

Unveiling Secondary Mutations in Blended Phenotypes: Dual ERCC4 and OTOA Pathogenic Variants Through WES Analysis
 ,
,  ,
,  and
and
Abstract
1. Introduction
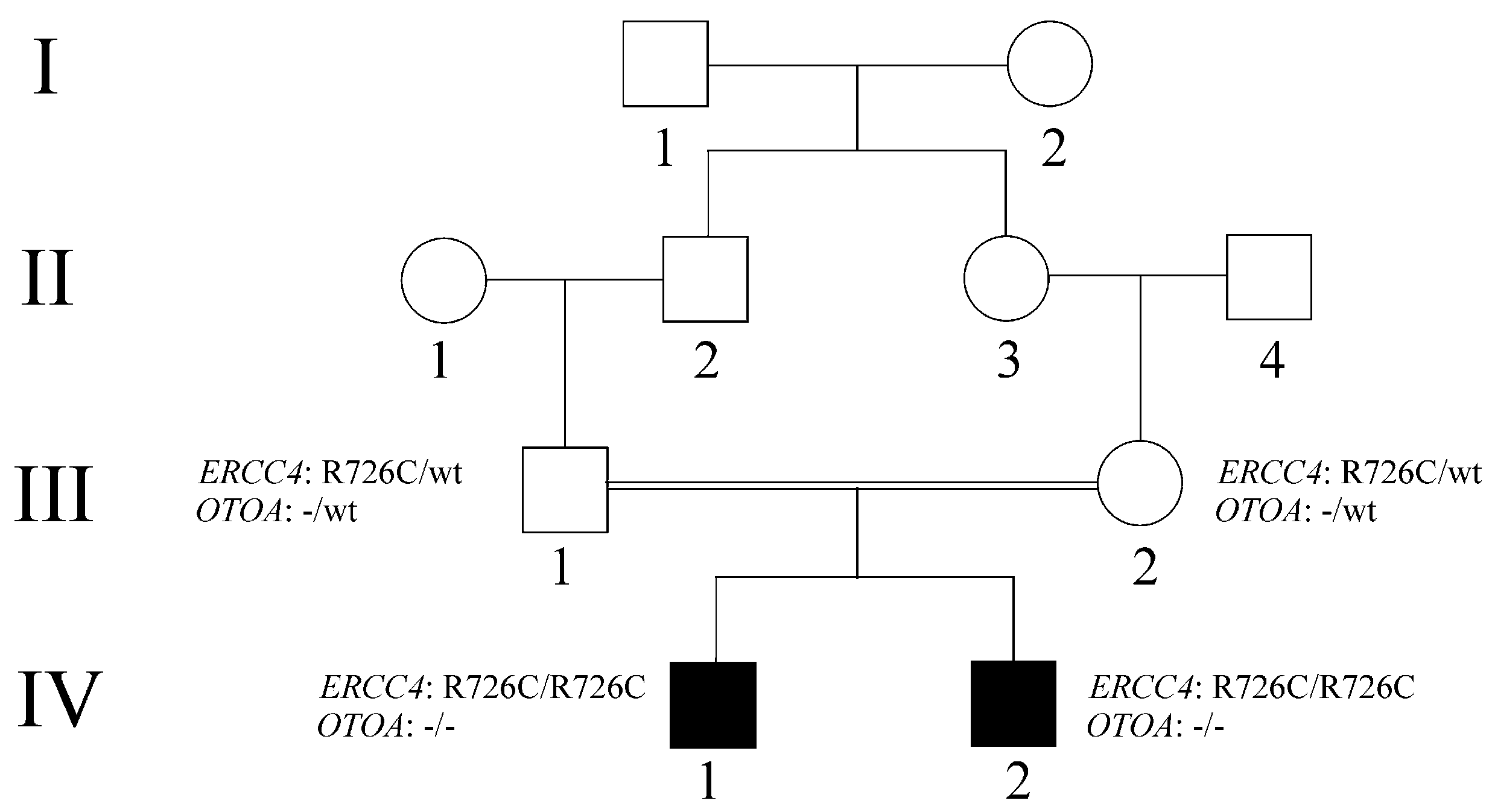
2. Detailed Case Description
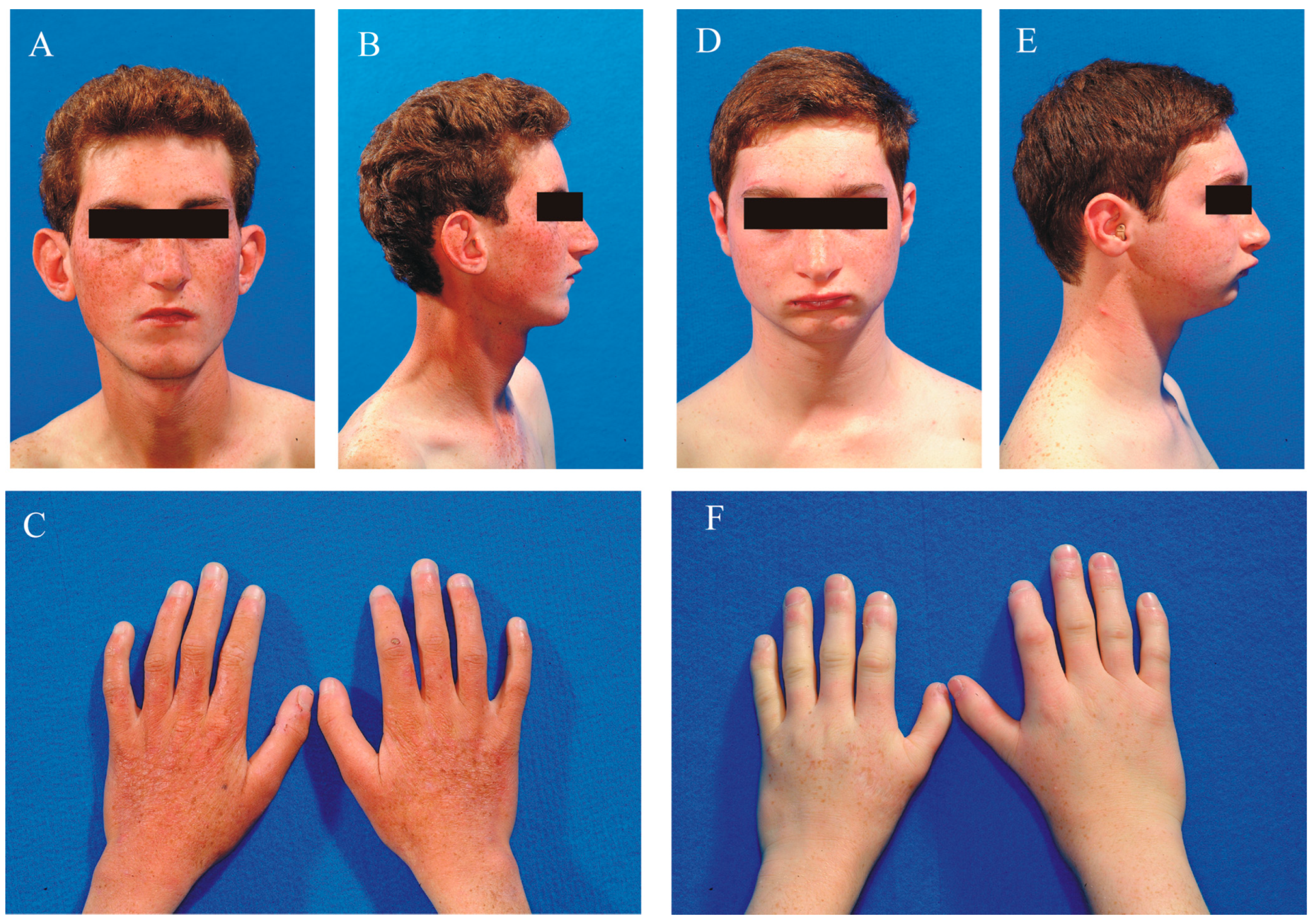
2.1. Clinical Data
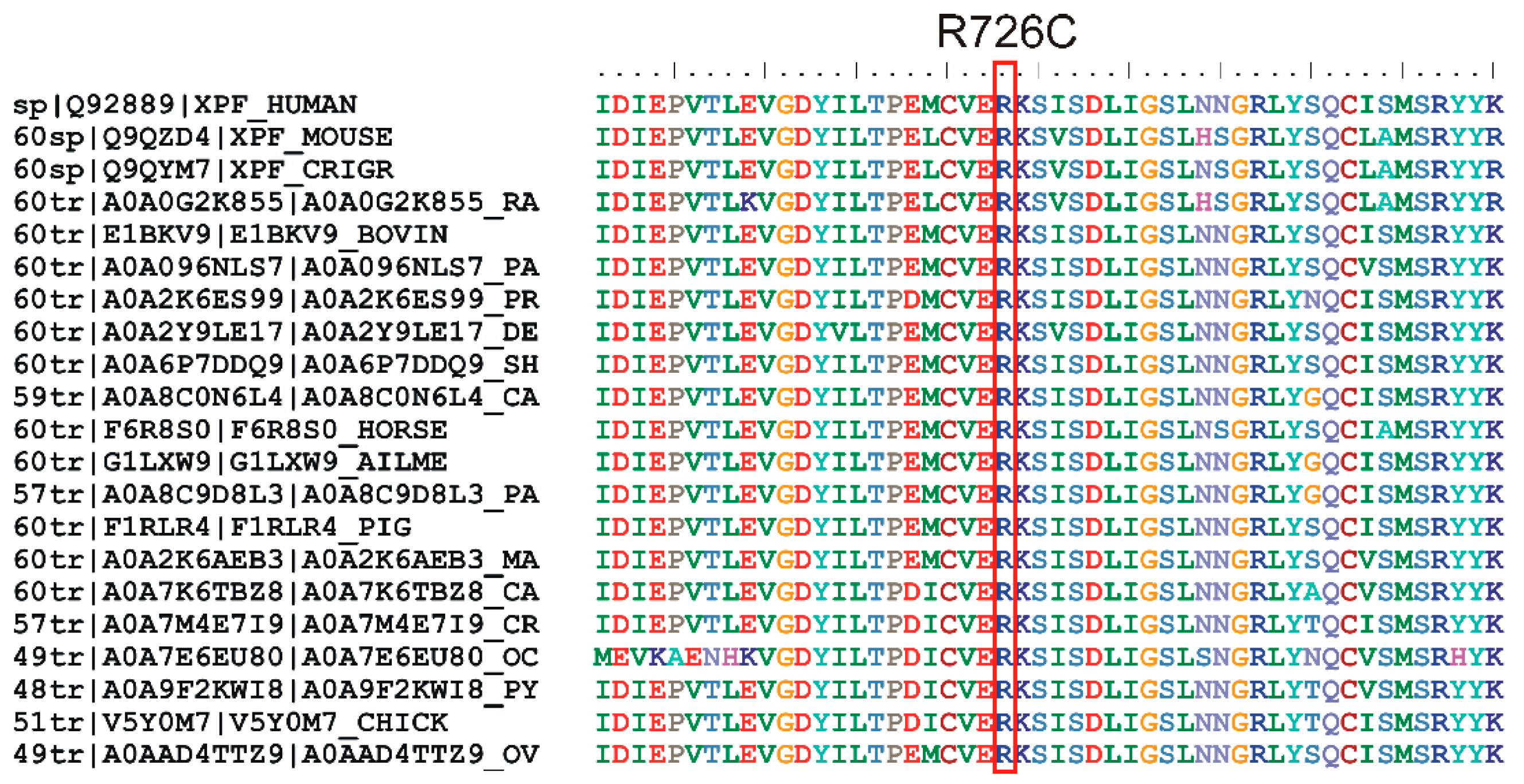
2.2. WES Analysis
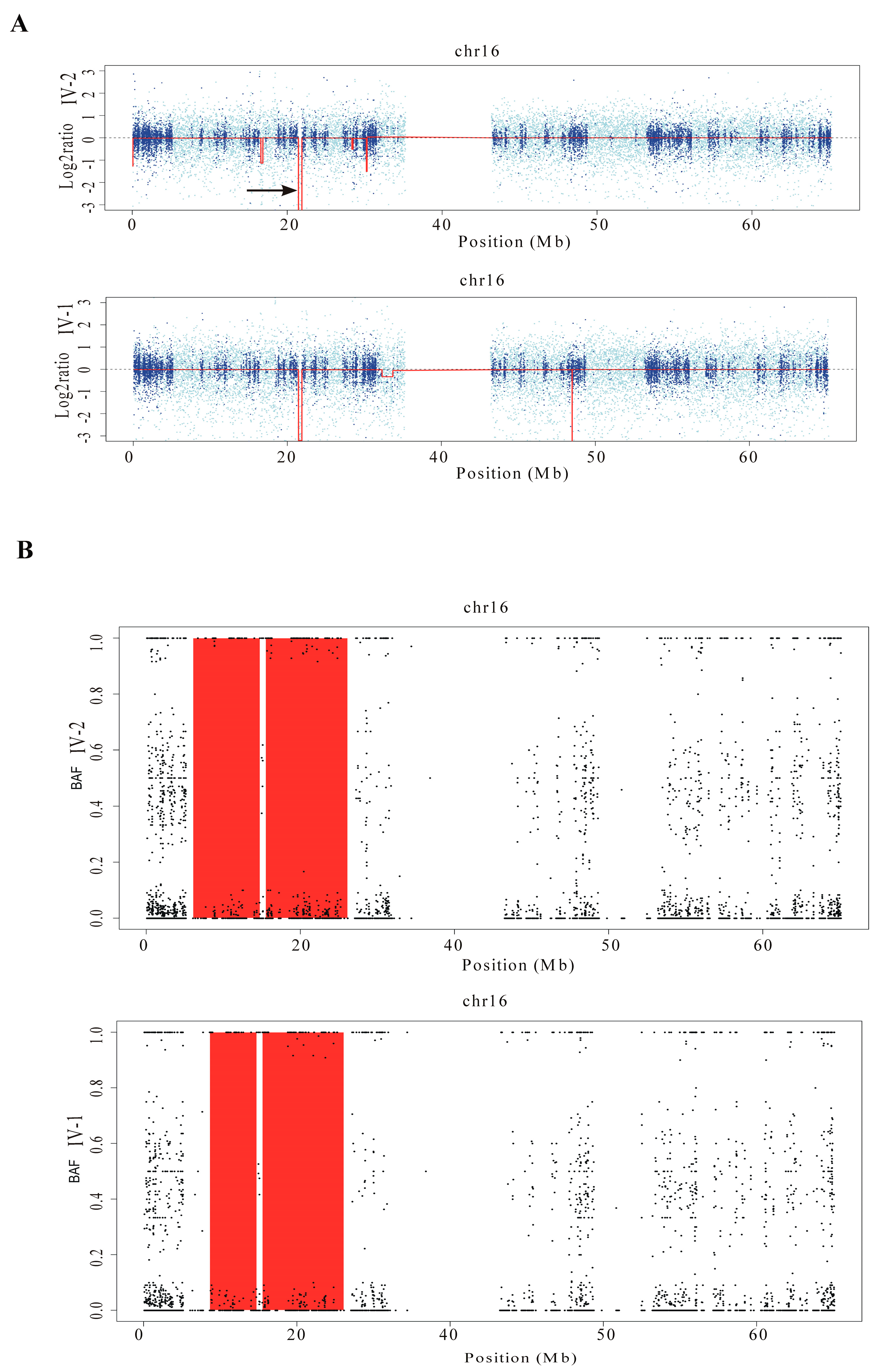
2.3. Excavator2 Analysis
2.4. H3M2 Analysis
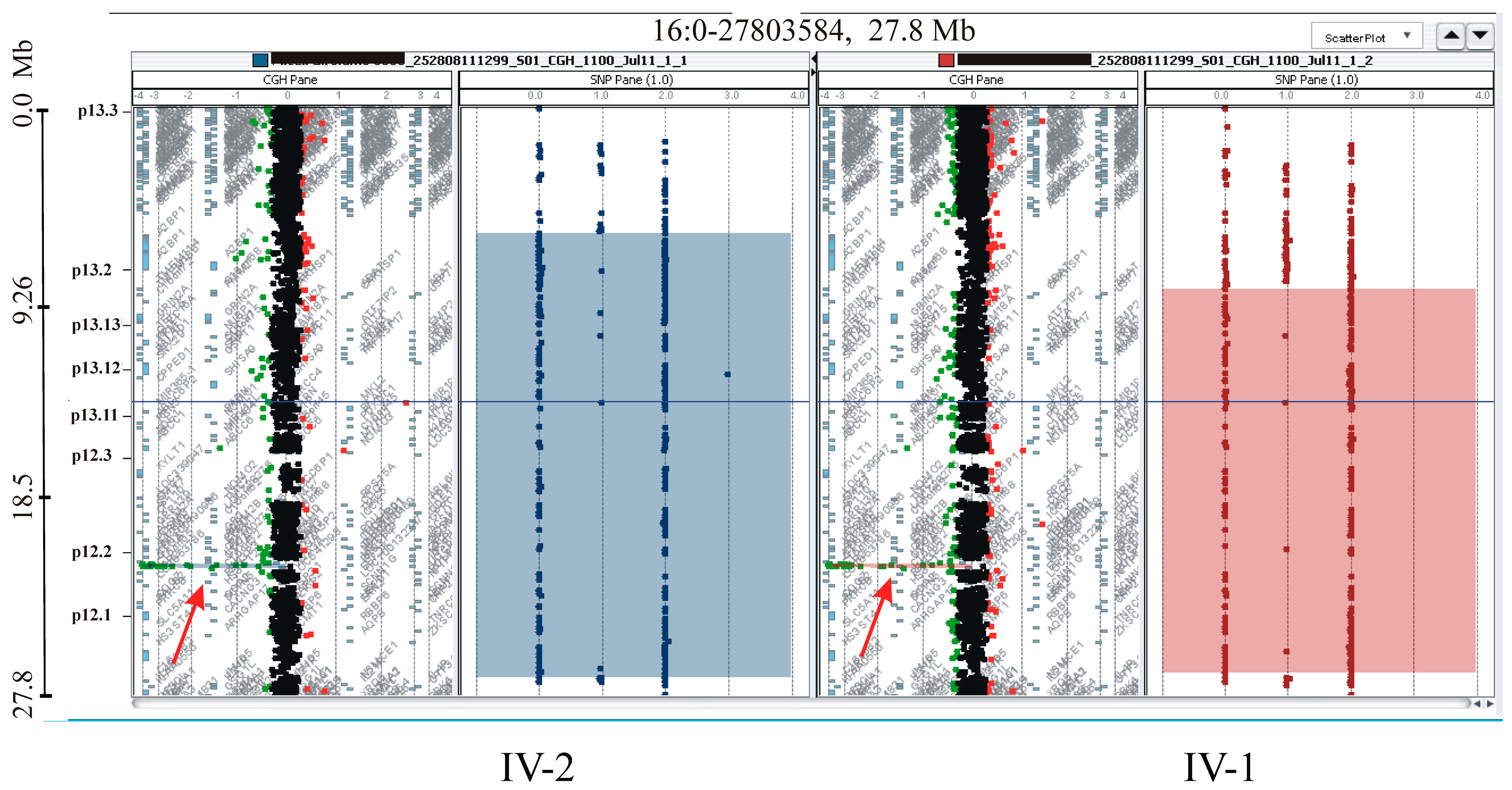
2.5. CGH/SNP ARRAY Analysis
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Rosina, E.; Pezzani, L.; Pezzoli, L.; Marchetti, D.; Bellini, M.; Pilotta, A.; Calabrese, O.; Nicastro, E.; Cirillo, F.; Cereda, A.; et al. Atypical, Composite, or Blended Phenotypes: How Different Molecular Mechanisms Could Associate in Double-Diagnosed Patients. Genes 2022, 13, 1275. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Weber, J.L. Long Homozygous Chromosomal Segments in Reference Families from the Centre d’Etude Du Polymorphisme Humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of Homozygosity: Windows into Population History and Trait Architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdottir, H.; Turner, D.; Mesirov, J.P. Igv.Js: An Embeddable JavaScript Implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 2023, 39, btac830. [Google Scholar] [CrossRef]
- Marín, M.; Ramírez, M.J.; Carmona, M.A.; Jia, N.; Ogi, T.; Bogliolo, M.; Surrallés, J. Functional Comparison of XPF Missense Mutations Associated to Multiple DNA Repair Disorders. Genes 2019, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- D’Aurizio, R.; Pippucci, T.; Tattini, L.; Giusti, B.; Pellegrini, M.; Magi, A. Enhanced Copy Number Variants Detection from Whole-Exome Sequencing Data Using EXCAVATOR2. Nucleic Acids Res. 2016, 44, e154. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Tattini, L.; Palombo, F.; Benelli, M.; Gialluisi, A.; Giusti, B.; Abbate, R.; Seri, M.; Gensini, G.F.; Romeo, G.; et al. H3M2: Detection of Runs of Homozygosity from Whole-Exome Sequencing Data. Bioinformatics 2014, 30, 2852–2859. [Google Scholar] [CrossRef]
- Sijbers, A.M.; de Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; de Jong, M.C.; et al. Xeroderma Pigmentosum Group F Caused by a Defect in a Structure-Specific DNA Repair Endonuclease. Cell 1996, 86, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.D.; Barrett, S.F.; Robbins, J.H. Xeroderma Pigmentosum Neurological Abnormalities Correlate with Colony-Forming Ability after Ultraviolet Radiation. Proc. Natl. Acad. Sci. USA 1978, 75, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Gregg, S.Q.; Robinson, A.R.; Niedernhofer, L.J. Physiological Consequences of Defects in ERCC1-XPF DNA Repair Endonuclease. DNA Repair. 2011, 10, 781–791. [Google Scholar] [CrossRef]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of Nuclease ERCC1-XPF Results in Diverse Clinical Manifestations and Causes Cockayne Syndrome, Xeroderma Pigmentosum, and Fanconi Anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar] [CrossRef]
- Bogliolo, M.; Schuster, B.; Stoepker, C.; Derkunt, B.; Su, Y.; Raams, A.; Trujillo, J.P.; Minguillón, J.; Ramírez, M.J.; Pujol, R.; et al. Mutations in ERCC4, Encoding the DNA-Repair Endonuclease XPF, Cause Fanconi Anemia. Am. J. Hum. Genet. 2013, 92, 800–806. [Google Scholar] [CrossRef]
- Rapin, I.; Weidenheim, K.; Lindenbaum, Y.; Rosenbaum, P.; Merchant, S.N.; Krishna, S.; Dickson, D.W. Cockayne Syndrome in Adults: Review with Clinical and Pathologic Study of a New Case. J. Child Neurol. 2006, 21, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Zwaenepoel, I.; Mustapha, M.; Leibovici, M.; Verpy, E.; Goodyear, R.; Liu, X.Z.; Nouaille, S.; Nance, W.E.; Kanaan, M.; Avraham, K.B.; et al. Otoancorin, an Inner Ear Protein Restricted to the Interface between the Apical Surface of Sensory Epithelia and Their Overlying Acellular Gels, Is Defective in Autosomal Recessive Deafness DFNB22. Proc. Natl. Acad. Sci. USA 2002, 99, 6240–6245. [Google Scholar] [CrossRef]
- Sugiyama, K.; Moteki, H.; Kitajiri, S.-I.; Kitano, T.; Nishio, S.-Y.; Yamaguchi, T.; Wakui, K.; Abe, S.; Ozaki, A.; Motegi, R.; et al. Mid-Frequency Hearing Loss Is Characteristic Clinical Feature of OTOA-Associated Hearing Loss. Genes 2019, 10, 715. [Google Scholar] [CrossRef] [PubMed]
- Blair, D.R.; Lyttle, C.S.; Mortensen, J.M.; Bearden, C.F.; Jensen, A.B.; Khiabanian, H.; Melamed, R.; Rabadan, R.; Bernstam, E.V.; Brunak, S.; et al. A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell 2013, 155, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Blair, D.R.; Hoffmann, T.J.; Shieh, J.T. Common genetic variation associated with Mendelian disease severity revealed through cryptic phenotype analysis. Nat. Commun. 2022, 13, 3675. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, Y.; Chen, R.; Qi, Q.; Zhang, X.; Zhao, S.; Liu, C.; Wang, W.; Li, Y.; Sun, G.; et al. Clinical Efficiency of Simultaneous CNV-Seq and Whole-Exome Sequencing for Testing Fetal Structural Anomalies. J. Transl. Med. 2022, 20, 10. [Google Scholar] [CrossRef] [PubMed]
- Correia-Costa, G.R.; Dos Santos, A.M.; de Leeuw, N.; Rigatto, S.Z.P.; Belangero, V.M.S.; Steiner, C.E.; Gil-da-Silva-Lopes, V.L.; Vieira, T.P. Dual Molecular Diagnoses of Recessive Disorders in a Child from Consanguineous Parents: Case Report and Literature Review. Genes 2022, 13, 2377. [Google Scholar] [CrossRef] [PubMed]
- Matis, T.; Michaud, V.; Van-Gils, J.; Raclet, V.; Plaisant, C.; Fergelot, P.; Lasseaux, E.; Arveiler, B.; Trimouille, A. Triple Diagnosis of Wiedemann-Steiner, Waardenburg and DLG3-Related Intellectual Disability Association Found by WES: A Case Report. J. Gene Med. 2020, 22, e3197. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Koyano, S.; Miyatake, S.; Nakajima, S.; Nakazawa, Y.; Kunii, M.; Tomita-Katsumoto, A.; Oda, K.; Yamaguchi, Y.; Fukai, R.; et al. Cerebellar ataxia-dominant phenotype in patients with ERCC4 mutations. J. Hum. Genet. 2018, 63, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Geschwind, M.D.; DiGiovanna, J.J.; Groden, C.; Godfrey, R.; Yousefzadeh, M.J.; Wade, E.A.; Niedernhofer, L.J.; Malicdan, M.C.V.; Kraemer, K.H.; et al. Neurodegeneration as the presenting symptom in 2 adults with xeroderma pigmentosum complementation group F. Neurol. Genet. 2018, 4, e240. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Fassihi, H.; Sethi, M.; Fawcett, H.; Wing, J.; Chandler, N.; Mohammed, S.; Craythorne, E.; Morley, A.M.S.; Lim, R.; Turner, S.; et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc. Natl. Acad. Sci. USA 2016, 113, E1236–E1245. [Google Scholar] [CrossRef] [PubMed]
- Carré, G.; Marelli, C.; Anheim, M.; Geny, C.; Renaud, M.; Rezvani, H.; Koenig, M.; Guissart, C.; Tranchant, C. Xeroderma pigmentosum complementation group F: A rare cause of cerebellar ataxia with chorea. J. Neurol. Sci. 2017, 376, 198–201. [Google Scholar] [CrossRef]
- Marelli, C.; Guissart, C.; Hubsch, C.; Renaud, M.; Villemin, J.-P.; Larrieu, L.; Charles, P.; Ayrignac, X.; Sacconi, S.; Collignon, P.; et al. Mini-Exome Coupled to Read-Depth Based Copy Number Variation Analysis in Patients with Inherited Ataxias. Hum. Mutat. 2016, 37, 1340–1353. [Google Scholar] [CrossRef]
- Nabouli, I.; Chikhaoui, A.; Othman, H.; Elouej, S.; Jones, M.; Lagarde, A.; Ben Rekaya, M.; Messaoud, O.; Zghal, M.; Delague, V.; et al. Case Report: Identification of Novel Variants in ERCC4 and DDB2 Genes in Two Tunisian Patients with Atypical Xeroderma Pigmentosum Phenotype. Front. Genet. 2021, 12, 650639. [Google Scholar] [CrossRef] [PubMed]
- Popp, I.; Punekar, M.; Telford, N.; Stivaros, S.; Chandler, K.; Minnis, M.; Castleton, A.; Higham, C.; Hopewell, L.; Evans, D.G.; et al. Fanconi anemia with sun-sensitivity caused by a Xeroderma pigmentosum-associated missense mutation in XPF. BMC Med. Genet. 2018, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.Y.; Wang, H.; Lin, Z.; Xu, G.; Ma, Z.; Zhao, J.; Feng, C.; Duo, L.; Yin, J.; Yang, Y. Clinical and molecular epidemiological study of xeroderma pigmentosum in China: A case series of 19 patients. J. Dermatol. 2017, 44, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Nishigori, C.; Yagi, T.; Imamura, S.; Takebe, H. Characterization of molecular defects in xeroderma pigmentosum group F in relation to its clinically mild symptoms. Hum. Mol. Genet. 1998, 7, 969–974. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Score | Prediction |
| SIFT v6.2.1 | 0 | deleterious |
| Polyphen-2 | 1 | probably damaging |
| CADD 1.7 | 28 | moderate pathogenic |
| REVEL v1.3 | 0.774 | likely disease-causing |
| MetaLR v0.14.7 | 0.584 | damaging |
| Mutation Assessor v4 | 0.965 | high pathogenic |
| AlphaMissense v2.0 | 0.968 | deleterious |
| Protein stability | ΔΔG (kcal/mol) | |
| MUpro v1.0 | −0.883 | destabilizing |
| MAESTROweb v1.2.35 | 0.184 | neutral |
| DynaMut v2 | −0.97 | destabilizing |
| DDGun v2 | −1.3 | destabilizing |
| mCSM | −1.907 | destabilizing |
| CUPSAT | −1.68 | destabilizing |
| Clinical Features | Patient IV-1 | Patient IV-2 |
|---|---|---|
| Abnormality of the eye | no | no |
| Abnormality of the integument | ||
| Cutaneous photosensitivity | yes | yes |
| Freckling | yes | yes |
| Abnormality of the musculoskeletal system | ||
| Flexion contracture | yes | yes |
| Microcephaly | yes | yes |
| Scoliosis | yes | no |
| Abnormality of the nervous system | ||
| Cerebellar ataxia | no | no |
| Dementia | no | no |
| Intellectual disability | yes | yes |
| Morphological central nervous system abnormality | Agenesis of the corpus callosum, dilation of the lateral ventricles | no |
| Hyperreflexia | yes | yes |
| Hearing impairment | yes | yes |
| Growth abnormality | ||
| Decreased body weight | yes | yes |
| Short stature | yes | yes |
| Neoplasm | no | no |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Failla, P.; Saccuzzo, L.; Galesi, O.; Greco, D.; Barresi, V.; Amata, S.; Romano, C.; Fichera, M. Unveiling Secondary Mutations in Blended Phenotypes: Dual ERCC4 and OTOA Pathogenic Variants Through WES Analysis. Int. J. Mol. Sci. 2024, 25, 13471. https://doi.org/10.3390/ijms252413471
Failla P, Saccuzzo L, Galesi O, Greco D, Barresi V, Amata S, Romano C, Fichera M. Unveiling Secondary Mutations in Blended Phenotypes: Dual ERCC4 and OTOA Pathogenic Variants Through WES Analysis. International Journal of Molecular Sciences. 2024; 25(24):13471. https://doi.org/10.3390/ijms252413471
Chicago/Turabian StyleFailla, Pinella, Lucia Saccuzzo, Ornella Galesi, Donatella Greco, Vincenza Barresi, Silvestra Amata, Corrado Romano, and Marco Fichera. 2024. "Unveiling Secondary Mutations in Blended Phenotypes: Dual ERCC4 and OTOA Pathogenic Variants Through WES Analysis" International Journal of Molecular Sciences 25, no. 24: 13471. https://doi.org/10.3390/ijms252413471
APA StyleFailla, P., Saccuzzo, L., Galesi, O., Greco, D., Barresi, V., Amata, S., Romano, C., & Fichera, M. (2024). Unveiling Secondary Mutations in Blended Phenotypes: Dual ERCC4 and OTOA Pathogenic Variants Through WES Analysis. International Journal of Molecular Sciences, 25(24), 13471. https://doi.org/10.3390/ijms252413471