Expression, Inflammatory Gene Induction, and LPS Response in a Mouse Model of Hypoxic Retinopathy. International Journal of Molecular Sciences. 2025; 26(3):1131. https://doi.org/10.3390/ijms26031131
Chicago/Turabian StyleRorex, Colin, Sandra M. Cardona, Kaira A. Church, Derek Rodriguez, Difernando Vanegas, Reina A. Saldivar, Amira El-Sheikh, Yufeng Wang, Stefka Gyoneva, Anne C. Cotleur, and et al. 2025. "CX3CR1–Fractalkine Dysregulation Affects Retinal GFAP Expression, Inflammatory Gene Induction, and LPS Response in a Mouse Model of Hypoxic Retinopathy" International Journal of Molecular Sciences 26, no. 3: 1131. https://doi.org/10.3390/ijms26031131
APA StyleRorex, C., Cardona, S. M., Church, K. A., Rodriguez, D., Vanegas, D., Saldivar, R. A., El-Sheikh, A., Wang, Y., Gyoneva, S., Cotleur, A. C., & Cardona, A. E. (2025). CX3CR1–Fractalkine Dysregulation Affects Retinal GFAP Expression, Inflammatory Gene Induction, and LPS Response in a Mouse Model of Hypoxic Retinopathy. International Journal of Molecular Sciences, 26(3), 1131. https://doi.org/10.3390/ijms26031131