
Unraveling the Immunopathological Landscape of Celiac Disease: A Comprehensive Review
 , , , and
, , , and
Abstract
:1. Introduction
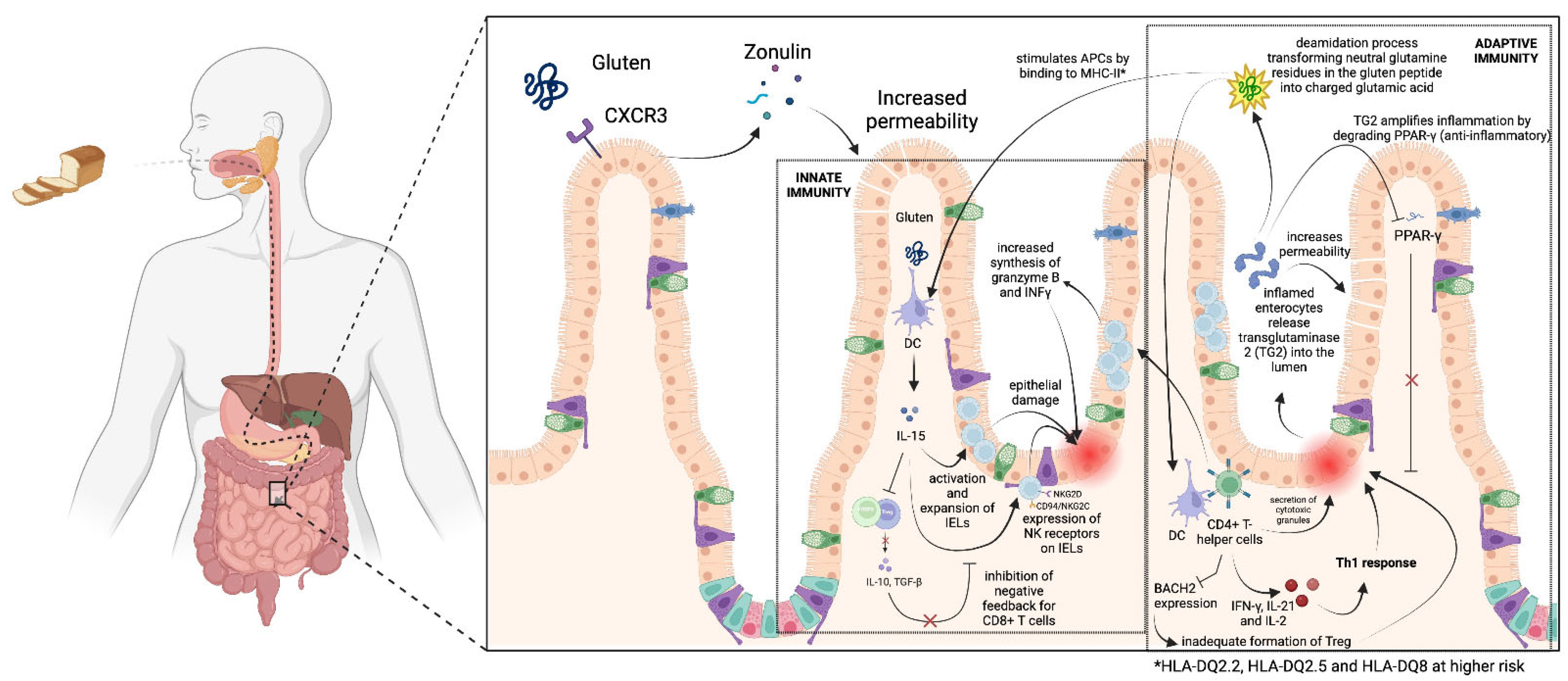
2. Immunological Cascade in Celiac Disease
2.1. Normal Immune Responses in the GI Tract
2.2. Innate Immune System
2.3. Adaptive Immune System
3. Refractory Celiac Disease: From Diagnosis to Lymphoma Progression
4. Extraintestinal Manifestations
5. Celiac Disease and Other Autoimmune Disorders and Some Common Pathways
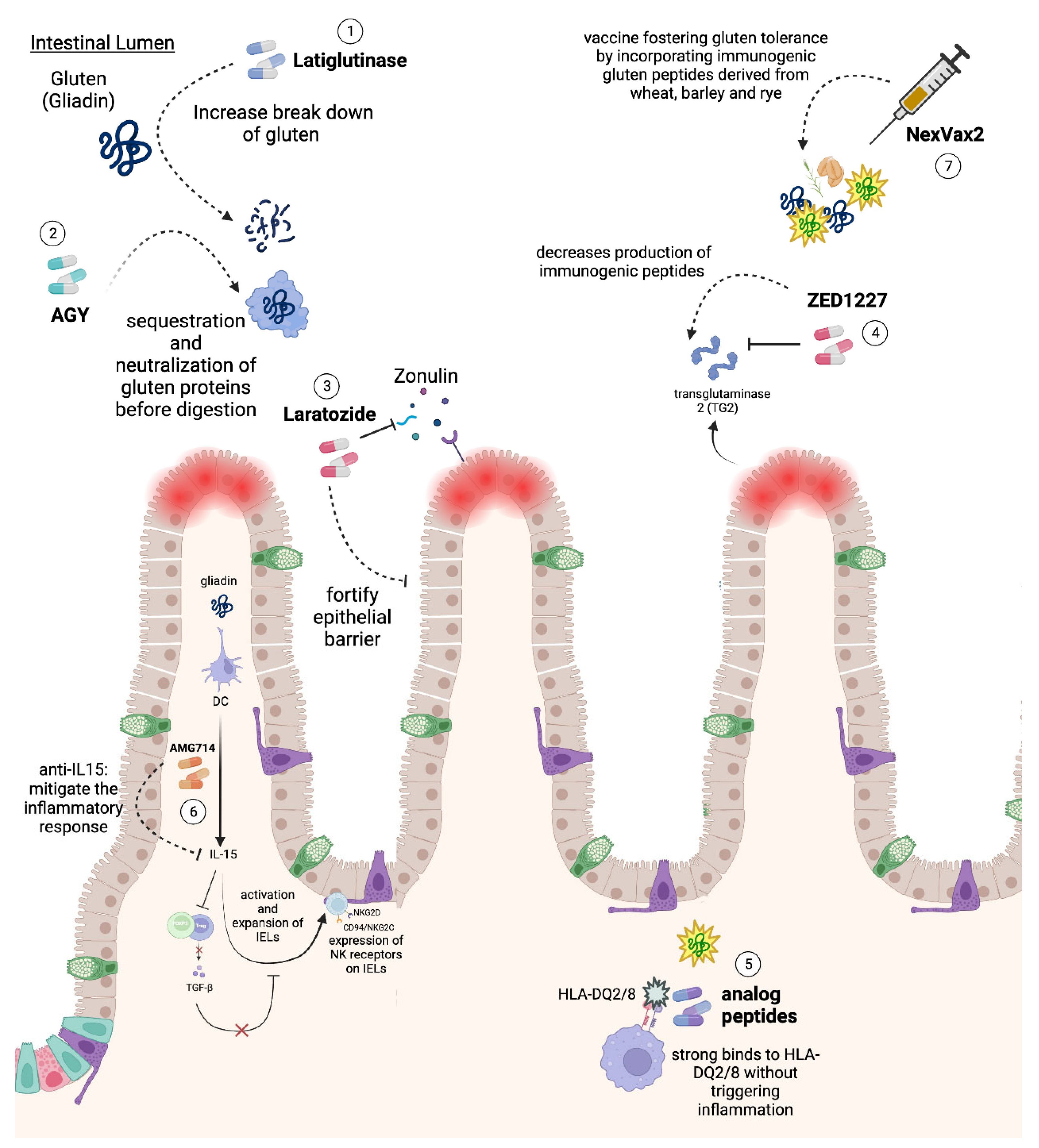
6. Novel Therapies

{kind=link}
{kind=link}
| Mechanism | Main Investigated Agent | Therapy Description |
|---|---|---|
| Gluten degradation | Latiglutenase (ALV003) [98,113] | Utilizes oral exogenous endopeptidases to more efficiently break down gluten proteins rich in glutamine and proline. |
| Gluten sequestration and neutralization | AGY (An oral egg yolk-derived anti-gliadin antibody) [101] | Engages in the preliminary neutralization and sequestration of gluten proteins before they undergo digestion, averting the generation of immunogenic peptides. |
| Enhancing intestinal epithelium integrity | Larazotide (AT1001) | A potential zonulin receptor antagonist aiming to fortify the epithelial barrier function by alleviating compromised tight junctions between epithelial cells |
| TG2 inhibition | ZED1227 [107,114] | An oral agent that selectively inhibits TG-2, a protein involved in the production of immunogenic peptides that are recognized by specific HLA markers on APCs |
| HLA-DQ2/8 binding | Analog peptides (molecules are currently in the preclinical research) [109,115] | A preclinical strategy that is focused on the development of analog peptides capable of strong binding to HLA-DQ2/8 without triggering inflammatory responses |
| Targeting IL-15 | AMG714 [110] | A therapeutic strategy leveraging an anti-IL-15 monoclonal antibody to potentially mitigate the inflammatory response central to CD pathogenesis |
| Gluten tolerance vaccine | Nexvax2 [116] | A vaccine strategy working to foster gluten tolerance by incorporating immunogenic gluten peptides derived from wheat, barley, and rye |
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ludvigsson, J.F.; Leffler, D.A.; Bai, J.C.; Biagi, F.; Fasano, A.; Green, P.H.R.; Hadjivassiliou, M.; Kaukinen, K.; Kelly, C.P.; Leonard, J.N.; et al. The Oslo definitions for coeliac disease and related terms. Gut 2013, 62, 43–52. [Google Scholar] [CrossRef]
- Sahin, Y. Celiac disease in children: A review of the literature. World J. Clin. Pediatr. 2021, 10, 53–71. [Google Scholar] [CrossRef]
- Vilppula, A.; Kaukinen, K.; Luostarinen, L.; Krekelä, I.; Patrikainen, H.; Valve, R.; Mäki, M.; Collin, P. Increasing prevalence and high incidence of celiac disease in elderly people: A population-based study. BMC Gastroenterol. 2009, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Caio, G.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Fasano, A. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Levescot, A.; Malamut, G.; Cerf-Bensussan, N. Immunopathogenesis and environmental triggers in coeliac disease. Gut 2022, 71, 2337–2349. [Google Scholar] [CrossRef]
- Raiteri, A.; Granito, A.; Giamperoli, A.; Catenaro, T.; Negrini, G.; Tovoli, F. Current guidelines for the management of celiac disease: A systematic review with comparative analysis. World J. Gastroenterol. 2022, 28, 154–176. [Google Scholar] [CrossRef]
- Catassi, C.; Fasano, A. Celiac disease diagnosis: Simple rules are better than complicated algorithms. Am. J. Med. 2010, 123, 691–693. [Google Scholar] [CrossRef] [PubMed]
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119. [Google Scholar] [CrossRef]
- Biesiekierski, J.R. What is gluten? J. Gastroenterol. Hepatol. 2017, 32, 78–81. [Google Scholar] [CrossRef]
- Garside, P.; Mowat, A.M.I. Oral tolerance. Semin. Immunol. 2001, 13, 177–185. [Google Scholar] [CrossRef]
- Commins, S.P. Mechanisms of Oral Tolerance. Pediatr. Clin. North Am. 2015, 62, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, B.; Ludvigsson, J.F.; Green, P.H.R. Celiac disease and non-celiac gluten sensitivity. BMJ 2015, 351, h4347. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann. N. Y. Acad. Sci. 2012, 1258, 25. [Google Scholar] [CrossRef]
- Stepniak, D.; Koning, F. Celiac disease—Sandwiched between innate and adaptive immunity. Hum. Immunol. 2006, 67, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Huang, X.; Yang, Y. Direct Action of Type I IFN on NK Cells Is Required for Their Activation in Response to Vaccinia Viral Infection In Vivo. J. Immunol. 2008, 180, 1592–1597. [Google Scholar] [CrossRef] [PubMed]
- Abadie, V.; Discepolo, V.; Jabri, B. Intraepithelial lymphocytes in celiac disease immunopathology. Semin. Immunopathol. 2012, 34, 551–556. [Google Scholar] [CrossRef]
- Benahmed, M.; Meresse, B.; Arnulf, B.; Barbe, U.; Mention, J.J.; Verkarre, V.; Allez, M.; Cellier, C.; Hermine, O.; Cerf–Bensussan, N. Inhibition of TGF-beta signaling by IL-15: A new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 2007, 132, 994–1008. [Google Scholar] [CrossRef]
- Ahmed, M.B.; Belhadj Hmida, N.; Moes, N.; Buyse, S.; Abdeladhim, M.; Louzir, H.; Cerf-Bensussan, N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J. Immunol. 2009, 182, 6763–6770. [Google Scholar] [CrossRef]
- Iversen, R.; Amundsen, S.F.; Kleppa, L.; du Pré, M.F.; Stamnaes, J.; Sollid, L.M. Evidence That Pathogenic Transglutaminase 2 in Celiac Disease Derives From Enterocytes. Gastroenterology 2020, 159, 788–790. [Google Scholar] [CrossRef]
- Molberg, Ø.; Mcadam, S.N.; Körner, R.; Quarsten, H.; Kristiansen, C.; Madsen, L.; Fugger, L.; Scott, H.; Norén, O.; Roepstorff, P.; et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998, 4, 713–717. [Google Scholar] [CrossRef]
- Qiao, S.W.; Bergseng, E.; Molberg, Ø.; Xia, J.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J. Immunol. 2004, 173, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Kårhus, L.L.; Thuesen, B.H.; Skaaby, T.; Rumessen, J.J.; Linneberg, A. The distribution of HLA DQ2 and DQ8 haplotypes and their association with health indicators in a general Danish population. United Eur. Gastroenterol. J. 2018, 6, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Mearin, M.L.; Biemond, I.; Pena, A.S.; Polanco, I.; Vazquez, C.; Schreuder, G.T.; de Vries, R.R.; van Rood, J.J. HLA-DR phenotypes in Spanish coeliac children: Their contribution to the understanding of the genetics of the disease. Gut 1983, 24, 532–537. [Google Scholar] [CrossRef]
- Bodd, M.; Ráki, M.; Tollefsen, S.; E Fallang, L.; Bergseng, E.; Lundin, K.E.A.; Sollid, L.M. HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol. 2010, 3, 594–601. [Google Scholar] [CrossRef] [PubMed]
- De Re, V.; Magris, R.; Cannizzaro, R. New Insights into the Pathogenesis of Celiac Disease. Front. Med. 2017, 4, 137. [Google Scholar] [CrossRef] [PubMed]
- Setty, M.; Discepolo, V.; Abadie, V.; Kamhawi, S.; Mayassi, T.; Kent, A.; Ciszewski, C.; Maglio, M.; Kistner, E.; Bhagat, G.; et al. Distinct and Synergistic Contributions of Epithelial Stress and Adaptive Immunity to Functions of Intraepithelial Killer Cells and Active Celiac Disease. Gastroenterology 2015, 149, 681–691.e10. [Google Scholar] [CrossRef] [PubMed]
- Eagar, T.N.; Miller, S.D. Helper T-Cell Subsets and Control of the Inflammatory Response. In Clinical Immunology: Principles and Practice, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 151–161. [Google Scholar]
- Klöck, C.; DiRaimondo, T.R.; Khosla, C. Role of Transglutaminase 2 in Celiac Disease Pathogenesis. Semin. Immunopathol. 2012, 34, 513–522. [Google Scholar] [CrossRef]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef]
- Brusca, I. Overview of Biomarkers for Diagnosis and Monitoring of Celiac Disease. Adv. Clin. Chem. 2015, 68, 1–55. [Google Scholar]
- Singh, P.M.; Kurray, L.D.; Agnihotri, A.M.; Das, P.; Verma, A.K.M.; Sreenivas, V.; Dattagupta, S.; Makharia, G.K.D. Titers of anti-tissue transglutaminase antibody correlate well with severity of villous abnormalities in celiac disease. J. Clin. Gastroenterol. 2015, 49, 212–217. [Google Scholar] [CrossRef]
- Aboulaghras, S.; Piancatelli, D.; Oumhani, K.; Balahbib, A.; Bouyahya, A.; Taghzouti, K. Pathophysiology and immunogenetics of celiac disease. Clin. Chim. Acta 2022, 528, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, B.; Qiao, S.-W.; Larsen, M.R.; Jung, G.; Roepstorff, P.; Sollid, L.M. Molecular characterization of covalent complexes between tissue transglutaminase and gliadin peptides. J. Biol. Chem. 2004, 279, 17607–17616. [Google Scholar] [CrossRef] [PubMed]
- Du Pré, M.F.; Sollid, L.M. T-cell and B-cell immunity in celiac disease. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Cellier, C.; Mulder, C.J.J. Refractory coeliac disease. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 413–424. [Google Scholar] [CrossRef]
- Patey-Mariaud De Serre, N.; Cellier, C.; Jabri, B.; Delabesse, E.; Verkarre, V.; Roche, B.; Lavergne, A.; Brière, J.; Mauvieux, L.; Leborgne, M.; et al. Distinction between coeliac disease and refractory sprue: A simple immunohistochemical method. Histopathology 2000, 37, 70–77. [Google Scholar] [CrossRef]
- Malamut, G.; Meresse, B.; Cellier, C.; Cerf-Bensussan, N. Refractory celiac disease: From bench to bedside. Semin. Immunopathol. 2012, 34, 601–613. [Google Scholar] [CrossRef]
- Malamut, G.; El Machhour, R.; Montcuquet, N.; Martin-Lannerée, S.; Dusanter-Fourt, I.; Verkarre, V.; Mention, J.J.; Rahmi, G.; Kiyono, H.; Butz, E.A.; et al. IL-15 triggers an antiapoptotic pathway in human intraepithelial lymphocytes that is a potential new target in celiac disease–associated inflammation and lymphomagenesis. J. Clin. Investig. 2010, 120, 2131–2143. [Google Scholar] [CrossRef]
- Malamut, G.; Afchain, P.; Verkarre, V.; Lecomte, T.; Amiot, A.; Damotte, D.; Bouhnik, Y.; Colombel, J.F.; Delchier, J.C.; Allez, M.; et al. Presentation and Long-Term Follow-up of Refractory Celiac Disease: Comparison of Type I with Type II. Gastroenterology 2009, 136, 81–90. [Google Scholar] [CrossRef]
- Meresse, B.; Curran, S.A.; Ciszewski, C.; Orbelyan, G.; Setty, M.; Bhagat, G.; Lee, L.; Tretiakova, M.; Semrad, C.; Kistner, E.; et al. Reprogramming of CTLs into natural killer–like cells in celiac disease. J. Exp. Med. 2006, 203, 1343–1355. [Google Scholar] [CrossRef]
- Rubio-Tapia, A.; Murray, J.A. Classification and management of refractory coeliac disease. Gut 2010, 59, 547–557. [Google Scholar] [CrossRef]
- Liu, H.; Brais, R.; Lavergne-Slove, A.; Jeng, Q.; Payne, K.; Ye, H.; Liu, Z.; Carreras, J.; Huang, Y.; Bacon, C.M.; et al. Continual monitoring of intraepithelial lymphocyte immunophenotype and clonality is more important than snapshot analysis in the surveillance of refractory coeliac disease. Gut 2010, 59, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Cording, S.; Lhermitte, L.; Malamut, G.; Berrabah, S.; Trinquand, A.; Guegan, N.; Villarese, P.; Kaltenbach, S.; Meresse, B.; Khater, S.; et al. Oncogenetic landscape of lymphomagenesis in coeliac disease. Gut 2021, 71, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, M.; Ferro, A.; Brascugli, I.; Mattivi, S.; Fagoonee, S.; Pellicano, R. Extra-Intestinal Manifestations of Celiac Disease: What Should We Know in 2022? J. Clin. Med. 2022, 11, 258. [Google Scholar] [CrossRef]
- Parzanese, I.; Qehajaj, D.; Patrinicola, F.; Aralica, M.; Chiriva-Internati, M.; Stifter, S.; Elli, L.; Grizzi, F. Celiac disease: From pathophysiology to treatment. World J. Gastrointest. Pathophysiol. 2017, 8, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Paez, M.A.; Gramelspacher, A.M.; Sinacore, J.; Winterfield, L.; Venu, M. Delay in Diagnosis of Celiac Disease in Patients Without Gastrointestinal Complaints. Am. J. Med. 2017, 130, 1318–1323. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.L.; Bardella, M.T. Bone in celiac disease. Osteoporos. Int. 2008, 19, 1705–1716. [Google Scholar] [CrossRef]
- Collin, P.; Salmi, T.T.; Hervonen, K.; Kaukinen, K.; Reunala, T. Dermatitis herpetiformis: A cutaneous manifestation of coeliac disease. Ann. Med. 2016, 49, 23–31. [Google Scholar] [CrossRef]
- Molteni, N.; Bardella, M.T.; Bianchi, P.A. Obstetric and gynecological problems in women with untreated celiac sprue. J. Clin. Gastroenterol. 1990, 12, 37–39. [Google Scholar] [CrossRef]
- Emilsson, L.; Andersson, B.; Elfström, P.; Green, P.H.; Ludvigsson, J.F.; James, S.; Askling, J.; Stenestrand, U.; Ingelsson, E.; Rusconi, P.; et al. Risk of idiopathic dilated cardiomyopathy in 29 000 patients with celiac disease. J. Am. Heart Assoc. 2012, 1, e001594. [Google Scholar] [CrossRef]
- Freeman, H.J. Endocrine manifestations in celiac disease. World J. Gastroenterol. 2016, 22, 8472–8479. [Google Scholar] [CrossRef]
- Wills, A.J. The neurology and neuropathology of coeliac disease. Neuropathol. Appl. Neurobiol. 2000, 26, 493–496. [Google Scholar] [CrossRef]
- Ungprasert, P.; Wijarnpreecha, K.; Kittanamongkolchai, W. Psoriasis and Risk of Celiac Disease: A Systematic Review and Meta-analysis. Indian J. Dermatol. 2017, 62, 41. [Google Scholar] [CrossRef]
- Persechino, F.; Galli, G.; Persechino, S.; Valitutti, F.; Zenzeri, L.; Mauro, A.; Corleto, V.D.; Parisi, P.; Ziparo, C.; Evangelisti, M.; et al. Skin Manifestations and Coeliac Disease in Paediatric Population. Nutrients 2021, 13, 3611. [Google Scholar] [CrossRef]
- Leffler, D.A.; Green, P.H.R.; Fasano, A. Extraintestinal manifestations of coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Caproni, M.; Antiga, E.; Melani, L.; Fabbri, P.; The Italian Group for Cutaneous Immunopathology. Guidelines for the diagnosis and treatment of dermatitis herpetiformis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Sárdy, M.; Kárpáti, S.; Merkl, B.; Paulsson, M.; Smyth, N. Epidermal Transglutaminase (TGase 3) Is the Autoantigen of Dermatitis Herpetiformis. J. Exp. Med. 2002, 195, 747–757. [Google Scholar] [CrossRef]
- Kárpáti, S.; Sárdy, M.; Németh, K.; Mayer, B.; Smyth, N.; Paulsson, M.; Traupe, H. Transglutaminases in autoimmune and inherited skin diseases: The phenomena of epitope spreading and functional compensation. Exp. Dermatol. 2018, 27, 807–814. [Google Scholar] [CrossRef]
- Didona, D.; Di Zenzo, G. Humoral epitope spreading in autoimmune bullous diseases. Front. Immunol. 2018, 9, 779. [Google Scholar] [CrossRef] [PubMed]
- Antiga, E.; Maglie, R.; Quintarelli, L.; Verdelli, A.; Bonciani, D.; Bonciolini, V.; Caproni, M. Dermatitis Herpetiformis: Novel Perspectives. Front. Immunol. 2019, 10, 1290. [Google Scholar] [CrossRef]
- Reunala, T.; Helin, H.; Pasternack, A.; Linder, E.; Kalimo, K. Renal involvement and circulating immune complexes in dermatitis herpetiformis. J. Am. Acad. Dermatol. 1983, 9, 219–223. [Google Scholar] [CrossRef]
- Hall, R.P.; Benbenisty, K.M.; Mickle, C.; Takeuchi, F.; Streilein, R.D. Serum IL-8 in Patients with Dermatitis Herpetiformis is Produced in Response to Dietary Gluten. J. Investig. Dermatol. 2007, 127, 2158–2165.e2. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Streilein, R.D.; Hall, R.P. Neutrophil CD11b, L-selectin and Fc IgA receptors in patients with dermatitis herpetiformis. Br. J. Dermatol. 2002, 147, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.P.; Takeuchi, F.; Benbenisty, K.M.; Streilein, R.D. Cutaneous Endothelial Cell Activation in Normal Skin of Patients with Dermatitis Herpetiformis Associated with Increased Serum Levels of IL-8, sE-Selectin, and TNF-α. Investig. Dermatol. 2006, 126, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Russo, V.; Klein, T.; Lim, D.J.; Solis, N.; Machado, Y.; Hiroyasu, S.; Nabai, L.; Shen, Y.; Zeglinski, M.R.; Zhao, H.; et al. Granzyme B is elevated in autoimmune blistering diseases and cleaves key anchoring proteins of the dermal-epidermal junction. Sci. Rep. 2018, 8, 9690. [Google Scholar] [CrossRef] [PubMed]
- Cooke, W.T.; Smith, W.T. Neurological Disorders Associated with Adult Celiac Disease. Brain 1966, 89, 683–722. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Croall, I.D.; Zis, P.; Sarrigiannis, P.G.; Sanders, D.S.; Aeschlimann, P.; Grünewald, R.A.; Armitage, P.A.; Connolly, D.; Aeschlimann, D.; et al. Neurologic Deficits in Patients with Newly Diagnosed Celiac Disease Are Frequent and Linked With Autoimmunity to Transglutaminase 6. Clin. Gastroenterol. Hepatol. 2019, 17, 2678–2686.e2. [Google Scholar] [CrossRef]
- Rouvroye, M.D.; Zis, P.; Van Dam, A.-M.; Rozemuller, A.J.M.; Bouma, G.; Hadjivassiliou, M. The Neuropathology of Gluten-Related Neurological Disorders: A Systematic Review. Nutrients 2020, 12, 822. [Google Scholar] [CrossRef]
- Pennisi, M.; Bramanti, A.; Cantone, M.; Pennisi, G.; Bella, R.; Lanza, G. Neurophysiology of the ‘celiac brain’: Disentangling gut-brain connections. Front. Neurosci. 2017, 11, 291045. [Google Scholar] [CrossRef]
- Cervio, E.; Volta, U.; Verri, M.; Boschi, F.; Pastoris, O.; Granito, A.; Barbara, G.; Parisi, C.; Felicani, C.; Tonini, M.; et al. Sera of patients with celiac disease and neurologic disorders evoke a mitochondrial-dependent apoptosis in vitro. Gastroenterology 2007, 133, 195–206. [Google Scholar] [CrossRef]
- Granito, A.; Tovoli, F.; Raiteri, A.; Volta, U. Anti-ganglioside antibodies and celiac disease. Allergy Asthma Clin. Immunol. 2021, 17, 53. [Google Scholar] [CrossRef]
- Cutillo, G.; Saariaho, A.-H.; Meri, S. Physiology of gangliosides and the role of antiganglioside antibodies in human diseases. Cell. Mol. Immunol. 2020, 17, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Aeschlimann, P.; Strigun, A.; Sanders, D.S.; Woodroofe, N.; Aeschlimann, D. Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann. Neurol. 2008, 64, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, M.; Gazzin, S.; Zoratti, C.; Llido, J.P.; Lanza, G.; Tiribelli, C.; Moretti, R. Celiac Disease and Neurological Manifestations: From Gluten to Neuroinflammation. Int. J. Mol. Sci. 2022, 23, 15564. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.A. The role of the thalamus in motor control. Curr. Opin. Neurobiol. 2003, 13, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Nanri, K.; Shibuya, M.; Taguchi, T.; Hasegawa, A.; Tanaka, N. Selective loss of Purkinje cells in a patient with anti-gliadin-antibody-positive autoimmune cerebellar ataxia. Diagn. Pathol. 2011, 6, 14. [Google Scholar] [CrossRef]
- Lai, T.-S.; Lindberg, R.A.; Zhou, H.-L.; Haroon, Z.A.; Dewhirst, M.W.; Hausladen, A.; Juang, Y.-L.; Stamler, J.S.; Greenberg, C.S. Endothelial cell-surface tissue transglutaminase inhibits neutrophil adhesion by binding and releasing nitric oxide. Sci. Rep. 2017, 7, 16163. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Grünewald, R.; Sanders, D.S.; Zis, P.; Croall, I.; Shanmugarajah, P.D.; Sarrigiannis, P.G.; Trott, N.; Wild, G.; Hoggard, N. The Significance of Low Titre Antigliadin Antibodies in the Diagnosis of Gluten Ataxia. Nutrients 2018, 10, 1444. [Google Scholar] [CrossRef]
- Volta, U.; Granito, A.; De Franceschi, L.; Petrolini, N.; Bianchi, F. Anti tissue transglutaminase antibodies as predictors of silent coeliac disease in patients with hypertransaminasaemia of unknown origin. Dig. Liver Dis. 2001, 33, 420–425. [Google Scholar] [CrossRef]
- Schuppan, D.; Ciccocioppo, R. Coeliac disease and secondary autoimmunity. Dig. Liver Dis. 2002, 34, 13–15. [Google Scholar] [CrossRef]
- Granito, A.; Muratori, P.; Cassani, F.; Pappas, G.; Muratori, L.; Agostinelli, D.; Veronesi, L.; Bortolotti, R.; Petrolini, N.; Bianchi, F.B.; et al. Anti-actin IgA antibodies in severe coeliac disease. Clin. Exp. Immunol. 2004, 137, 386–392. [Google Scholar] [CrossRef]
- Sollid, L.M.; Jabri, B. Celiac disease and transglutaminase 2: A model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr. Opin. Immunol. 2011, 23, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Baker, F.J.; Lee, M.; Chien, Y.-H.; Davis, M.M. Restricted islet-cell reactive T cell repertoire of early pancreatic islet infiltrates in NOD mice. Proc. Natl. Acad. Sci. USA 2002, 99, 9374–9379. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.R.; Rebelo, J.F.; Maximiano, C.; Gomes, M.M.; Martins, V.; Meireles, C.; Antunes, H.; Martins, S. HLA DQ2/DQ8 haplotypes and anti-transglutaminase antibodies as celiac disease markers in a pediatric population with type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2022, 66, 229–236. [Google Scholar] [CrossRef]
- Zauli, D.; Grassi, A.; Granito, A.; Foderaro, S.; De Franceschi, L.; Ballardini, G.; Bianchi, F.; Volta, U. Prevalence of silent coeliac disease in atopics. Dig. Liver Dis. 2000, 32, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Ashok, T.; Patni, N.; Fatima, M.; Lamis, A.; Siddiqui, S.W. Celiac Disease and Autoimmune Thyroid Disease: The Two Peas in a Pod. Cureus 2022, 14, e26243. [Google Scholar] [CrossRef]
- Collin, P.; Salmi, J.; Hällström, O.; Reunala, T.; Pasternack, A. Autoimmune thyroid disorders and coeliac disease. Eur. J. Endocrinol. 1994, 130, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Volta, U.; Ravaglia, G.; Granito, A.; Forti, P.; Maioli, F.; Petrolini, N.; Zoli, M.; Bianchi, F.B. Coeliac disease in patients with autoimmune thyroiditis. Digestion 2001, 64, 61–65. [Google Scholar] [CrossRef]
- Volta, U.; Rodrigo, L.; Granito, A.; Petrolini, N.; Muratori, P.; Muratori, L.; Linares, A.; Veronesi, L.; Fuentes, D.; Zauli, D.; et al. Celiac disease in autoimmune cholestatic liver disorders. Am. J. Gastroenterol. 2002, 97, 2609–2613. [Google Scholar] [CrossRef]
- Ashorn, S.; Raukola, H.; Välineva, T.; Ashorn, M.; Wei, B.; Braun, J.; Rantala, I.; Kaukinen, K.; Luukkaala, T.; Collin, P.; et al. Elevated serum anti-Saccharomyces cerevisiae, anti-I2 and anti-OmpW antibody levels in patients with suspicion of celiac disease. J. Clin. Immunol. 2008, 28, 486–494. [Google Scholar] [CrossRef]
- Granito, A.; Zauli, D.; Muratori, P.; Muratori, L.; Grassi, A.; Bortolotti, R.; Petrolini, N.; Veronesi, L.; Gionchetti, P.; Bianchi, F.B.; et al. Anti-Saccharomyces cerevisiae and perinuclear anti-neutrophil cytoplasmic antibodies in coeliac disease before and after gluten-free diet. Aliment. Pharmacol. Ther. 2005, 21, 881–887. [Google Scholar] [CrossRef]
- Granito, A.; Muratori, L.; Muratori, P.; Guidi, M.; Lenzi, M.; Bianchi, F.B.; Volta, U. Anti-saccharomyces cerevisiae antibodies (ASCA) in coeliac disease. Gut 2006, 55, 296. [Google Scholar]
- Machado, M.V. New Developments in Celiac Disease Treatment. Int. J. Mol. Sci. 2023, 24, 945. [Google Scholar] [CrossRef] [PubMed]
- Hall, N.J.; Rubin, G.; Charnock, A. Systematic review: Adherence to a gluten-free diet in adult patients with coeliac disease. Aliment. Pharmacol. Ther. 2009, 30, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Hollon, J.R.; Cureton, P.A.; Martin, M.L.; Puppa, E.L.L.; Fasano, A. Trace gluten contamination may play a role in mucosal and clinical recovery in a subgroup of diet-adherent non-responsive celiac disease patients. BMC Gastroenterol. 2013, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Rowinski, S.A.; Christensen, E. Epidemiologic and therapeutic aspects of refractory coeliac disease—A systematic review. Dan. Med. J. 2016, 63, A5307. [Google Scholar]
- Kivelä, L.; Caminero, A.; Leffler, D.A.; Pinto-Sanchez, M.I.; Tye-Din, J.A.; Lindfors, K. Current and emerging therapies for coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 18, 181–195. [Google Scholar] [CrossRef]
- Murray, J.A.; Kelly, C.P.; Green, P.H.; Marcantonio, A.; Wu, T.-T.; Mäki, M.; Adelman, D.C.; Ansari, S.; Ayub, K.; Basile, A.; et al. No Difference Between Latiglutenase and Placebo in Reducing Villous Atrophy or Improving Symptoms in Patients With Symptomatic Celiac Disease. Gastroenterology 2017, 152, 787–798.e2. [Google Scholar] [CrossRef]
- Lähdeaho, M.-L.; Kaukinen, K.; Laurila, K.; Vuotikka, P.; Koivurova, O.-P.; Kärjä-Lahdensuu, T.; Marcantonio, A.; Adelman, D.C.; Mäki, M. Glutenase ALV003 Attenuates Gluten-Induced Mucosal Injury in Patients With Celiac Disease. Gastroenterology 2014, 146, 1649–1658. [Google Scholar] [CrossRef]
- Murray, J.A.; Syage, J.A.; Wu, T.-T.; Dickason, M.A.; Ramos, A.G.; Van Dyke, C.; Horwath, I.; Lavin, P.T.; Mäki, M.; Hujoel, I.; et al. Latiglutenase Protects the Mucosa and Attenuates Symptom Severity in Patients With Celiac Disease Exposed to a Gluten Challenge. Gastroenterology 2022, 163, 1510–1521.e6. [Google Scholar] [CrossRef]
- Sample, D.A.; Sunwoo, H.H.; Huynh, H.Q.; Rylance, H.L.; Robert, C.L.; Xu, B.-W.; Kang, S.H.; Gujral, N.; Dieleman, L.A. AGY, a Novel Egg Yolk-Derived Anti-gliadin Antibody, Is Safe for Patients with Celiac Disease. Dig. Dis. Sci. 2016, 62, 1277–1285. [Google Scholar] [CrossRef]
- Yoosuf, S.; Makharia, G.K. Evolving therapy for celiac disease. Front. Pediatr. 2019, 7, 441843. [Google Scholar] [CrossRef] [PubMed]
- Paterson, B.M.; Lammers, K.M.; Arrieta, M.C.; Fasano, A.; Meddings, J.B. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: A proof of concept study. Aliment. Pharmacol. Ther. 2007, 26, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Hoilat, G.J.; Altowairqi, A.K.; Ayas, M.F.; Alhaddab, N.T.; Alnujaidi, R.A.; Alharbi, H.A.; Alyahyawi, N.; Kamal, A.; Alhabeeb, H.; Albazee, E.; et al. Larazotide acetate for treatment of celiac disease: A systematic review and meta-analysis of randomized controlled trials. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101782. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, S.; Durai, M.; Kitchens, K.; Tamiz, A.P.; Somerville, R.; Ginski, M.; Paterson, B.M.; Murray, J.A.; Verdu, E.F.; Alkan, S.S.; et al. Larazotide acetate regulates epithelial tight junctions in vitro and in vivo. Peptides 2012, 35, 86–94. [Google Scholar] [CrossRef]
- Valvano, M.; Fabiani, S.; Monaco, S.; Calabrò, M.; Mancusi, A.; Frassino, S.; Rolandi, C.; Mosca, M.; Faenza, S.; Sgamma, E.; et al. Old and New Adjunctive Therapies in Celiac Disease and Refractory Celiac Disease: A Review. Int. J. Mol. Sci. 2023, 24, 12800. [Google Scholar] [CrossRef]
- Schuppan, D.; Mäki, M.; Lundin, K.E.; Isola, J.; Friesing-Sosnik, T.; Taavela, J.; Popp, A.; Koskenpato, J.; Langhorst, J.; Hovde, Ø.; et al. A Randomized Trial of a Transglutaminase 2 Inhibitor for Celiac Disease. N. Engl. J. Med. 2021, 385, 35–45. [Google Scholar] [CrossRef]
- Xia, J.; Bergseng, E.; Fleckenstein, B.; Siegel, M.; Kim, C.-Y.; Khosla, C.; Sollid, L.M. Cyclic and dimeric gluten peptide analogues inhibiting DQ2-mediated antigen presentation in celiac disease. Bioorg. Med. Chem. 2007, 15, 6565–6573. [Google Scholar] [CrossRef]
- Kapoerchan, V.V.; Wiesner, M.; Hillaert, U.; Drijfhout, J.W.; Overhand, M.; Alard, P.; van der Marel, G.A.; Overkleeft, H.S.; Koning, F. Design, synthesis and evaluation of high-affinity binders for the celiac disease associated HLA-DQ2 molecule. Mol. Immunol. 2010, 47, 1091–1097. [Google Scholar] [CrossRef]
- Lähdeaho, M.-L.; Scheinin, M.; Vuotikka, P.; Taavela, J.; Popp, A.; Laukkarinen, J.; Koffert, J.; Koivurova, O.-P.; Pesu, M.; Kivelä, L.; et al. Safety and efficacy of AMG 714 in adults with coeliac disease exposed to gluten challenge: A phase 2a, randomised, double-blind, placebo-controlled study. Lancet Gastroenterol. Hepatol. 2019, 4, 948–959. [Google Scholar] [CrossRef]
- Daveson, A.J.M.; Ee, H.C.; Andrews, J.M.; King, T.; Goldstein, K.E.; Dzuris, J.L.; MacDougall, J.A.; Williams, L.J.; Treohan, A.; Cooreman, M.P.; et al. Epitope-Specific Immunotherapy Targeting CD4-Positive T Cells in Celiac Disease: Safety, Pharmacokinetics, and Effects on Intestinal Histology and Plasma Cytokines with Escalating Dose Regimens of Nexvax2 in a Randomized, Double-Blind, Placebo-Controlled Phase 1 Study. EBioMedicine 2017, 26, 78–90. [Google Scholar]
- Goel, G.; King, T.; Daveson, A.J.; Andrews, J.M.; Krishnarajah, J.; Krause, R.; Brown, G.J.E.; Fogel, R.; Barish, C.F.; Epstein, R.; et al. Epitope-specific immunotherapy targeting CD4-positive T cells in coeliac disease: Two randomised, double-blind, placebo-controlled phase 1 studies. Lancet Gastroenterol. Hepatol. 2017, 2, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.; Garber, M.E.; Spencer, A.G.; Botwick, W.; Kumar, P.; Williams, R.N.; Kozuka, K.; Shreeniwas, R.; Pratha, V.; Adelman, D.C. Safety, tolerability, and activity of ALV003: Results from two phase 1 single, escalating-dose clinical trials. Dig. Dis. Sci. 2011, 57, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Büchold, C.; Hils, M.; Gerlach, U.; Weber, J.; Pelzer, C.; Heil, A.; Aeschlimann, D.; Pasternack, R. Features of ZED1227: The First-In-Class Tissue Transglutaminase Inhibitor Undergoing Clinical Evaluation for the Treatment of Celiac Disease. Cells 2022, 11, 1667. [Google Scholar] [CrossRef] [PubMed]
- Huan, J.; Meza-Romero, R.; Mooney, J.L.; Vandenbark, A.A.; Offner, H.; Burrows, G.G. Single-chain recombinant HLA-DQ2.5/peptide molecules block α2-gliadin-specific pathogenic CD4+ T-cell proliferation and attenuate production of inflammatory cytokines: A potential therapy for celiac disease. Mucosal Immunol. 2011, 4, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Truitt, K.E.; Daveson, A.J.M.; Ee, H.C.; Goel, G.; MacDougall, J.; Neff, K.; Anderson, R.P. Randomised clinical trial: A placebo-controlled study of subcutaneous or intradermal NEXVAX2, an investigational immunomodulatory peptide therapy for coeliac disease. Aliment. Pharmacol. Ther. 2019, 50, 547–555. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patt, Y.S.; Lahat, A.; David, P.; Patt, C.; Eyade, R.; Sharif, K. Unraveling the Immunopathological Landscape of Celiac Disease: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 15482. https://doi.org/10.3390/ijms242015482
Patt YS, Lahat A, David P, Patt C, Eyade R, Sharif K. Unraveling the Immunopathological Landscape of Celiac Disease: A Comprehensive Review. International Journal of Molecular Sciences. 2023; 24(20):15482. https://doi.org/10.3390/ijms242015482
Chicago/Turabian StylePatt, Yonatan Shneor, Adi Lahat, Paula David, Chen Patt, Rowand Eyade, and Kassem Sharif. 2023. "Unraveling the Immunopathological Landscape of Celiac Disease: A Comprehensive Review" International Journal of Molecular Sciences 24, no. 20: 15482. https://doi.org/10.3390/ijms242015482