
The Killer’s Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
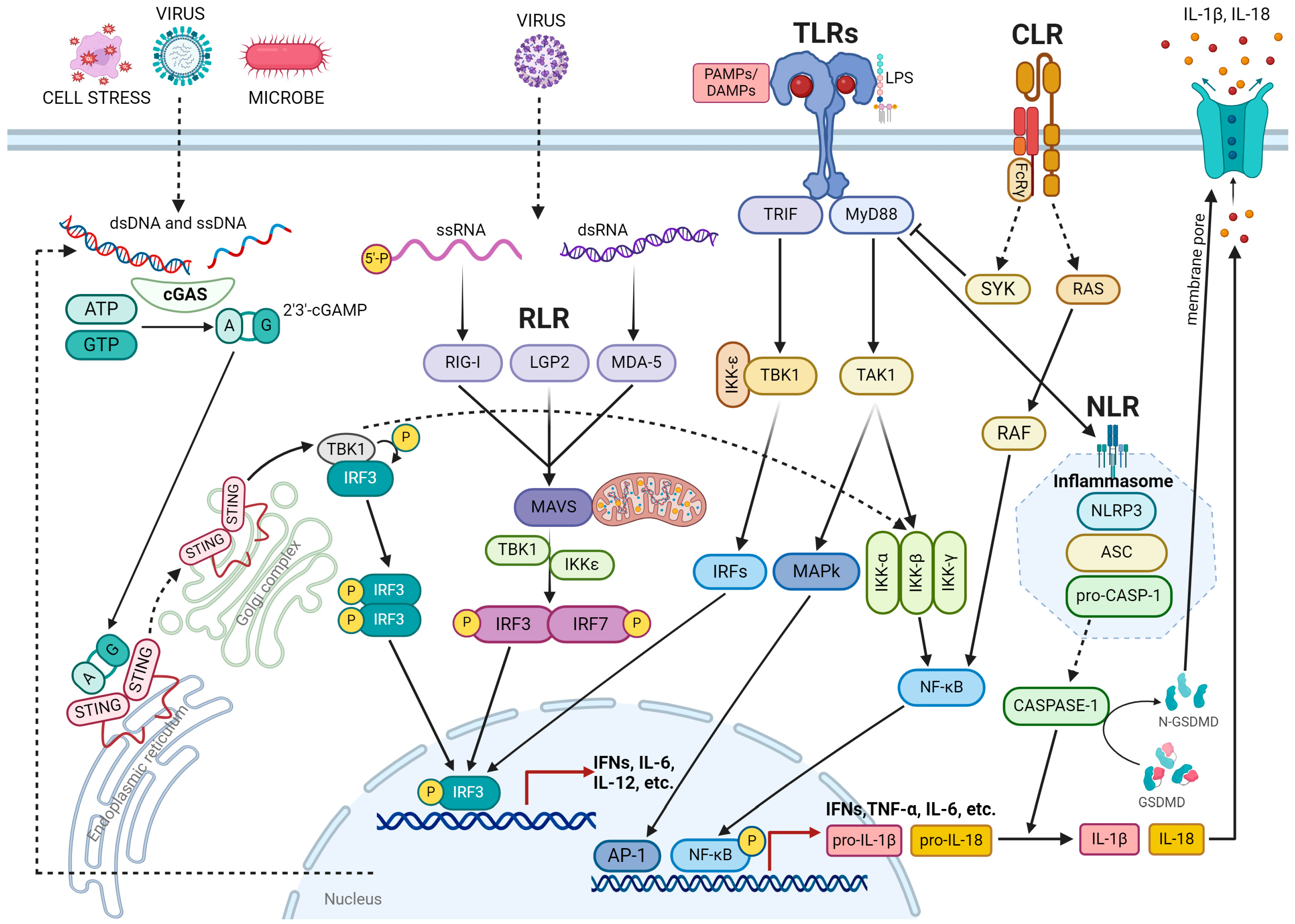
2. Inflammation
3. DNA Damage Response in Inflammation
3.1. Mechanism Underlying DNA Damage-Induced Inflammation
3.2. DNA Repair Deficiency Disorder and Inflammation
3.3. Inflammation Promoted by Transposable Element (TE) Instability
3.4. Inflammation Triggered by R-Loops Resolving Defects
3.5. Anti-Tumor Cytotoxicity Mediated by Inflammatory Response
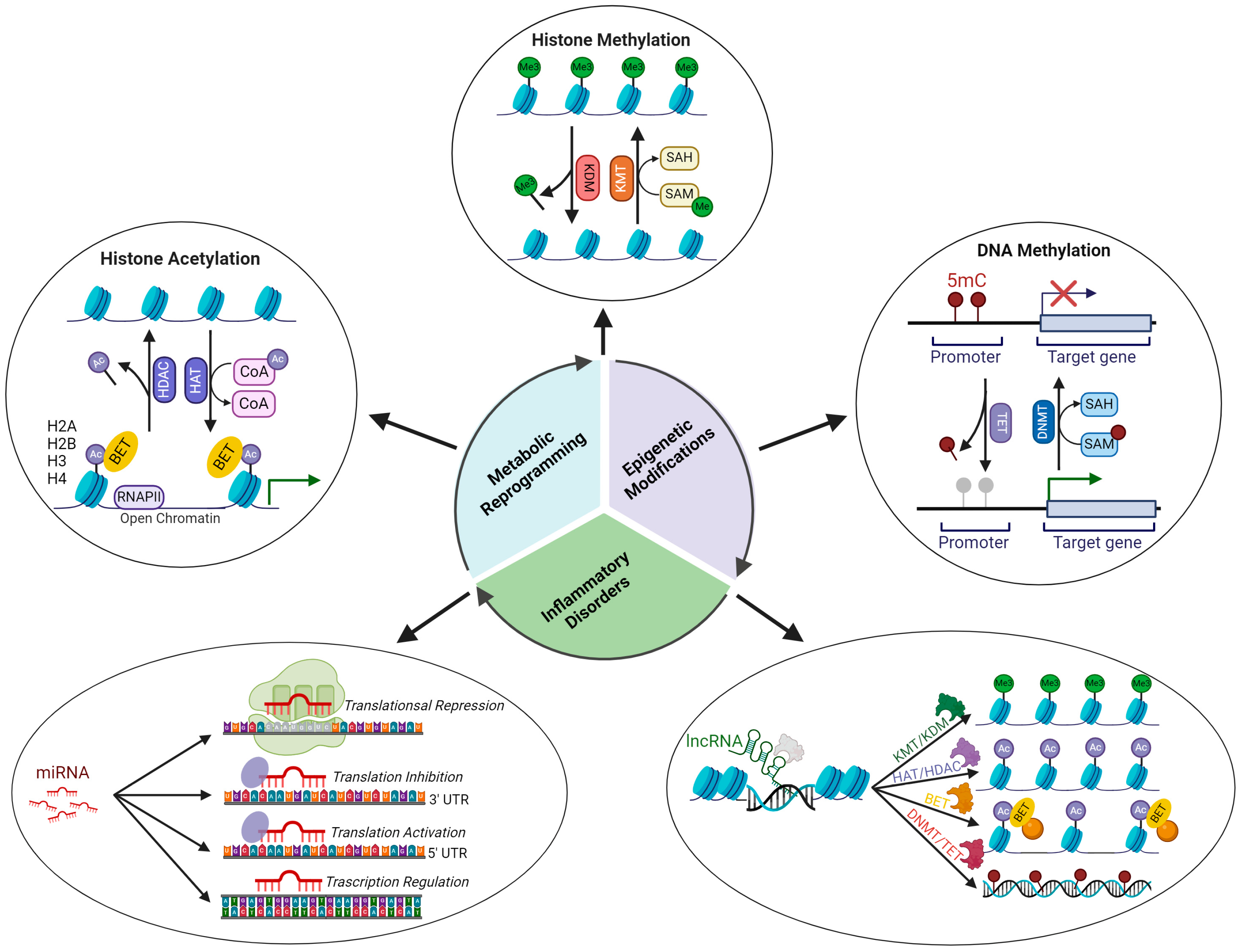
4. Epigenetics and Inflammation
4.1. Epigenetic Regulatory Mechanisms
4.2. Epigenetic Signatures Underlying Inflammation
4.3. Epigenetic Modulation of the Immune Function
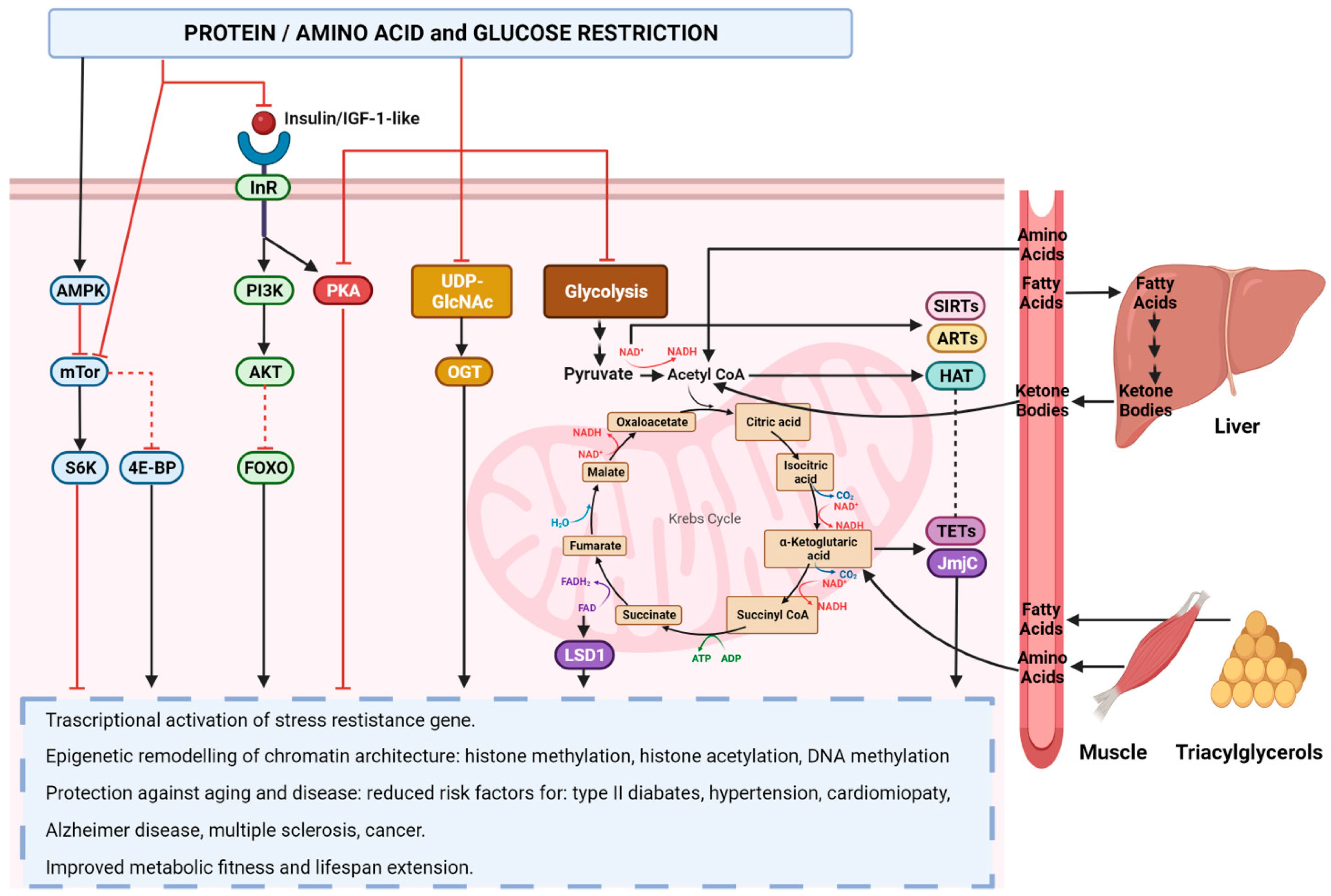
5. Nutrition, Inflammation, and Epigenetics
5.1. Role of Nutrition in Inflammation
5.2. Role of Nutrition in Epigenetic Reprogramming
6. Inflammation, Epigenetics, and Cancer
6.1. Inflammation in Tumor Onset and Progression
6.2. Epigenetic Alterations in Cancer
6.3. Inflammation-Induced Epigenetic Alterations in Tumor Immune Microenvironment
6.4. Nutrition and Cancer
7. Cancer Therapies and Future Directions
7.1. Anti-Inflammatory Drugs
7.2. DNA Damage Response-Targeted Therapy
7.3. Epigenetic-Targeted Therapy
7.4. Dietary Interventions as Adjuvant Therapy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA damage and repair in age-related inflammation. Nat. Rev. Immunol. 2023, 23, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Longo, V.D. Metabolites and Immune Response in Tumor Microenvironments. Cancers 2023, 15, 3898. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Garay, C.; Djouder, N. Dietary interventions and precision nutrition in cancer therapy. Trends Mol. Med. 2023, 29, 489–511. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Balka, K.R.; De Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef]
- Imaizumi, T.; Aratani, S.; Nakajima, T.; Carlson, M.; Matsumiya, T.; Tanji, K.; Ookawa, K.; Yoshida, H.; Tsuchida, S.; McIntyre, T.M.; et al. Retinoic acid-inducible gene-I is induced in endothelial cells by LPS and regulates expression of COX-2. Biochem. Biophys. Res. Commun. 2002, 292, 274–279. [Google Scholar] [CrossRef]
- Imaizumi, T.; Hatakeyama, M.; Yamashita, K.; Yoshida, H.; Ishikawa, A.; Taima, K.; Satoh, K.; Mori, F.; Wakabayashi, K. Interferon-gamma induces retinoic acid-inducible gene-I in endothelial cells. Endothelium 2004, 11, 169–173. [Google Scholar] [CrossRef]
- Imaizumi, T.; Matsumiya, T.; Yoshida, H.; Naraoka, T.; Uesato, R.; Ishibashi, Y.; Ota, K.; Toh, S.; Fukuda, S.; Satoh, K. Tumor-necrosis factor-alpha induces retinoic acid-inducible gene-I in rheumatoid fibroblast-like synoviocytes. Immunol. Lett. 2009, 122, 89–93. [Google Scholar] [CrossRef]
- Sakaki, H.; Imaizumi, T.; Matsumiya, T.; Kusumi, A.; Nakagawa, H.; Kubota, K.; Nishi, N.; Nakamura, T.; Hirashima, M.; Satoh, K.; et al. Retinoic acid-inducible gene-I is induced by interleukin-1beta in cultured human gingival fibroblasts. Oral. Microbiol. Immunol. 2005, 20, 47–50. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Wu, J.; Dobbs, N.; Yang, K.; Yan, N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020, 53, 115–126.e5. [Google Scholar] [CrossRef]
- Gazi, U.; Martinez-Pomares, L. Influence of the mannose receptor in host immune responses. Immunobiology 2009, 214, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; de Wet, B.J.; Martinez-Pomares, L.; Radcliffe, C.M.; Dwek, R.A.; Rudd, P.M.; Gordon, S. The mannose receptor mediates dengue virus infection of macrophages. PLoS Pathog. 2008, 4, e17. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; den Dunnen, J.; Litjens, M.; van Het Hof, B.; van Kooyk, Y.; Geijtenbeek, T.B. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 2007, 26, 605–616. [Google Scholar] [CrossRef]
- Hodges, A.; Sharrocks, K.; Edelmann, M.; Baban, D.; Moris, A.; Schwartz, O.; Drakesmith, H.; Davies, K.; Kessler, B.; McMichael, A.; et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat. Immunol. 2007, 8, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Rothfuchs, A.G.; Bafica, A.; Feng, C.G.; Egen, J.G.; Williams, D.L.; Brown, G.D.; Sher, A. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J. Immunol. 2007, 179, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Van Kooyk, Y.; Rabinovich, G.A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 2008, 9, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Richard, M.; Thibault, N.; Veilleux, P.; Gareau-Pagé, G.; Beaulieu, A.D. Granulocyte macrophage-colony stimulating factor reduces the affinity of SHP-2 for the ITIM of CLECSF6 in neutrophils: A new mechanism of action for SHP-2. Mol. Immunol. 2006, 43, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Koseki, T.; Lin, J.; del Peso, L.; Lucas, P.C.; Chen, F.F.; Ogura, Y.; Núñez, G. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J. Biol. Chem. 2000, 275, 27823–27831. [Google Scholar] [CrossRef] [PubMed]
- Abbott, D.W.; Wilkins, A.; Asara, J.M.; Cantley, L.C. The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr. Biol. 2004, 14, 2217–2227. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Chan, A.H.; Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 2020, 217, e20190314. [Google Scholar] [CrossRef]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhu, F.; Xu, Z.; Xiong, J. Role of Inflammasomes in Kidney Diseases via Both Canonical and Non-canonical Pathways. Front. Cell Dev. Biol. 2020, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol. 2011, 187, 4835–4843. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Van Vugt, M.A.; Parkes, E.E. When breaks get hot: Inflammatory signaling in BRCA1/2-mutant cancers. Trends Cancer 2022, 8, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes. Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Hinz, M.; Stilmann, M.; Arslan, S.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-κB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef]
- Fang, L.; Choudhary, S.; Zhao, Y.; Edeh, C.B.; Yang, C.; Boldogh, I.; Brasier, A.R. ATM regulates NF-κB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res. 2014, 42, 8416–8432. [Google Scholar] [CrossRef]
- Sun, X.; Liu, T.; Zhao, J.; Xia, H.; Xie, J.; Guo, Y.; Zhong, L.; Li, M.; Yang, Q.; Peng, C.; et al. DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat. Commun. 2020, 11, 6182. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef] [PubMed]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Coquel, F.; Silva, M.J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Gratia, M.; Rodero, M.P.; Conrad, C.; Bou Samra, E.; Maurin, M.; Rice, G.I.; Duffy, D.; Revy, P.; Petit, F.; Dale, R.C.; et al. Bloom syndrome protein restrains innate immune sensing of micronuclei by cGAS. J. Exp. Med. 2019, 216, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Gul, E.; Sayar, E.H.; Gungor, B.; Eroglu, F.K.; Surucu, N.; Keles, S.; Guner, S.N.; Findik, S.; Alpdundar, E.; Ayanoglu, I.C.; et al. Type I IFN-related NETosis in ataxia telangiectasia and Artemis deficiency. J. Allergy Clin. Immunol. 2018, 142, 246–257. [Google Scholar] [CrossRef]
- Song, X.; Ma, F.; Herrup, K. Accumulation of Cytoplasmic DNA Due to ATM Deficiency Activates the Microglial Viral Response System with Neurotoxic Consequences. J. Neurosci. 2019, 39, 6378–6394. [Google Scholar] [CrossRef]
- Davies, S.L.; North, P.S.; Hickson, I.D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007, 14, 677–679. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes. Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Majora, M.; Sondenheimer, K.; Knechten, M.; Uthe, I.; Esser, C.; Schiavi, A.; Ventura, N.; Krutmann, J. HDAC inhibition improves autophagic and lysosomal function to prevent loss of subcutaneous fat in a mouse model of Cockayne syndrome. Sci. Transl. Med. 2018, 10, eaam7510. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Goewey Ruiz, J.A.; Sebastian, M.; Kidane, D. Defective DNA polymerase beta invoke a cytosolic DNA mediated inflammatory response. Front. Immunol. 2022, 13, 1039009. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Kajikawa, M.; Okada, N. LINE retrotransposition and host DNA repair machinery. Mob. Genet. Elem. 2015, 5, 92–97. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Kazazian, H.H.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mita, P.; McKerrow, W.; Fenyö, D.; Boeke, J.D.; Linker, S.B.; Gage, F.H.; Kreiling, J.A.; Petrashen, A.P.; et al. The role of retrotransposable elements in ageing and age-associated diseases. Nature 2021, 596, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.N.; Victorelli, S.G.; Salmonowicz, H.; Dasgupta, N.; Liu, T.; Passos, J.F.; Adams, P.D. Cytoplasmic DNA: Sources, sensing, and role in aging and disease. Cell 2021, 184, 5506–5526. [Google Scholar] [CrossRef]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Goulielmaki, E.; Tsekrekou, M.; Batsiotos, N.; Ascensão-Ferreira, M.; Ledaki, E.; Stratigi, K.; Chatzinikolaou, G.; Topalis, P.; Kosteas, T.; Altmüller, J.; et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat. Commun. 2021, 12, 3153. [Google Scholar] [CrossRef]
- Chang, E.Y.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef] [PubMed]
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Sanchez, M.; Pichierri, P.; Franchitto, A. ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res. 2019, 47, 3485–3502. [Google Scholar] [CrossRef] [PubMed]
- Barnhoorn, S.; Uittenboogaard, L.M.; Jaarsma, D.; Vermeij, W.P.; Tresini, M.; Weymaere, M.; Menoni, H.; Brandt, R.M.; de Waard, M.C.; Botter, S.M.; et al. Cell-autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genet. 2014, 10, e1004686. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat. Rev. Clin. Oncol. 2011, 8, 151–160. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Anti-tumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E.; Yang, Y.G.; Robins, P. Biochemical properties of mammalian TREX1 and its association with DNA replication and inherited inflammatory disease. Biochem. Soc. Trans. 2009, 37, 535–538. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Anti-tumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar]
- Cheung, P.; Lau, P. Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol. 2005, 19, 563–573. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Narlikar, G.J.; Fan, H.Y.; Kingston, R.E. Cooperation between complexes that regulate chromatin structure and transcription. Cell 2002, 108, 475–487. [Google Scholar] [CrossRef]
- Youn, H.D. Methylation and demethylation of DNA and histones in chromatin: The most complicated epigenetic marker. Exp. Mol. Med. 2017, 49, e321. [Google Scholar] [CrossRef]
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034. [Google Scholar] [CrossRef]
- Shi, Y.G.; Tsukada, Y. The discovery of histone demethylases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017947. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Flores, K.B.; Wolschin, F.; Amdam, G.V. The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 2013, 53, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.J.; Kim, K.H.; Kim, J.Y.; Jeong, S.; Kim, N. Identification of DNA-Methylated CpG Islands Associated with Gene Silencing in the Adult Body Tissues of the Ogye Chicken Using RNA-Seq and Reduced Representation Bisulfite Sequencing. Front. Genet. 2019, 10, 346. [Google Scholar] [CrossRef]
- Rollins, R.A.; Haghighi, F.; Edwards, J.R.; Das, R.; Zhang, M.Q.; Ju, J.; Bestor, T.H. Large-scale structure of genomic methylation patterns. Genome Res. 2006, 16, 157–163. [Google Scholar] [CrossRef]
- Pappalardo, X.G.; Barra, V. Losing DNA methylation at repetitive elements and breaking bad. Epigenetics Chromatin 2021, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef]
- Bodega, B.; Orlando, V. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr. Opin. Cell Biol. 2014, 31, 67–73. [Google Scholar] [CrossRef]
- Shapiro, J.A.; von Sternberg, R. Why repetitive DNA is essential to genome function. Biol. Rev. Camb. Philos. Soc. 2005, 80, 227–250. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5 mC to 5 hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Xu, J.; Sannai, M.; Moore, R.; Caretti, E.; Cigliano, A.; Le Coz, M.; Devarajan, K.; Wessels, A.; Soprano, D.; et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Turner, D.; Masciullo, V.; Schepis, F.; Albino, D.; Daniel, R.; Skalka, A.M.; Meropol, N.J.; Alberti, C.; Larue, L.; et al. The base excision repair enzyme MED1 mediates DNA damage response to anti-tumor drugs and is associated with mismatch repair system integrity. Proc. Natl. Acad. Sci. USA 2003, 100, 15071–15076. [Google Scholar] [CrossRef]
- Du, Q.; Luu, P.L.; Stirzaker, C.; Clark, S.J. Methyl-CpG-binding domain proteins: Readers of the epigenome. Epigenomics 2015, 7, 1051–1073. [Google Scholar] [CrossRef] [PubMed]
- Steinacher, R.; Barekati, Z.; Botev, P.; Kuśnierczyk, A.; Slupphaug, G.; Schär, P. SUMOylation coordinates BERosome assembly in active DNA demethylation during cell differentiation. EMBO J. 2019, 38, e99242. [Google Scholar] [CrossRef]
- Sannai, M.; Doneddu, V.; Giri, V.; Seeholzer, S.; Nicolas, E.; Yip, S.C.; Bassi, M.R.; Mancuso, P.; Cortellino, S.; Cigliano, A.; et al. Modification of the base excision repair enzyme MBD4 by the small ubiquitin-like molecule SUMO1. DNA Repair 2019, 82, 102687. [Google Scholar] [CrossRef]
- Boyes, J.; Bird, A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell 1991, 64, 1123–1134. [Google Scholar] [CrossRef]
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312. [Google Scholar] [CrossRef]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [CrossRef]
- Geiman, T.M.; Sankpal, U.T.; Robertson, A.K.; Zhao, Y.; Robertson, K.D. DNMT3B interacts with hSNF2H chromatin remodeling enzyme, HDACs 1 and 2, and components of the histone methylation system. Biochem. Biophys. Res. Commun. 2004, 318, 544–555. [Google Scholar] [CrossRef]
- Cervoni, N.; Szyf, M. Demethylase activity is directed by histone acetylation. J. Biol. Chem. 2001, 276, 40778–40787. [Google Scholar] [CrossRef]
- Szyf, M.; Eliasson, L.; Mann, V.; Klein, G.; Razin, A. Cellular and viral DNA hypomethylation associated with induction of Epstein-Barr virus lytic cycle. Proc. Natl. Acad. Sci. USA 1985, 82, 8090–8094. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Hacisuleyman, E.; Shukla, C.J.; Weiner, C.L.; Rinn, J.L. Function and evolution of local repeats in the Firre locus. Nat. Commun. 2016, 7, 11021. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Sotomayor, E.M.; Seto, E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 2010, 29, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Calao, M.; Burny, A.; Quivy, V.; Dekoninck, A.; Van Lint, C. A pervasive role of histone acetyltransferases and deacetylases in an NF-kappaB-signaling code. Trends Biochem. Sci. 2008, 33, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, M.; Bai, S.; Kong, L.; Su, X. Identification of histone acetylation in a murine model of allergic asthma by proteomic analysis. Exp. Biol. Med. 2021, 246, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Stender, J.D.; Pascual, G.; Liu, W.; Kaikkonen, M.U.; Do, K.; Spann, N.J.; Boutros, M.; Perrimon, N.; Rosenfeld, M.G.; Glass, C.K. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 2012, 48, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Yu, Y.; Huang, H.; Fan, H.; Yin, C.; Li, K.; Fulton, D.J.; Chen, F. Epigenetic Regulation of Interleukin 6 by Histone Acetylation in Macrophages and Its Role in Paraquat-Induced Pulmonary Fibrosis. Front. Immunol. 2016, 7, 696. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, S.; Wang, C.; Huang, Y.; Li, H.; Wang, Y.; Zhu, Z.; Tang, J.; Yan, M. Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 2017, 7, 43592. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Suliman, B.A.; Yu, L.; Yuan, X.; Wang, D.; Irving, A.T.; Sarvestani, S.T.; Banerjee, A.; Mansell, A.S.; Liu, J.P.; et al. The acetyltransferase HAT1 moderates the NF-κB response by regulating the transcription factor PLZF. Nat. Commun. 2015, 6, 6795. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef]
- Bayarsaihan, D. Epigenetic mechanisms in inflammation. J. Dent. Res. 2011, 90, 9–17. [Google Scholar] [CrossRef]
- Qu, L.; Yin, T.; Zhao, Y.; Lv, W.; Liu, Z.; Chen, C.; Liu, K.; Shan, S.; Zhou, R.; Li, X.; et al. Histone demethylases in the regulation of immunity and inflammation. Cell Death Discov. 2023, 9, 188. [Google Scholar] [CrossRef]
- Katakia, Y.T.; Thakkar, N.P.; Thakar, S.; Sakhuja, A.; Goyal, R.; Sharma, H.; Dave, R.; Mandloi, A.; Basu, S.; Nigam, I.; et al. Dynamic alterations of H3K4me3 and H3K27me3 at ADAM17 and Jagged-1 gene promoters cause an inflammatory switch of endothelial cells. J. Cell Physiol. 2022, 237, 992–1012. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cao, J.; Cai, W.L.; Lang, S.M.; Horton, J.R.; Jansen, D.J.; Liu, Z.Z.; Chen, J.F.; Zhang, M.; Mott, B.T.; et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018, 16, e2006134. [Google Scholar] [CrossRef] [PubMed]
- Balada, E.; Castro-Marrero, J.; Felip, L.; Ordi-Ros, J.; Vilardell-Tarrés, M. Associations between the expression of epigenetically regulated genes and the expression of DNMTs and MBDs in systemic lupus erythematosus. PLoS ONE 2012, 7, e45897. [Google Scholar] [CrossRef] [PubMed]
- Vucic, E.A.; Chari, R.; Thu, K.L.; Wilson, I.M.; Cotton, A.M.; Kennett, J.Y.; Zhang, M.; Lonergan, K.M.; Steiling, K.; Brown, C.J.; et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am. J. Respir. Cell Mol. Biol. 2014, 50, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Karatzas, P.S.; Mantzaris, G.J.; Safioleas, M.; Gazouli, M. DNA methylation profile of genes involved in inflammation and autoimmunity in inflammatory bowel disease. Medicine 2014, 93, e309. [Google Scholar] [CrossRef]
- Brandt, D.; Sohr, E.; Pablik, J.; Schnabel, A.; Kapplusch, F.; Mäbert, K.; Girschick, J.H.; Morbach, H.; Thielemann, F.; Hofmann, S.R.; et al. CD14. Clin. Immunol. 2018, 196, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Akbaba, T.H.; Akkaya-Ulum, Y.Z.; Tavukcuoglu, Z.; Bilginer, Y.; Ozen, S.; Balci-Peynircioglu, B. Inflammation-related differentially expressed common miRNAs in systemic autoinflammatory disorders patients can regulate the clinical course. Clin. Exp. Rheumatol. 2021, 39 (Suppl. S132), 109–117. [Google Scholar] [CrossRef]
- Hussain, N.; Zhu, W.; Jiang, C.; Xu, J.; Wu, X.; Geng, M.; Hussain, S.; Cai, Y.; Xu, K.; Xu, P.; et al. Down-regulation of miR-10a-5p in synoviocytes contributes to TBX5-controlled joint inflammation. J. Cell Mol. Med. 2018, 22, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Aznaourova, M.; Janga, H.; Sefried, S.; Kaufmann, A.; Dorna, J.; Volkers, S.M.; Georg, P.; Lechner, M.; Hoppe, J.; Dökel, S.; et al. Noncoding RNA. Proc. Natl. Acad. Sci. USA 2020, 117, 9042–9053. [Google Scholar] [CrossRef] [PubMed]
- Agliano, F.; Fitzgerald, K.A.; Vella, A.T.; Rathinam, V.A.; Medvedev, A.E. Long Non-coding RNA LincRNA-EPS Inhibits Host Defense Against. Front. Cell Infect. Microbiol. 2019, 9, 481. [Google Scholar]
- IIott, N.E.; Heward, J.A.; Roux, B.; Tsitsiou, E.; Fenwick, P.S.; Lenzi, L.; Goodhead, I.; Hertz-Fowler, C.; Heger, A.; Hall, N.; et al. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat. Commun. 2014, 5, 3979. [Google Scholar] [PubMed]
- Krawczyk, M.; Emerson, B.M. p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-κB complexes. eLife 2014, 3, e01776. [Google Scholar] [CrossRef]
- Zhang, Q.; Chao, T.C.; Patil, V.S.; Qin, Y.; Tiwari, S.K.; Chiou, J.; Dobin, A.; Tsai, C.M.; Li, Z.; Dang, J.; et al. The long noncoding RNA. EMBO J. 2019, 38, e100041. [Google Scholar] [CrossRef]
- Atianand, M.K.; Hu, W.; Satpathy, A.T.; Shen, Y.; Ricci, E.P.; Alvarez-Dominguez, J.R.; Bhatta, A.; Schattgen, S.A.; McGowan, J.D.; Blin, J.; et al. A Long Noncoding RNA lincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell 2016, 165, 1672–1685. [Google Scholar] [CrossRef]
- Zou, Y.; Xu, H. Involvement of long noncoding RNAs in the pathogenesis of autoimmune diseases. J. Transl. Autoimmun. 2020, 3, 100044. [Google Scholar] [CrossRef]
- Kersh, E.N.; Fitzpatrick, D.R.; Murali-Krishna, K.; Shires, J.; Speck, S.H.; Boss, J.M.; Ahmed, R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J. Immunol. 2006, 176, 4083–4093. [Google Scholar] [CrossRef]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Collins, P.L.; Aune, T.M. T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J. Immunol. 2008, 181, 8372–8381. [Google Scholar] [CrossRef] [PubMed]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Scharer, C.D.; Barwick, B.G.; Youngblood, B.A.; Ahmed, R.; Boss, J.M. Global DNA methylation remodeling accompanies CD8 T cell effector function. J. Immunol. 2013, 191, 3419–3429. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P.; Araki, Y.; Subedi, K. The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 2012, 12, 306–315. [Google Scholar] [CrossRef]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar] [CrossRef]
- Huang, D.; Chen, J.; Yang, L.; Ouyang, Q.; Li, J.; Lao, L.; Zhao, J.; Liu, J.; Lu, Y.; Xing, Y.; et al. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat. Immunol. 2018, 19, 1112–1125. [Google Scholar] [CrossRef]
- Pyfrom, S.C.; Quinn, C.C.; Dorando, H.K.; Luo, H.; Payton, J.E. BCALM (AC099524.1) Is a Human B Lymphocyte-Specific Long Noncoding RNA That Modulates B Cell Receptor-Mediated Calcium Signaling. J. Immunol. 2020, 205, 595–607. [Google Scholar] [CrossRef]
- Li, J.; Tian, J.; Lu, J.; Wang, Z.; Ling, J.; Wu, X.; Yang, F.; Xia, Y. LncRNA GAS5 inhibits Th17 differentiation and alleviates immune thrombocytopenia via promoting the ubiquitination of STAT3. Int. Immunopharmacol. 2020, 80, 106127. [Google Scholar] [CrossRef]
- Fu, J.; Shi, H.; Wang, B.; Zhan, T.; Shao, Y.; Ye, L.; Wu, S.; Yu, C.; Zheng, L. LncRNA PVT1 links Myc to glycolytic metabolism upon CD4. J. Autoimmun. 2020, 107, 102358. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.A.; Wapinski, O.L.; Yang, Y.W.; Bureau, J.F.; Gopinath, S.; Monack, D.M.; Chang, H.Y.; Brahic, M.; Kirkegaard, K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 2013, 152, 743–754. [Google Scholar] [CrossRef]
- Spurlock, C.F.; Tossberg, J.T.; Guo, Y.; Collier, S.P.; Crooke, P.S.; Aune, T.M. Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat. Commun. 2015, 6, 6932. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Sears, B.; Lawren, B. The Zone: A Dietary Road Map; ReganBooks: New York, NY, USA, 1995; p. xvii. 328p. [Google Scholar]
- Brenner, R.R. Hormonal modulation of delta6 and delta5 desaturases: Case of diabetes. Prostaglandins Leukot. Essent. Fatty Acids 2003, 68, 151–162. [Google Scholar] [CrossRef]
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor Necrosis Factor-Alpha: Role in Development of Insulin Resistance and Pathogenesis of Type 2 Diabetes Mellitus. J. Cell Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.C.; Parsons, H.G. Modulation in delta 9, delta 6, and delta 5 fatty acid desaturase activity in the human intestinal CaCo-2 cell line. J. Lipid Res. 1995, 36, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef]
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J.; et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- McArthur, S.; Juban, G.; Gobbetti, T.; Desgeorges, T.; Theret, M.; Gondin, J.; Toller-Kawahisa, J.E.; Reutelingsperger, C.P.; Chazaud, B.; Perretti, M.; et al. Annexin A1 drives macrophage skewing to accelerate muscle regeneration through AMPK activation. J. Clin. Investig. 2020, 130, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell-based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef]
- Traba, J.; Kwarteng-Siaw, M.; Okoli, T.C.; Li, J.; Huffstutler, R.D.; Bray, A.; Waclawiw, M.A.; Han, K.; Pelletier, M.; Sauve, A.A.; et al. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J. Clin. Investig. 2015, 125, 4592–4600. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef]
- Cortellino, S.; Raveane, A.; Chiodoni, C.; Delfanti, G.; Pisati, F.; Spagnolo, V.; Visco, E.; Fragale, G.; Ferrante, F.; Magni, S.; et al. Fasting renders immunotherapy effective against low-immunogenic breast cancer while reducing side effects. Cell Rep. 2022, 40, 111256. [Google Scholar] [CrossRef]
- Longo, V.D.; Cortellino, S. Fasting, dietary restriction, and immunosenescence. J. Allergy Clin. Immunol. 2020, 146, 1002–1004. [Google Scholar] [CrossRef]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef]
- Vernieri, C.; Fucà, G.; Ligorio, F.; Huber, V.; Vingiani, A.; Iannelli, F.; Raimondi, A.; Rinchai, D.; Frigè, G.; Belfiore, A.; et al. Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Anti-tumor Immunity in Patients with Cancer. Cancer Discov. 2022, 12, 90–107. [Google Scholar] [CrossRef]
- Fontana, L. The scientific basis of caloric restriction leading to longer life. Curr. Opin. Gastroenterol. 2009, 25, 144–150. [Google Scholar] [CrossRef]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L. Promoting health and longevity through diet: From model organisms to humans. Cell 2015, 161, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Ghezzi, L.; Cross, A.H.; Piccio, L. Effects of dietary restriction on neuroinflammation in neurodegenerative diseases. J. Exp. Med. 2021, 218, e20190086. [Google Scholar] [CrossRef] [PubMed]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.H.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, S.; Jia, W. Calorie restriction and its impact on gut microbial composition and global metabolism. Front. Med. 2018, 12, 634–644. [Google Scholar] [CrossRef]
- Tanca, A.; Abbondio, M.; Palomba, A.; Fraumene, C.; Marongiu, F.; Serra, M.; Pagnozzi, D.; Laconi, E.; Uzzau, S. Caloric restriction promotes functional changes involving short-chain fatty acid biosynthesis in the rat gut microbiota. Sci. Rep. 2018, 8, 14778. [Google Scholar] [CrossRef] [PubMed]
- Roopchand, D.E.; Carmody, R.N.; Kuhn, P.; Moskal, K.; Rojas-Silva, P.; Turnbaugh, P.J.; Raskin, I. Dietary Polyphenols Promote Growth of the Gut Bacterium Akkermansia muciniphila and Attenuate High-Fat Diet-Induced Metabolic Syndrome. Diabetes 2015, 64, 2847–2858. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef]
- Keating, S.T.; El-Osta, A. Epigenetics and metabolism. Circ. Res. 2015, 116, 715–736. [Google Scholar] [CrossRef]
- Pietrocola, F.; Demont, Y.; Castoldi, F.; Enot, D.; Durand, S.; Semeraro, M.; Baracco, E.E.; Pol, J.; Bravo-San Pedro, J.M.; Bordenave, C.; et al. Metabolic effects of fasting on human and mouse blood in vivo. Autophagy 2017, 13, 567–578. [Google Scholar] [CrossRef]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363, eaau0135. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Li, W.; Zhou, X. Lactylation, an emerging hallmark of metabolic reprogramming: Current progress and open challenges. Front. Cell Dev. Biol. 2022, 10, 972020. [Google Scholar] [CrossRef]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Büttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef] [PubMed]
- Maurice, N.J.; Berner, J.; Taber, A.K.; Zehn, D.; Prlic, M. Inflammatory signals are sufficient to elicit TOX expression in mouse and human CD8+ T cells. JCI Insight 2021, 6, e150744. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Drutman, S.B.; Haerynck, F.; Zhong, F.L.; Hum, D.; Hernandez, N.J.; Belkaya, S.; Rapaport, F.; de Jong, S.J.; Creytens, D.; Tavernier, S.J.; et al. Homozygous. Proc. Natl. Acad. Sci. USA 2019, 116, 19055–19063. [Google Scholar] [CrossRef]
- Zhong, F.L.; Mamaï, O.; Sborgi, L.; Boussofara, L.; Hopkins, R.; Robinson, K.; Szeverényi, I.; Takeichi, T.; Balaji, R.; Lau, A.; et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016, 167, 187–202.e17. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerbäck, C.; Rosdahl, I.; Söderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment. Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.L.; Fock, K.M.; Mitchell, H.M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: A case-control study and gene expression analyses. PLoS ONE 2015, 10, e0117870. [Google Scholar] [CrossRef] [PubMed]
- Miskiewicz, A.; Szparecki, G.; Durlik, M.; Rydzewska, G.; Ziobrowski, I.; Górska, R. The Q705K and F359L Single-Nucleotide Polymorphisms of NOD-Like Receptor Signaling Pathway: Association with Chronic Pancreatitis, Pancreatic Cancer, and Periodontitis. Arch. Immunol. Ther. Exp. 2015, 63, 485–494. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, C.; Hua, M.; Wang, R.; Zhong, C.; Yu, J.; Han, F.; He, N.; Zhao, Y.; Liu, G.; et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 2017, 8, 108571–108583. [Google Scholar] [CrossRef]
- Chai, D.; Zhang, Z.; Shi, S.Y.; Qiu, D.; Zhang, C.; Wang, G.; Fang, L.; Li, H.; Tian, H.; Zheng, J. Absent in melanoma 2-mediating M1 macrophages facilitate tumor rejection in renal carcinoma. Transl. Oncol. 2021, 14, 101018. [Google Scholar] [CrossRef]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell-Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef]
- Huang, Q.; Lan, F.; Wang, X.; Yu, Y.; Ouyang, X.; Zheng, F.; Han, J.; Lin, Y.; Xie, Y.; Xie, F.; et al. IL-1β-induced activation of p38 promotes metastasis in gastric adenocarcinoma via upregulation of AP-1/c-fos, MMP2 and MMP9. Mol. Cancer 2014, 13, 18. [Google Scholar] [CrossRef]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ko, Y.S.; Kim, H.J. P2Y2R-mediated inflammasome activation is involved in tumor progression in breast cancer cells and in radiotherapy-resistant breast cancer. Int. J. Oncol. 2018, 53, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lei, J.H.; Bao, J.; Wang, H.; Hao, W.; Li, L.; Peng, C.; Masuda, T.; Miao, K.; Xu, J.; et al. BRCA1 Deficiency Impairs Mitophagy and Promotes Inflammasome Activation and Mammary Tumor Metastasis. Adv. Sci. 2020, 7, 1903616. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 81. [Google Scholar] [CrossRef]
- Deswaerte, V.; Nguyen, P.; West, A.; Browning, A.F.; Yu, L.; Ruwanpura, S.M.; Balic, J.; Livis, T.; Girard, C.; Preaudet, A.; et al. Inflammasome Adaptor ASC Suppresses Apoptosis of Gastric Cancer Cells by an IL18-Mediated Inflammation-Independent Mechanism. Cancer Res. 2018, 78, 1293–1307. [Google Scholar] [CrossRef]
- Liu, W.; Luo, Y.; Dunn, J.H.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J. Investig. Dermatol. 2013, 133, 518–527. [Google Scholar] [CrossRef]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef]
- Gasparoto, T.H.; de Oliveira, C.E.; de Freitas, L.T.; Pinheiro, C.R.; Hori, J.I.; Garlet, G.P.; Cavassani, K.A.; Schillaci, R.; da Silva, J.S.; Zamboni, D.S.; et al. Inflammasome activation is critical to the protective immune response during chemically induced squamous cell carcinoma. PLoS ONE 2014, 9, e107170. [Google Scholar] [CrossRef]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Dagenais, M.; Dupaul-Chicoine, J.; Douglas, T.; Champagne, C.; Morizot, A.; Saleh, M. The Interleukin (IL)-1R1 pathway is a critical negative regulator of PyMT-mediated mammary tumorigenesis and pulmonary metastasis. Oncoimmunology 2017, 6, e1287247. [Google Scholar] [CrossRef]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: In vivo analysis of tumor-stromal interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef]
- Jia, D.; Jolly, M.K.; Kulkarni, P.; Levine, H. Phenotypic Plasticity and Cell Fate Decisions in Cancer: Insights from Dynamical Systems Theory. Cancers 2017, 9, 70. [Google Scholar] [CrossRef]
- Sacchetti, A.; Teeuwssen, M.; Verhagen, M.; Joosten, R.; Xu, T.; Stabile, R.; van der Steen, B.; Watson, M.M.; Gusinac, A.; Kim, W.K.; et al. Phenotypic plasticity underlies local invasion and distant metastasis in colon cancer. eLife 2021, 10, e61461. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nat. Rev. Cancer 2021, 21, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef]
- Avvakumov, N.; Côté, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef]
- Huntly, B.J.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef]
- Modena, P.; Lualdi, E.; Facchinetti, F.; Galli, L.; Teixeira, M.R.; Pilotti, S.; Sozzi, G. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005, 65, 4012–4019. [Google Scholar] [CrossRef] [PubMed]
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhang, Z.M.; Xia, Y.; Liao, G.Q.; Pan, Y.; Liu, S.; Zhang, Y.; Yan, Z.S. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br. J. Cancer 2013, 109, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef]
- Zhou, M.; Venkata, P.P.; Viswanadhapalli, S.; Palacios, B.; Alejo, S.; Chen, Y.; He, Y.; Pratap, U.P.; Liu, J.; Zou, Y.; et al. KDM1A inhibition is effective in reducing stemness and treating triple negative breast cancer. Breast Cancer Res. Treat. 2021, 185, 343–357. [Google Scholar] [CrossRef]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/ΚDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef]
- Belkina, A.C.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef]
- De Koning, L.; Savignoni, A.; Boumendil, C.; Rehman, H.; Asselain, B.; Sastre-Garau, X.; Almouzni, G. Heterochromatin protein 1alpha: A hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009, 1, 178–191. [Google Scholar] [CrossRef]
- Coles, A.H.; Jones, S.N. The ING gene family in the regulation of cell growth and tumorigenesis. J. Cell Physiol. 2009, 218, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, R.; Cortellino, S.; Riccio, A.; Jagmohan-Changur, S.; Van der Klift, H.; Wijnen, J.; Turner, D.; Ventura, A.; Rovella, V.; Percesepe, A.; et al. Involvement of MBD4 inactivation in mismatch repair-deficient tumorigenesis. Oncotarget 2015, 6, 42892–42904. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Aaltonen, L.A.; Godwin, A.K.; Loukola, A.; Percesepe, A.; Salovaara, R.; Masciullo, V.; Genuardi, M.; Paravatou-Petsotas, M.; Bassi, D.E.; et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat. Genet. 1999, 23, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Nejati-Koshki, K.; Roberts, C.T.; Babaei, G.; Rastegar, M. The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers 2023, 15, 2683. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Allis, C.D.; Chi, P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol. Med. 2007, 13, 373–380. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Alonso-Curbelo, D.; Morris, J.P.; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a restrains Kras-dependent changes in acinar cell identity. eLife 2018, 7, e35216. [Google Scholar] [CrossRef]
- Treger, T.D.; Chowdhury, T.; Pritchard-Jones, K.; Behjati, S. The genetic changes of Wilms tumour. Nat. Rev. Nephrol. 2019, 15, 240–251. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2018, 33, 151–152. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Xu, J.; Cortellino, S.; Tricarico, R.; Chang, W.C.; Scher, G.; Devarajan, K.; Slifker, M.; Moore, R.; Bassi, M.R.; Caretti, E.; et al. Is involved in the pathogenesis of intestinal tumors with reduced. Oncotarget 2017, 8, 89988–89997. [Google Scholar] [CrossRef][Green Version]
- Tricarico, R.; Madzo, J.; Scher, G.; Cohen, M.; Jelinek, J.; Maegawa, S.; Nagarathinam, R.; Scher, C.; Chang, W.C.; Nicolas, E.; et al. TET1 and TDG Suppress Inflammatory Response in Intestinal Tumorigenesis: Implications for Colorectal Tumors with the CpG Island Methylator Phenotype. Gastroenterology 2023, 164, 921–936.e1. [Google Scholar] [CrossRef]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10; 11)(q22; q23). Leukemia 2003, 17, 637–641. [Google Scholar] [CrossRef]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem. Cell 2011, 9, 193–204. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Brenne, S.S.; Madsen, P.H.; Pedersen, I.S.; Hveem, K.; Skorpen, F.; Krarup, H.B.; Giskeødegård, G.F.; Laugsand, E.A. Colorectal cancer detected by liquid biopsy 2 years prior to clinical diagnosis in the HUNT study. Br. J. Cancer 2023, 129, 861–868. [Google Scholar] [CrossRef]
- Jedi, M.; Young, G.P.; Pedersen, S.K.; Symonds, E.L. Methylation and Gene Expression of. Clin. Med. Insights Oncol. 2018, 12, 1179554918775064. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Ma, Q.; Wu, X.; Dai, X.; Peng, Q.; Cai, H. Detection of DNA Methylation in Gene Loci ASTN1, DLX1, ITGA4, RXFP3, SOX17, and ZNF671 for Diagnosis of Cervical Cancer. Cancer Manag. Res. 2023, 15, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Luttmer, R.; De Strooper, L.M.; Dijkstra, M.G.; Berkhof, J.; Snijders, P.J.; Steenbergen, R.D.; van Kemenade, F.J.; Rozendaal, L.; Helmerhorst, T.J.; Verheijen, R.H.; et al. FAM19A4 methylation analysis in self-samples compared with cervical scrapes for detecting cervical (pre)cancer in HPV-positive women. Br. J. Cancer 2016, 115, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Davalos, V.; Esteller, M. Cancer epigenetics in clinical practice. CA Cancer J. Clin. 2023, 73, 376–424. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Chen, X.; Huang, M.; Wang, H.; Chen, J.; Liang, Z.; Zhang, J.; Yu, Y.; Chen, S.; Xu, S.; et al. A urine-based DNA methylation assay to facilitate early detection and risk stratification of bladder cancer. Clin. Epigenet. 2021, 13, 91. [Google Scholar] [CrossRef] [PubMed]
- Moinova, H.R.; LaFramboise, T.; Lutterbaugh, J.D.; Chandar, A.K.; Dumot, J.; Faulx, A.; Brock, W.; De la Cruz Cabrera, O.; Guda, K.; Barnholtz-Sloan, J.S.; et al. Identifying DNA methylation biomarkers for non-endoscopic detection of Barrett’s esophagus. Sci. Transl. Med. 2018, 10, eaao5848. [Google Scholar] [CrossRef] [PubMed]
- Jerónimo, C.; Henrique, R.; Hoque, M.O.; Mambo, E.; Ribeiro, F.R.; Varzim, G.; Oliveira, J.; Teixeira, M.R.; Lopes, C.; Sidransky, D. A quantitative promoter methylation profile of prostate cancer. Clin. Cancer Res. 2004, 10, 8472–8478. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing—The challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Calin, G.A.; Cimmino, A.; Fabbri, M.; Ferracin, M.; Wojcik, S.E.; Shimizu, M.; Taccioli, C.; Zanesi, N.; Garzon, R.; Aqeilan, R.I.; et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 5166–5171. [Google Scholar] [CrossRef] [PubMed]
- Kogo, R.; Mimori, K.; Tanaka, F.; Komune, S.; Mori, M. Clinical significance of miR-146a in gastric cancer cases. Clin. Cancer Res. 2011, 17, 4277–4284. [Google Scholar] [CrossRef]
- Moridikia, A.; Mirzaei, H.; Sahebkar, A.; Salimian, J. MicroRNAs: Potential candidates for diagnosis and treatment of colorectal cancer. J. Cell Physiol. 2018, 233, 901–913. [Google Scholar] [CrossRef]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer-Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef]
- Ma, L. Role of miR-10b in breast cancer metastasis. Breast Cancer Res. 2010, 12, 210. [Google Scholar] [CrossRef]
- Subramanian, N.; Kanwar, J.R.; Kanwar, R.K.; Krishnakumar, S. Blocking the maturation of OncomiRNAs using pri-miRNA-17∼92 aptamer in retinoblastoma. Nucleic Acid. Ther. 2015, 25, 47–52. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Li, Z.X.; Zhu, Q.N.; Zhang, H.B.; Hu, Y.; Wang, G.; Zhu, Y.S. MALAT1: A potential biomarker in cancer. Cancer Manag. Res. 2018, 10, 6757–6768. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, L.; Wang, Y.; Li, H.; Ren, X.; Wei, F.; Yu, W.; Wang, X.; Yu, J.; Hao, X. Long noncoding RNA HOTAIR involvement in cancer. Tumour Biol. 2014, 35, 9531–9538. [Google Scholar] [CrossRef]
- Redis, R.S.; Sieuwerts, A.M.; Look, M.P.; Tudoran, O.; Ivan, C.; Spizzo, R.; Zhang, X.; de Weerd, V.; Shimizu, M.; Ling, H.; et al. CCAT2, a novel long non-coding RNA in breast cancer: Expression study and clinical correlations. Oncotarget 2013, 4, 1748–1762. [Google Scholar] [CrossRef]
- Rampal, R.; Alkalin, A.; Madzo, J.; Vasanthakumar, A.; Pronier, E.; Patel, J.; Li, Y.; Ahn, J.; Abdel-Wahab, O.; Shih, A.; et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014, 9, 1841–1855. [Google Scholar] [CrossRef]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton-Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef] [PubMed]
- Tovy, A.; Spiro, A.; McCarthy, R.; Shipony, Z.; Aylon, Y.; Allton, K.; Ainbinder, E.; Furth, N.; Tanay, A.; Barton, M.; et al. p53 is essential for DNA methylation homeostasis in naïve embryonic stem cells, and its loss promotes clonal heterogeneity. Genes. Dev. 2017, 31, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.H.; Shah, S.R.; Kim, H.N.; Kshitiz; Kim, P.; Quiñones-Hinojosa, A.; Levchenko, A. Switch-like enhancement of epithelial-mesenchymal transition by YAP through feedback regulation of WT1 and Rho-family GTPases. Nat. Commun. 2019, 10, 2797. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.; da Costa, L.T.; Pinto-de-Sousa, J.; Duluc, I.; Freund, J.N.; David, L.; Almeida, R. CDX2 autoregulation in human intestinal metaplasia of the stomach: Impact on the stability of the phenotype. Gut 2011, 60, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Del Poggetto, E.; Ho, I.L.; Balestrieri, C.; Yen, E.Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.Y.; et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 2021, 373, eabj0486. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Rodríguez-Ubreva, J.; Ballestar, E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin. Immunol. 2018, 196, 64–71. [Google Scholar] [CrossRef]
- Stoitzner, P.; Green, L.K.; Jung, J.Y.; Price, K.M.; Atarea, H.; Kivell, B.; Ronchese, F. Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol. Immunother. 2008, 57, 1665–1673. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, H.; Xie, R.; Wang, H.; Li, S.; Xu, Q.; Xu, N.; Cheng, Q.; Qian, Y.; Huang, R.; et al. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol. Oncol. 2019, 13, 873–893. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Rosenzweig, J.M.; Glenn, J.D.; Calabresi, P.A.; Whartenby, K.A. KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J. Biol. Chem. 2013, 288, 23868–23874. [Google Scholar] [CrossRef]
- Choi, Y.E.; Yu, H.N.; Yoon, C.H.; Bae, Y.S. Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur. J. Immunol. 2009, 39, 858–868. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013, 34, 216–223. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Zhao, J.; Olson, J.; Adams, A.; Taylor, R.; Bridges, B.; Marshall, L.; Roberts, B.; Weinman, S.A. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor γ-dependent macrophage differentiation. J. Biol. Chem. 2017, 292, 6882–6894. [Google Scholar] [CrossRef] [PubMed]
- Mullican, S.E.; Gaddis, C.A.; Alenghat, T.; Nair, M.G.; Giacomin, P.R.; Everett, L.J.; Feng, D.; Steger, D.J.; Schug, J.; Artis, D.; et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes. Dev. 2011, 25, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wei, J.; Zhong, L.; Shi, M.; Zhou, P.; Zuo, S.; Wu, K.; Zhu, M.; Huang, X.; Yu, Y.; et al. Cross talk between histone deacetylase 4 and STAT6 in the transcriptional regulation of arginase 1 during mouse dendritic cell differentiation. Mol. Cell Biol. 2015, 35, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Dunddel, D.; Pan, F.; Zeng, Q.; Gorbounov, M.; Albesiano, E.; Fu, J.; Blosser, R.L.; Tam, A.J.; Bruno, T.; Zhang, H.; et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J. Clin. Investig. 2013, 123, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Lienlaf, M.; Perez-Villarroel, P.; Wang, H.W.; Lee, C.; Woan, K.; Woods, D.; Knox, T.; Bergman, J.; Pinilla-Ibarz, J.; et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 2014, 60, 44–53. [Google Scholar] [CrossRef]
- De Almeida Nagata, D.E.; Chiang, E.Y.; Jhunjhunwala, S.; Caplazi, P.; Arumugam, V.; Modrusan, Z.; Chan, E.; Merchant, M.; Jin, L.; Arnott, D.; et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019, 27, 269–281.e4. [Google Scholar] [CrossRef]
- Youn, J.I.; Kumar, V.; Collazo, M.; Nefedova, Y.; Condamine, T.; Cheng, P.; Villagra, A.; Antonia, S.; McCaffrey, J.C.; Fishman, M.; et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat. Immunol. 2013, 14, 211–220. [Google Scholar] [CrossRef]
- Zhao, N.H.; Qian, Y.; Wu, C.S.; Wang, J.W.; Fang, Y.; Fan, X.P.; Gao, S.; Fan, Y.C.; Wang, K. Diagnostic value of NKG2D promoter methylation in hepatitis B virus-associated hepatocellular carcinoma. Biomark. Med. 2019, 13, 1093–1105. [Google Scholar] [CrossRef]
- Bugide, S.; Green, M.R.; Wajapeyee, N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl. Acad. Sci. USA 2018, 115, E3509–E3518. [Google Scholar] [CrossRef]
- Zhao, D.; Zhang, Q.; Liu, Y.; Li, X.; Zhao, K.; Ding, Y.; Li, Z.; Shen, Q.; Wang, C.; Li, N.; et al. H3K4me3 Demethylase Kdm5a Is Required for NK Cell Activation by Associating with p50 to Suppress SOCS1. Cell Rep. 2020, 30, 2460. [Google Scholar] [CrossRef] [PubMed]
- Stephen, T.L.; Payne, K.K.; Chaurio, R.A.; Allegrezza, M.J.; Zhu, H.; Perez-Sanz, J.; Perales-Puchalt, A.; Nguyen, J.M.; Vara-Ailor, A.E.; Eruslanov, E.B.; et al. SATB1 Expression Governs Epigenetic Repression of PD-1 in Tumor-Reactive T Cells. Immunity 2017, 46, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Cheng, S.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct epigenetic features of tumor-reactive CD8+ T cells in colorectal cancer patients revealed by genome-wide DNA methylation analysis. Genome Biol. 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Oncoimmunology 2018, 7, e1393596. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, R.; Deng, Y.; Qian, J.; Lu, Z.; Wang, Y.; Zhang, D.; Luo, F.; Chu, Y. MiR-15a/16 deficiency enhances anti-tumor immunity of glioma-infiltrating CD8+ T cells through targeting mTOR. Int. J. Cancer 2017, 141, 2082–2092. [Google Scholar] [CrossRef]
- Ban, Y.H.; Oh, S.C.; Seo, S.H.; Kim, S.M.; Choi, I.P.; Greenberg, P.D.; Chang, J.; Kim, T.D.; Ha, S.J. miR-150-Mediated Foxo1 Regulation Programs CD8. Cell Rep. 2017, 20, 2598–2611. [Google Scholar] [CrossRef]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef]
- Zhou, X.; Yu, S.; Zhao, D.M.; Harty, J.T.; Badovinac, V.P.; Xue, H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010, 33, 229–240. [Google Scholar] [CrossRef]
- DuPage, M.; Chopra, G.; Quiros, J.; Rosenthal, W.L.; Morar, M.M.; Holohan, D.; Zhang, R.; Turka, L.; Marson, A.; Bluestone, J.A. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 2015, 42, 227–238. [Google Scholar] [CrossRef]
- Marson, A.; Kretschmer, K.; Frampton, G.M.; Jacobsen, E.S.; Polansky, J.K.; MacIsaac, K.D.; Levine, S.S.; Fraenkel, E.; von Boehmer, H.; Young, R.A. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 2007, 445, 931–935. [Google Scholar] [CrossRef]
- Morikawa, H.; Ohkura, N.; Vandenbon, A.; Itoh, M.; Nagao-Sato, S.; Kawaji, H.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Forrest, A.R.; et al. Differential roles of epigenetic changes and Foxp3 expression in regulatory T cell-specific transcriptional regulation. Proc. Natl. Acad. Sci. USA 2014, 111, 5289–5294. [Google Scholar] [CrossRef]
- Key, T.J.; Allen, N.E.; Spencer, E.A.; Travis, R.C. The effect of diet on risk of cancer. Lancet 2002, 360, 861–868. [Google Scholar] [CrossRef]
- Newmark, H.L.; Yang, K.; Kurihara, N.; Fan, K.; Augenlicht, L.H.; Lipkin, M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: A preclinical model for human sporadic colon cancer. Carcinogenesis 2009, 30, 88–92. [Google Scholar] [CrossRef]
- Trichopoulou, A.; Bamia, C.; Lagiou, P.; Trichopoulos, D. Conformity to traditional Mediterranean diet and breast cancer risk in the Greek EPIC (European Prospective Investigation into Cancer and Nutrition) cohort. Am. J. Clin. Nutr. 2010, 92, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Ames, B.N. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutat. Res. 2001, 475, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Pietrocola, F.; Eisenberg, T.; Kong, Y.; Malik, S.A.; Andryushkova, A.; Schroeder, S.; Pendl, T.; Harger, A.; Niso-Santano, M.; et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 2014, 53, 710–725. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Doerner, S.K.; Reis, E.S.; Leung, E.S.; Ko, J.S.; Heaney, J.D.; Berger, N.A.; Lambris, J.D.; Nadeau, J.H. High-Fat Diet-Induced Complement Activation Mediates Intestinal Inflammation and Neoplasia, Independent of Obesity. Mol. Cancer Res. 2016, 14, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Lamas, O.; Marti, A.; Martínez, J.A. Obesity and immunocompetence. Eur. J. Clin. Nutr. 2002, 56 (Suppl. S3), S42–S45. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Delacre, M.; Abboud, G.; Ouk, T.S.; Delanoye, A.; Verwaerde, C.; Saule, P.; Wolowczuk, I. Impairment of dendritic cell functionality and steady-state number in obese mice. J. Immunol. 2006, 177, 5997–6006. [Google Scholar] [CrossRef]
- Conroy, M.J.; Dunne, M.R.; Donohoe, C.L.; Reynolds, J.V. Obesity-associated cancer: An immunological perspective. Proc. Nutr. Soc. 2016, 75, 125–138. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Mocellin, S.; Briarava, M.; Pilati, P. Vitamin B6 and Cancer Risk: A Field Synopsis and Meta-Analysis. J. Natl. Cancer Inst. 2017, 109, djw230. [Google Scholar] [CrossRef]
- Dou, R.; Ng, K.; Giovannucci, E.L.; Manson, J.E.; Qian, Z.R.; Ogino, S. Vitamin D and colorectal cancer: Molecular, epidemiological and clinical evidence. Br. J. Nutr. 2016, 115, 1643–1660. [Google Scholar] [CrossRef]
- Goodwin, P.J.; Ennis, M.; Pritchard, K.I.; Koo, J.; Hood, N. Prognostic effects of 25-hydroxyvitamin D levels in early breast cancer. J. Clin. Oncol. 2009, 27, 3757–3763. [Google Scholar] [CrossRef] [PubMed]
- Tretli, S.; Hernes, E.; Berg, J.P.; Hestvik, U.E.; Robsahm, T.E. Association between serum 25(OH)D and death from prostate cancer. Br. J. Cancer 2009, 100, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Stampfer, M.J.; Speizer, F.E.; Giovannucci, E.L. The influence of folate and multivitamin use on the familial risk of colon cancer in women. Cancer Epidemiol. Biomark. Prev. 2002, 11, 227–234. [Google Scholar]
- Shrubsole, M.J.; Jin, F.; Dai, Q.; Shu, X.O.; Potter, J.D.; Hebert, J.R.; Gao, Y.T.; Zheng, W. Dietary folate intake and breast cancer risk: Results from the Shanghai Breast Cancer Study. Cancer Res. 2001, 61, 7136–7141. [Google Scholar]
- Stolzenberg-Solomon, R.Z.; Pietinen, P.; Barrett, M.J.; Taylor, P.R.; Virtamo, J.; Albanes, D. Dietary and other methyl-group availability factors and pancreatic cancer risk in a cohort of male smokers. Am. J. Epidemiol. 2001, 153, 680–687. [Google Scholar] [CrossRef] [PubMed]
- Donnenfeld, M.; Deschasaux, M.; Latino-Martel, P.; Diallo, A.; Galan, P.; Hercberg, S.; Ezzedine, K.; Touvier, M. Prospective association between dietary folate intake and skin cancer risk: Results from the Supplémentation en Vitamines et Minéraux Antioxydants cohort. Am. J. Clin. Nutr. 2015, 102, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Sun, J.; Chen, F.; Wu, G. Potential effects of gut microbiota on host cancers: Focus on immunity, DNA damage, cellular pathways, and anticancer therapy. ISME J. 2023, 17, 1535–1551. [Google Scholar] [CrossRef]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef]
- Bonnesen, K.; Schmidt, M. Recategorization of Non-Aspirin Nonsteroidal Anti-inflammatory Drugs According to Clinical Relevance: Abandoning the Traditional NSAID Terminology. Can. J. Cardiol. 2021, 37, 1705–1707. [Google Scholar] [CrossRef]
- Sun, L.; Chen, K.; Jiang, Z.; Chen, X.; Ma, J.; Ma, Q.; Duan, W. Indometacin inhibits the proliferation and activation of human pancreatic stellate cells through the downregulation of COX-2. Oncol. Rep. 2018, 39, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Zhang, L.H.; Gao, J.H.; Zhao, C.; Tong, H.; Ye, C.; Huang, Z.Y.; Liu, R.; Tang, C.W. Suppressing growth and invasion of human hepatocellular carcinoma cells by celecoxib through inhibition of cyclooxygenase-2. Cancer Manag. Res. 2019, 11, 2831–2848. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/β-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Akrami, H.; Aminzadeh, S.; Fallahi, H. Inhibitory effect of ibuprofen on tumor survival and angiogenesis in gastric cancer cell. Tumour Biol. 2015, 36, 3237–3243. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Feng, J.; Liu, Y.; Wang, T.; Zhao, M.; Ye, L.; Zhang, X. Aspirin suppresses the abnormal lipid metabolism in liver cancer cells via disrupting an NFκB-ACSL1 signaling. Biochem. Biophys. Res. Commun. 2017, 486, 827–832. [Google Scholar] [CrossRef]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New aspects of an old drug--diclofenac targets MYC and glucose metabolism in tumor cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.; Palmer, J.R.; Zauber, A.G.; Warshauer, M.E.; Stolley, P.D.; Shapiro, S. A hypothesis: Nonsteroidal anti-inflammatory drugs reduce the incidence of large-bowel cancer. J. Natl. Cancer Inst. 1991, 83, 355–358. [Google Scholar] [CrossRef]
- Trelle, S.; Reichenbach, S.; Wandel, S.; Hildebrand, P.; Tschannen, B.; Villiger, P.M.; Egger, M.; Jüni, P. Cardiovascular safety of non-steroidal anti-inflammatory drugs: Network meta-analysis. BMJ 2011, 342, c7086. [Google Scholar] [CrossRef]
- Young, S.D.; Whissell, M.; Noble, J.C.; Cano, P.O.; Lopez, P.G.; Germond, C.J. Phase II clinical trial results involving treatment with low-dose daily oral cyclophosphamide, weekly vinblastine, and rofecoxib in patients with advanced solid tumors. Clin. Cancer Res. 2006, 12, 3092–3098. [Google Scholar] [CrossRef]
- Reyners, A.K.L.; de Munck, L.; Erdkamp, F.L.G.; Smit, W.M.; Hoekman, K.; Lalisang, R.I.; de Graaf, H.; Wymenga, A.N.M.; Polee, M.; Hollema, H.; et al. A randomized phase II study investigating the addition of the specific COX-2 inhibitor celecoxib to docetaxel plus carboplatin as first-line chemotherapy for stage IC to IV epithelial ovarian cancer, Fallopian tube or primary peritoneal carcinomas: The DoCaCel study. Ann. Oncol. 2012, 23, 2896–2902. [Google Scholar]
- Perroud, H.A.; Alasino, C.M.; Rico, M.J.; Mainetti, L.E.; Queralt, F.; Pezzotto, S.M.; Rozados, V.R.; Scharovsky, O.G. Metastatic breast cancer patients treated with low-dose metronomic chemotherapy with cyclophosphamide and celecoxib: Clinical outcomes and biomarkers of response. Cancer Chemother. Pharmacol. 2016, 77, 365–374. [Google Scholar] [CrossRef]
- Legge, F.; Paglia, A.; D’Asta, M.; Fuoco, G.; Scambia, G.; Ferrandina, G. Phase II study of the combination carboplatin plus celecoxib in heavily pre-treated recurrent ovarian cancer patients. BMC Cancer 2011, 11, 214. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ghonim, M.A.; Ibba, S.V.; Tarhuni, A.F.; Errami, Y.; Luu, H.H.; Dean, M.J.; El-Bahrawy, A.H.; Wyczechowska, D.; Benslimane, I.A.; Del Valle, L.; et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J. Immunother. Cancer 2021, 9, e001643. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Pusztai, L.; Yau, C.; Wolf, D.M.; Han, H.S.; Du, L.; Wallace, A.M.; String-Reasor, E.; Boughey, J.C.; Chien, A.J.; Elias, A.D.; et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 2021, 39, 989–998.e5. [Google Scholar] [CrossRef]
- Jin, M.H.; Nam, A.R.; Park, J.E.; Bang, J.H.; Bang, Y.J.; Oh, D.Y. Therapeutic Co-targeting of WEE1 and ATM Downregulates PD-L1 Expression in Pancreatic Cancer. Cancer Res. Treat. 2020, 52, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhou, M.; Bao, X.; Pan, D.; Jiao, M.; Liu, X.; Li, F.; Li, C.Y. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J. Clin. Investig. 2021, 131, e139333. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef]
- Schoonen, P.M.; Kok, Y.P.; Wierenga, E.; Bakker, B.; Foijer, F.; Spierings, D.C.J.; van Vugt, M.A.T.M. Premature mitotic entry induced by ATR inhibition potentiates olaparib inhibition-mediated genomic instability, inflammatory signaling, and cytotoxicity in BRCA2-deficient cancer cells. Mol. Oncol. 2019, 13, 2422–2440. [Google Scholar] [CrossRef]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Wang, G.; Li, W.; Zhang, L.; Wang, R.; Huang, Y.; Du, L.; Jiang, J.; Wu, C.; He, X.; et al. Histone deacetylase inhibitors prevent activation-induced cell death and promote anti-tumor immunity. Oncogene 2015, 34, 5960–5970. [Google Scholar] [CrossRef]
- Hicks, K.C.; Knudson, K.M.; Lee, K.L.; Hamilton, D.H.; Hodge, J.W.; Figg, W.D.; Ordentlich, P.; Jones, F.R.; Rabizadeh, S.; Soon-Shiong, P.; et al. Cooperative Immune-Mediated Mechanisms of the HDAC Inhibitor Entinostat, an IL15 Superagonist, and a Cancer Vaccine Effectively Synergize as a Novel Cancer Therapy. Clin. Cancer Res. 2020, 26, 704–716. [Google Scholar] [CrossRef]
- Loo Yau, H.; Bell, E.; Ettayebi, I.; de Almeida, F.C.; Boukhaled, G.M.; Shen, S.Y.; Allard, D.; Morancho, B.; Marhon, S.A.; Ishak, C.A.; et al. DNA hypomethylating agents increase activation and cytolytic activity of CD8. Mol. Cell 2021, 81, 1469–1483.e8. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenet. 2021, 13, 166. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Marampon, F.; Popov, V.M.; Pestell, R.G.; Zani, B.M.; Tombolini, V. Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol. Cancer 2010, 9, 305. [Google Scholar] [CrossRef]
- Nepali, K.; Liou, J.P. Recent developments in epigenetic cancer therapeutics: Clinical advancement and emerging trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Tiago, M.; Capparelli, C.; Erkes, D.A.; Purwin, T.J.; Heilman, S.A.; Berger, A.C.; Davies, M.A.; Aplin, A.E. Targeting BRD/BET proteins inhibits adaptive kinome upregulation and enhances the effects of BRAF/MEK inhibitors in melanoma. Br. J. Cancer 2020, 122, 789–800. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Pan, Z.; Zhao, Y.; Wang, X.; Xie, X.; Liu, M.; Zhang, K.; Wang, L.; Bai, D.; Foster, L.J.; Shu, R.; et al. Targeting bromodomain-containing proteins: Research advances of drug discovery. Mol. Biomed. 2023, 4, 13. [Google Scholar] [CrossRef]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.R.; Falcone, J.N.; Cantley, L.C.; Goncalves, M.D. Developing dietary interventions as therapy for cancer. Nat. Rev. Cancer 2022, 22, 452–466. [Google Scholar] [CrossRef]
- Kanarek, N.; Petrova, B.; Sabatini, D.M. Dietary modifications for enhanced cancer therapy. Nature 2020, 579, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Xia, S.; Xiang, J.; Li, Y.; Ross, M.O.; Lim, S.A.; Yang, F.; Tu, J.; Xie, L.; Dougherty, U.; et al. Trans-vaccenic acid reprograms CD8. Nature 2023, 623, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Fontana, L. Calorie restriction and cancer prevention: Metabolic and molecular mechanisms. Trends Pharmacol. Sci. 2010, 31, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Lee, C.; Safdie, F.M.; Wei, M.; Madia, F.; Bianchi, G.; Longo, V.D. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc. Natl. Acad. Sci. USA 2008, 105, 8215–8220. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Safdie, F.; Bianchi, G.; Dorff, T.; Fontana, L.; Longo, V.D. Fasting and differential chemotherapy protection in patients. Cell Cycle 2010, 9, 4474–4476. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Raffaghello, L.; Brandhorst, S.; Safdie, F.M.; Bianchi, G.; Martin-Montalvo, A.; Pistoia, V.; Wei, M.; Hwang, S.; Merlino, A.; et al. Fasting cycles retard growth of tumors and sensitize a range of cancer cell types to chemotherapy. Sci. Transl. Med. 2012, 4, 124ra127. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; Vander Heiden, M.G. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Salvadori, G.; Zanardi, F.; Iannelli, F.; Lobefaro, R.; Vernieri, C.; Longo, V.D. Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab. 2021, 33, 2247–2259.e6. [Google Scholar] [CrossRef]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e5. [Google Scholar] [CrossRef]
- Lu, Z.; Xie, J.; Wu, G.; Shen, J.; Collins, R.; Chen, W.; Kang, X.; Luo, M.; Zou, Y.; Huang, L.J.; et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat. Med. 2017, 23, 79–90. [Google Scholar] [CrossRef]
- Amelio, I.; Melino, G.; Frezza, C. Exploiting tumour addiction with a serine and glycine-free diet. Cell Death Differ. 2017, 24, 1311–1313. [Google Scholar] [CrossRef]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; de Braud, F.; et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 2332. [Google Scholar] [CrossRef]
- Komninou, D.; Leutzinger, Y.; Reddy, B.S.; Richie, J.P. Methionine restriction inhibits colon carcinogenesis. Nutr. Cancer 2006, 54, 202–208. [Google Scholar] [CrossRef]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Lozano, T.; Exposito, F.; Calvo, A.; Valencia, K.; Redrado, M.; Remírez, A.; Lecanda, F.; Alignani, D.; et al. Short-term starvation reduces IGF-1 levels to sensitize lung tumors to PD-1 immune checkpoint blockade. Nat. Cancer 2020, 1, 75–85. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Q.; Liao, Q.; Li, M.; Zhang, P.; Santos, H.O.; Kord-Varkaneh, H.; Abshirini, M. Effects of intermittent fasting diets on plasma concentrations of inflammatory biomarkers: A systematic review and meta-analysis of randomized controlled trials. Nutrition 2020, 79–80, 110974. [Google Scholar] [CrossRef]
- Cortellino, S.; Quagliariello, V.; Delfanti, G.; Blaževitš, O.; Chiodoni, C.; Maurea, N.; Di Mauro, A.; Tatangelo, F.; Pisati, F.; Shmahala, A.; et al. Fasting mimicking diet in mice delays cancer growth and reduces immunotherapy-associated cardiovascular and systemic side effects. Nat. Commun. 2023, 14, 5529. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.Q.; Huang, J.T.; Zhang, S.L.; Kong, C.; Li, Z.M.; Jing, H.; Chen, H.L.; Kong, C.Y.; Huang, S.H.; Cai, P.R.; et al. The antitumour effects of caloric restriction are mediated by the gut microbiome. Nat. Metab. 2023, 5, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fu, L.; Li, Y.; Wang, W.; Gong, M.; Zhang, J.; Dong, X.; Huang, J.; Wang, Q.; Mackay, C.R.; et al. Gut microbial metabolites facilitate anticancer therapy efficacy by modulating cytotoxic CD8. Cell Metab. 2021, 33, 988–1000.e7. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Pallavi, R.; Gatti, E.; Durfort, T.; Stendardo, M.; Ravasio, R.; Leonardi, T.; Falvo, P.; Duso, B.A.; Punzi, S.; Xieraili, A.; et al. Caloric restriction leads to druggable LSD1-dependent cancer stem cells expansion. Nat. Commun. 2024, 15, 828. [Google Scholar] [CrossRef] [PubMed]
- Da Prat, V.; Casirati, A.; Colombo, E.; Preziati, G.; Perrone, L.; Serra, F.; Caccialanza, R.; Pedrazzoli, P. The complex reality of malnutrition management in Oncology. Ann. Res. Oncol. 2023, 3, 59–62. [Google Scholar] [CrossRef]
- Pedrazzoli, P.; Rosti, G.; Caccialanza, R. A novel and definitive approach to nutrition in prevention and treatment of cancer: A provocative call. Ann. Res. Oncol. 2022, 2, 209–210. [Google Scholar] [CrossRef]
- Esposito, T.; Cortellino, S. Calorie restriction and periodic fasting from rodent to human: Lost in translation? Ann. Res. Oncol. 2024, in press. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mecca, M.; Picerno, S.; Cortellino, S. The Killer’s Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer. Int. J. Mol. Sci. 2024, 25, 2750. https://doi.org/10.3390/ijms25052750
Mecca M, Picerno S, Cortellino S. The Killer’s Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer. International Journal of Molecular Sciences. 2024; 25(5):2750. https://doi.org/10.3390/ijms25052750
Chicago/Turabian StyleMecca, Marisabel, Simona Picerno, and Salvatore Cortellino. 2024. "The Killer’s Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer" International Journal of Molecular Sciences 25, no. 5: 2750. https://doi.org/10.3390/ijms25052750
APA StyleMecca, M., Picerno, S., & Cortellino, S. (2024). The Killer’s Web: Interconnection between Inflammation, Epigenetics and Nutrition in Cancer. International Journal of Molecular Sciences, 25(5), 2750. https://doi.org/10.3390/ijms25052750