
Immune Fingerprint in Diabetes: Ocular Surface and Retinal Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
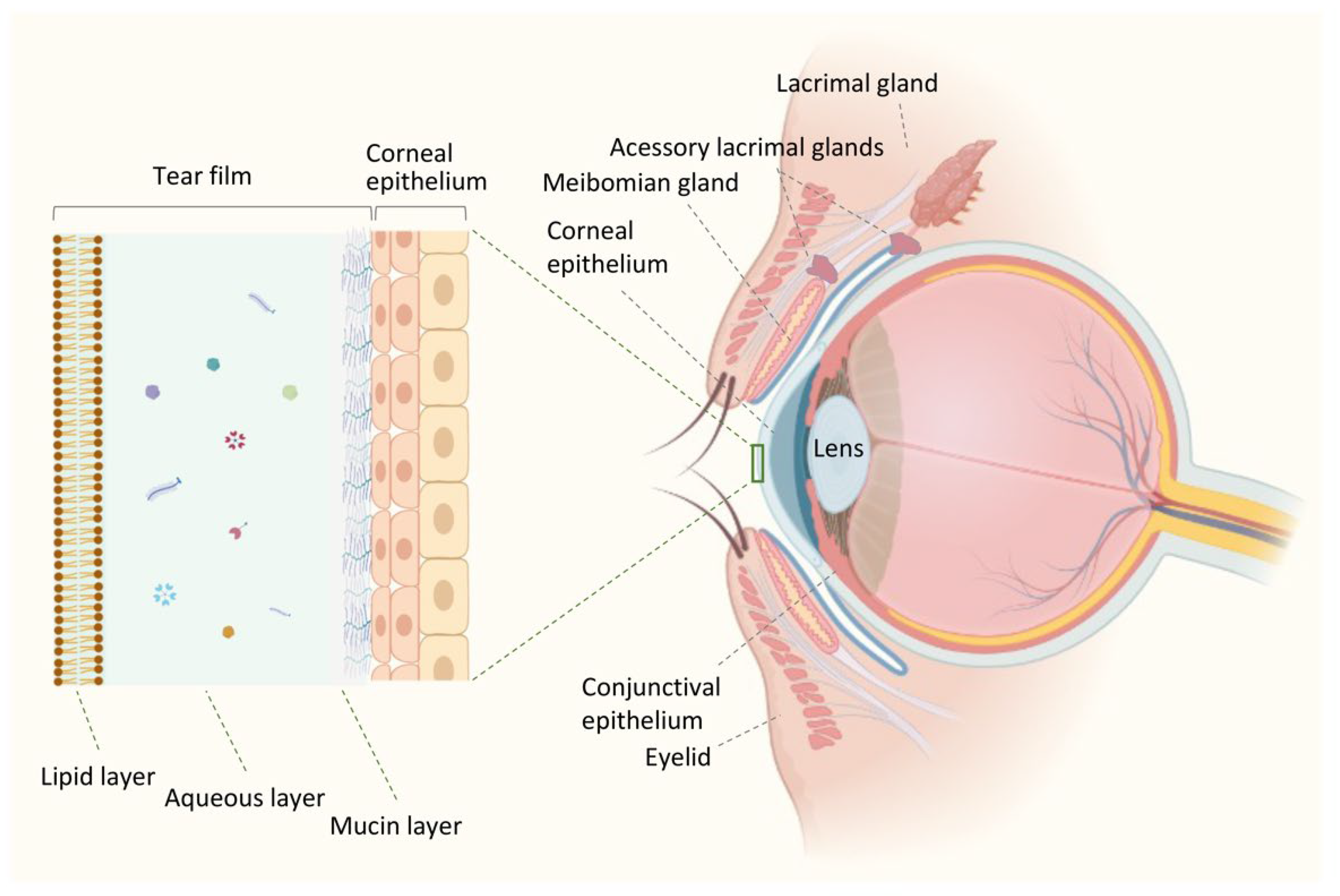
2. Ocular Surface Environment in Health
3. Ocular Surface Innate Immunity
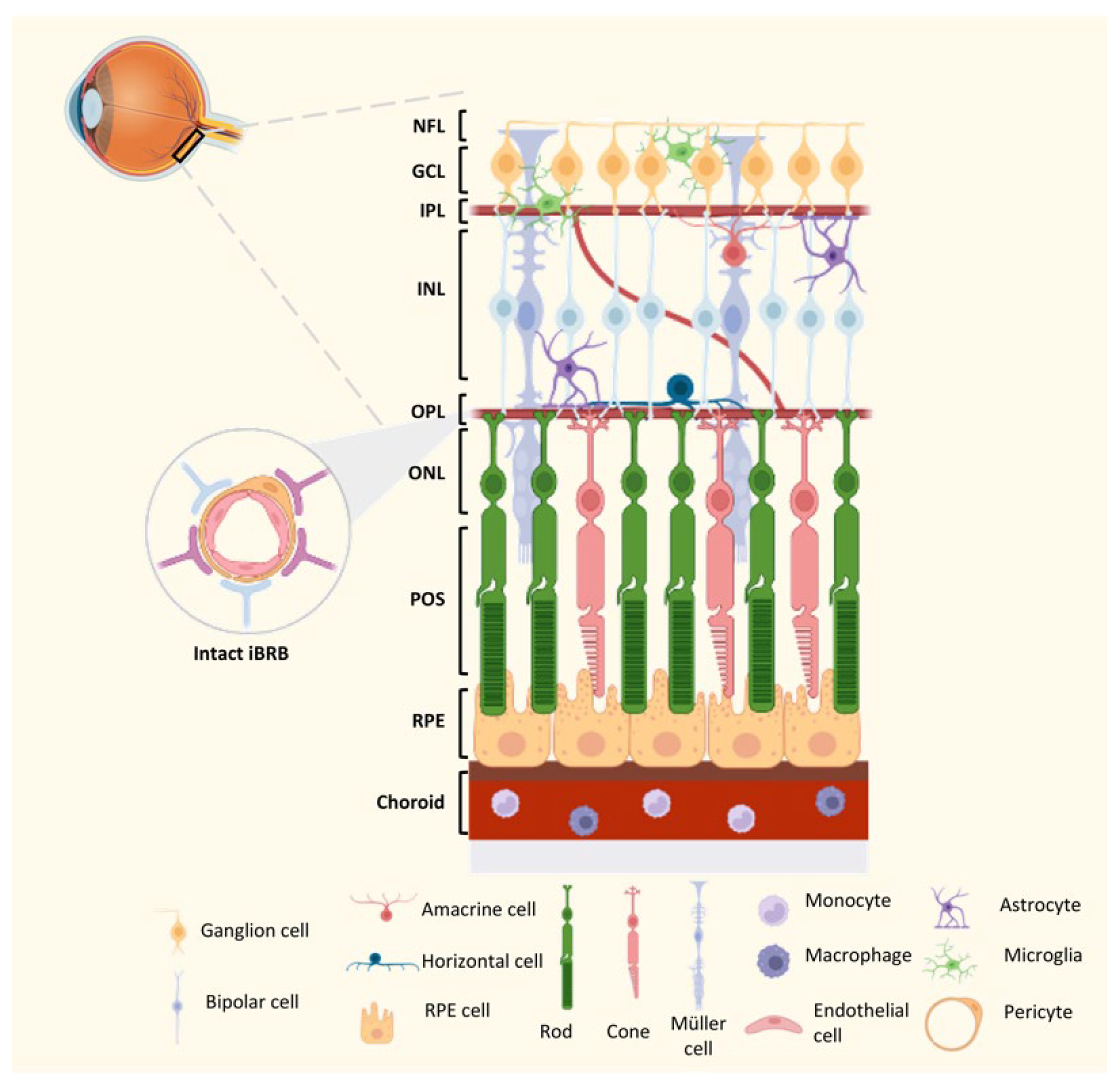
4. Homeostasis of the Innate Immune Network in the Retina
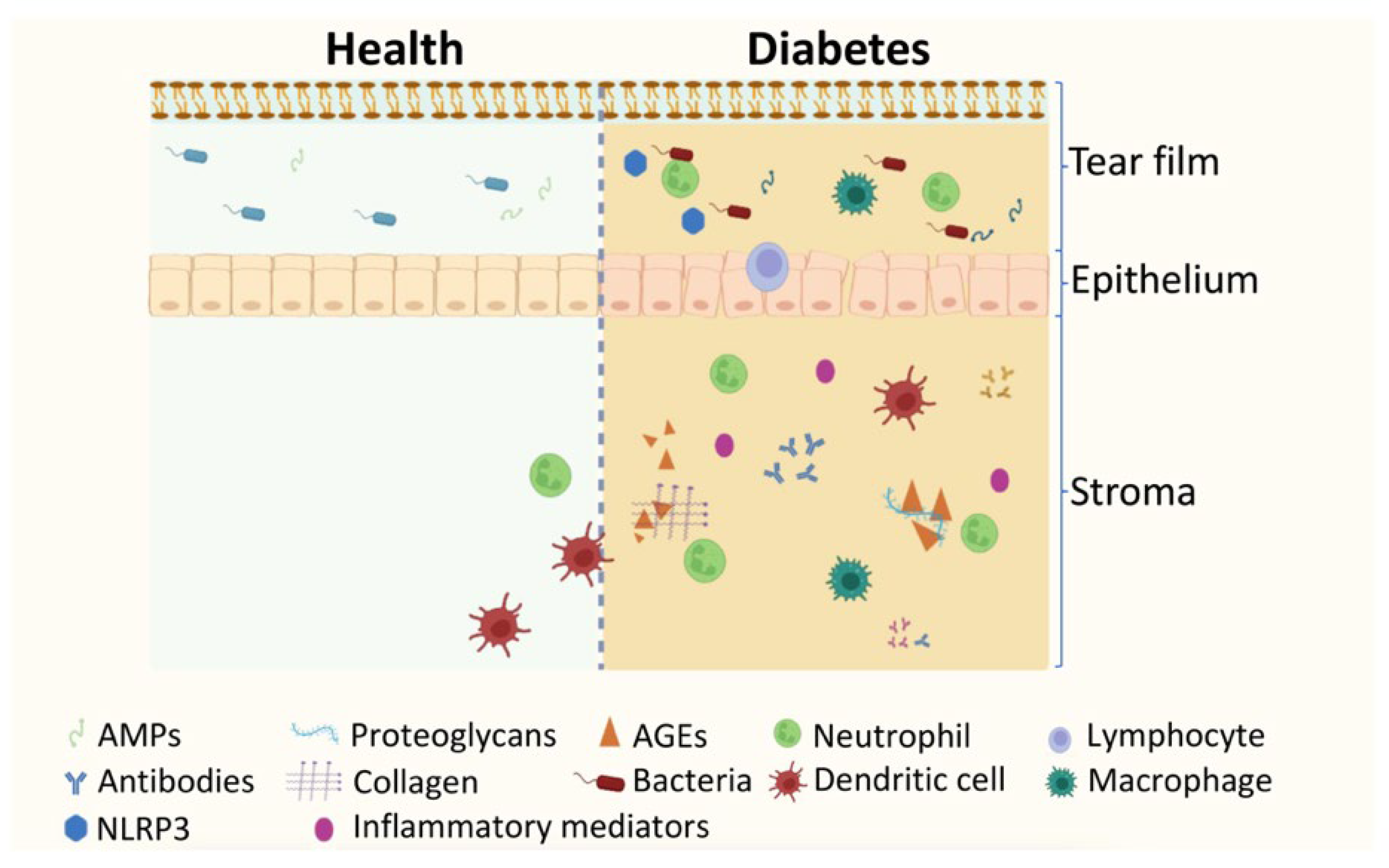
5. The Impact of Diabetes and DR on Ocular Surface and Tear Protein Profile
6. Evidence of Changes in Ocular Surface and Immune Response in Experimental Models of Diabetes
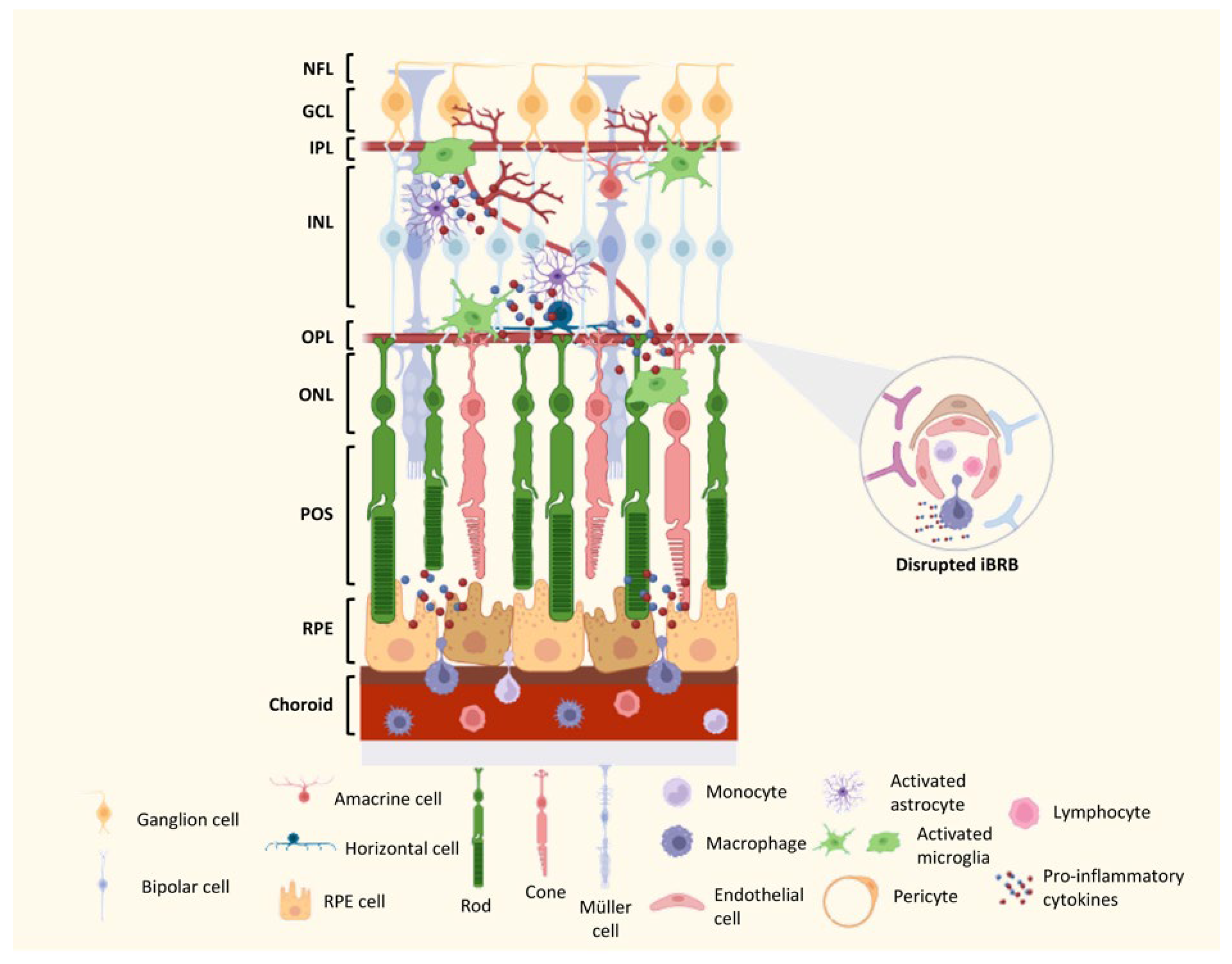
7. Mechanisms of DR Neurodegeneration and Vascular Lesions
8. The Contribution of the Immune Response to DR in Preclinical Models
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Magliano, D.J.; Boyko, E.J. IDF Diabetes Atlas, 10th ed.; IDF Diabetes Atlas: Brussels, Belgium, 2021. [Google Scholar]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Suwajanakorn, O.; Puangsricharern, V.; Kittipibul, T.; Chatsuwan, T. Ocular surface microbiome in diabetes mellitus. Sci. Rep. 2022, 12, 21527. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, L.; Wang, Q.; Li, Y.; Wei, C.; Xie, L. Mechanistic investigations of diabetic ocular surface diseases. Front. Endocrinol. 2022, 13, 1079541. [Google Scholar] [CrossRef] [PubMed]
- Ljubimov, A.V. Diabetic complications in the cornea. Vis. Res. 2017, 139, 138–152. [Google Scholar] [CrossRef]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kern, T.S.; Song, B.; Stuebe, C. Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am. J. Pathol. 2017, 187, 9–19. [Google Scholar] [CrossRef]
- Kinoshita, N.; Kakehashi, A.; Inoda, S.; Itou, Y.; Kuroki, M.; Yasu, T.; Kawakami, M.; Kanazawa, Y. Effective and selective prevention of retinal leukostasis in streptozotocin-induced diabetic rats using gliclazide. Diabetologia 2002, 45, 735–739. [Google Scholar] [CrossRef]
- Forrester, J.V.; Kuffova, L.; Delibegovic, M. The Role of Inflammation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 583687. [Google Scholar] [CrossRef]
- Li, X.; Yu, Z.W.; Wang, Y.; Fu, Y.H.; Gao, X.Y. MicroRNAs: Potential Targets in Diabetic Retinopathy. Horm. Metab. Res. 2020, 52, 142–148. [Google Scholar] [CrossRef]
- Grauslund, J.; Green, A.; Sjolie, A.K. Prevalence and 25 year incidence of proliferative retinopathy among Danish type 1 diabetic patients. Diabetologia 2009, 52, 1829–1835. [Google Scholar] [CrossRef]
- Broe, R.; Rasmussen, M.L.; Frydkjaer-Olsen, U.; Olsen, B.S.; Mortensen, H.B.; Peto, T.; Grauslund, J. The 16-year incidence, progression and regression of diabetic retinopathy in a young population-based Danish cohort with type 1 diabetes mellitus: The Danish cohort of pediatric diabetes 1987 (DCPD1987). Acta Diabetol. 2014, 51, 413–420. [Google Scholar] [CrossRef]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., 3rd; Klein, R.; American Diabetes, A. Diabetic retinopathy. Diabetes Care 2003, 26, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.J.; Liu, Z.; Khamaisi, M.; King, G.L.; Klein, R.; Klein, B.E.K.; Hughes, T.M.; Craft, S.; Freedman, B.I.; Bowden, D.W.; et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2017, 102, 4343–4410. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.W.; Lin, F.; Fort, P.E. The innate immune system in diabetic retinopathy. Prog. Retin. Eye Res. 2021, 84, 100940. [Google Scholar] [CrossRef] [PubMed]
- Gipson, I.K. The ocular surface: The challenge to enable and protect vision: The Friedenwald lecture. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4391–4398. [Google Scholar] [CrossRef]
- Bolanos-Jimenez, R.; Navas, A.; Lopez-Lizarraga, E.P.; de Ribot, F.M.; Pena, A.; Graue-Hernandez, E.O.; Garfias, Y. Ocular Surface as Barrier of Innate Immunity. Open Ophthalmol. J. 2015, 9, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W. Ocular immune privilege. Eye 2009, 23, 1885–1889. [Google Scholar] [CrossRef]
- Caspi, R.R. In this issue: Immunology of the eye--inside and out. Int. Rev. Immunol. 2013, 32, 750138. [Google Scholar] [CrossRef]
- Kinoshita, S.; Adachi, W.; Sotozono, C.; Nishida, K.; Yokoi, N.; Quantock, A.J.; Okubo, K. Characteristics of the human ocular surface epithelium. Prog. Retin. Eye Res. 2001, 20, 639–673. [Google Scholar] [CrossRef]
- Walcott, B.; Moore, L.C.; Birzgalis, A.; Claros, N.; Valiunas, V.; Ott, T.; Willecke, K.; Brink, P.R. Role of gap junctions in fluid secretion of lacrimal glands. Am. J. Physiol. Cell Physiol. 2002, 282, C501–C507. [Google Scholar] [CrossRef]
- Williams, K.; Watsky, M. Gap junctional communication in the human corneal endothelium and epithelium. Curr. Eye Res. 2002, 25, 29–36. [Google Scholar] [CrossRef]
- Mantelli, F.; Mauris, J.; Argueso, P. The ocular surface epithelial barrier and other mechanisms of mucosal protection: From allergy to infectious diseases. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 563–568. [Google Scholar] [CrossRef]
- Zhou, L.; Beuerman, R.W. Tear analysis in ocular surface diseases. Prog. Retin. Eye Res. 2012, 31, 527–550. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Hamrah, P.; Shimazaki, J. Bilateral Alterations in Corneal Nerves, Dendritic Cells, and Tear Cytokine Levels in Ocular Surface Disease. Cornea 2016, 35 (Suppl. S1), S65–S70. [Google Scholar] [CrossRef]
- Belmonte, C.; Aracil, A.; Acosta, M.C.; Luna, C.; Gallar, J. Nerves and sensations from the eye surface. Ocul. Surf. 2004, 2, 248–253. [Google Scholar] [CrossRef]
- Morris, C.A.; Holden, B.A.; Papas, E.; Griesser, H.J.; Bolis, S.; Anderton, P.; Carney, F. The ocular surface, the tear film, and the wettability of contact lenses. Adv. Exp. Med. Biol. 1998, 438, 717–722. [Google Scholar] [CrossRef]
- Holly, F.J. Basic Aspects of Tear Film Formation and Stability. In Physicochemical Hydrodynamics; NATO ASI Series; Springer: Boston, MA, USA, 1988; Volume 174. [Google Scholar]
- Willcox, M.D.P.; Argueso, P.; Georgiev, G.A.; Holopainen, J.M.; Laurie, G.W.; Millar, T.J.; Papas, E.B.; Rolland, J.P.; Schmidt, T.A.; Stahl, U.; et al. TFOS DEWS II Tear Film Report. Ocul. Surf. 2017, 15, 366–403. [Google Scholar] [CrossRef]
- Ambroziak, A.M.; Szaflik, J.; Szaflik, J.P.; Ambroziak, M.; Witkiewicz, J.; Skopinski, P. Immunomodulation on the ocular surface: A review. Cent. Eur. J. Immunol. 2016, 41, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Tamhane, M.; Cabrera-Ghayouri, S.; Abelian, G.; Viswanath, V. Review of Biomarkers in Ocular Matrices: Challenges and Opportunities. Pharm. Res. 2019, 36, 40. [Google Scholar] [CrossRef] [PubMed]
- Albarran, C.; Pons, A.M.; Lorente, A.; Montes, R.; Artigas, J.M. Influence of the tear film on optical quality of the eye. Contact Lens Anterior Eye 1997, 20, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Rentka, A.; Koroskenyi, K.; Harsfalvi, J.; Szekanecz, Z.; Szucs, G.; Szodoray, P.; Kemeny-Beke, A. Evaluation of commonly used tear sampling methods and their relevance in subsequent biochemical analysis. Ann. Clin. Biochem. 2017, 54, 521–529. [Google Scholar] [CrossRef]
- Stahl, U.; Willcox, M.; Stapleton, F. Osmolality and tear film dynamics. Clin. Exp. Optom. 2012, 95, 3–11. [Google Scholar] [CrossRef]
- Bachman, W.G.; Wilson, G. Essential ions for maintenance of the corneal epithelial surface. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1484–1488. [Google Scholar]
- King-Smith, P.E.; Hinel, E.A.; Nichols, J.J. Application of a novel interferometric method to investigate the relation between lipid layer thickness and tear film thinning. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2418–2423. [Google Scholar] [CrossRef]
- Green-Church, K.B.; Butovich, I.; Willcox, M.; Borchman, D.; Paulsen, F.; Barabino, S.; Glasgow, B.J. The international workshop on meibomian gland dysfunction: Report of the subcommittee on tear film lipids and lipid-protein interactions in health and disease. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1979–1993. [Google Scholar] [CrossRef]
- Van Haeringen, N.J. Clinical biochemistry of tears. Surv. Ophthalmol. 1981, 26, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Butovich, I.A. Lipidomics of human Meibomian gland secretions: Chemistry, biophysics, and physiological role of Meibomian lipids. Prog. Lipid Res. 2011, 50, 278–301. [Google Scholar] [CrossRef]
- Carreno, E.; Enriquez-de-Salamanca, A.; Teson, M.; Garcia-Vazquez, C.; Stern, M.E.; Whitcup, S.M.; Calonge, M. Cytokine and chemokine levels in tears from healthy subjects. Acta Ophthalmol. 2010, 88, e250–e258. [Google Scholar] [CrossRef]
- Gokcinar, N.B.; Karabulut, A.A.; Onaran, Z.; Yumusak, E.; Budak Yildiran, F.A. Elevated Tear Human Neutrophil Peptides 1-3, Human Beta Defensin-2 Levels and Conjunctival Cathelicidin LL-37 Gene Expression in Ocular Rosacea. Ocul. Immunol. Inflamm. 2019, 27, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, I.; Said, D.G.; Dua, H.S. Human antimicrobial peptides in ocular surface defense. Prog. Retin. Eye Res. 2017, 61, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Garreis, F.; Gottschalt, M.; Schlorf, T.; Glaser, R.; Harder, J.; Worlitzsch, D.; Paulsen, F.P. Expression and regulation of antimicrobial peptide psoriasin (S100A7) at the ocular surface and in the lacrimal apparatus. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4914–4922. [Google Scholar] [CrossRef] [PubMed]
- Garreis, F.; Gottschalt, M.; Paulsen, F.P. Antimicrobial peptides as a major part of the innate immune defense at the ocular surface. Dev. Ophthalmol. 2010, 45, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Findlay, F.; Pohl, J.; Svoboda, P.; Shakamuri, P.; McLean, K.; Inglis, N.F.; Proudfoot, L.; Barlow, P.G. Carbon Nanoparticles Inhibit the Antimicrobial Activities of the Human Cathelicidin LL-37 through Structural Alteration. J. Immunol. 2017, 199, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Guha, S.; Garg, P.; Roy, S. Differential expression of antimicrobial peptides in corneal infection and regulation of antimicrobial peptides and reactive oxygen species by type III secretion system of Pseudomonas aeruginosa. Pathog. Dis. 2018, 76, fty001. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Haynes, R.J.; Tighe, P.J.; Dua, H.S. Antimicrobial defensin peptides of the human ocular surface. Br. J. Ophthalmol. 1999, 83, 737–741. [Google Scholar] [CrossRef]
- Huang, L.C.; Jean, D.; Proske, R.J.; Reins, R.Y.; McDermott, A.M. Ocular surface expression and in vitro activity of antimicrobial peptides. Curr. Eye Res. 2007, 32, 595–609. [Google Scholar] [CrossRef]
- Sorensen, O.E.; Follin, P.; Johnsen, A.H.; Calafat, J.; Tjabringa, G.S.; Hiemstra, P.S.; Borregaard, N. Human cathelicidin, hCAP-18, is processed to the antimicrobial peptide LL-37 by extracellular cleavage with proteinase 3. Blood 2001, 97, 3951–3959. [Google Scholar] [CrossRef]
- McDermott, A.M.; Redfern, R.L.; Zhang, B.; Pei, Y.; Huang, L.; Proske, R.J. Defensin expression by the cornea: Multiple signalling pathways mediate IL-1beta stimulation of hBD-2 expression by human corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1859–1865. [Google Scholar] [CrossRef]
- Akpek, E.K.; Gottsch, J.D. Immune defense at the ocular surface. Eye 2003, 17, 949–956. [Google Scholar] [CrossRef]
- Niederkorn, J.Y.; Peeler, J.S.; Mellon, J. Phagocytosis of particulate antigens by corneal epithelial cells stimulates interleukin-1 secretion and migration of Langerhans cells into the central cornea. Reg. Immunol. 1989, 2, 83–90. [Google Scholar]
- Cubitt, C.L.; Lausch, R.N.; Oakes, J.E. Synthesis of type II interleukin-1 receptors by human corneal epithelial cells but not by keratocytes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 701–704. [Google Scholar]
- Hattori, T.; Chauhan, S.K.; Lee, H.; Ueno, H.; Dana, R.; Kaplan, D.H.; Saban, D.R. Characterization of Langerin-expressing dendritic cell subsets in the normal cornea. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4598–4604. [Google Scholar] [CrossRef]
- Hamrah, P.; Dana, M.R. Corneal antigen-presenting cells. Chem. Immunol. Allergy 2007, 92, 58–70. [Google Scholar] [CrossRef]
- Forrester, J.V.; Xu, H.; Kuffova, L.; Dick, A.D.; McMenamin, P.G. Dendritic cell physiology and function in the eye. Immunol. Rev. 2010, 234, 282–304. [Google Scholar] [CrossRef]
- Akhlaq, A.; Colon, C.; Cavalcanti, B.M.; Aggarwal, S.; Qazi, Y.; Cruzat, A.; Jersey, C.; Critser, D.B.; Watts, A.; Beyer, J.; et al. Density and distribution of dendritiform cells in the peripheral cornea of healthy subjects using in vivo confocal microscopy. Ocul. Surf. 2022, 26, 157–165. [Google Scholar] [CrossRef]
- Sichien, D.; Lambrecht, B.N.; Guilliams, M.; Scott, C.L. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal. Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef]
- Hamrah, P.; Huq, S.O.; Liu, Y.; Zhang, Q.; Dana, M.R. Corneal immunity is mediated by heterogeneous population of antigen-presenting cells. J. Leukoc. Biol. 2003, 74, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Mayer, W.J.; Irschick, U.M.; Moser, P.; Wurm, M.; Huemer, H.P.; Romani, N.; Irschick, E.U. Characterization of antigen-presenting cells in fresh and cultured human corneas using novel dendritic cell markers. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4459–4467. [Google Scholar] [CrossRef]
- Zhivov, A.; Stave, J.; Vollmar, B.; Guthoff, R. In vivo confocal microscopic evaluation of Langerhans cell density and distribution in the normal human corneal epithelium. Graefe Arch. Clin. Exp. Ophthalmol. 2005, 243, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, H.R.; Zhang, X.Y.; Wu, C.Y.; Downie, L.E. Corneal immune cell morphometry as an indicator of local and systemic pathology: A review. Clin. Exp. Ophthalmol. 2021, 49, 729–740. [Google Scholar] [CrossRef]
- Jamali, A.; Kenyon, B.; Ortiz, G.; Abou-Slaybi, A.; Sendra, V.G.; Harris, D.L.; Hamrah, P. Plasmacytoid dendritic cells in the eye. Prog. Retin. Eye Res. 2021, 80, 100877. [Google Scholar] [CrossRef]
- Hamrah, P.; Liu, Y.; Zhang, Q.; Dana, M.R. The corneal stroma is endowed with a significant number of resident dendritic cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 581–589. [Google Scholar] [CrossRef]
- Knickelbein, J.E.; Watkins, S.C.; McMenamin, P.G.; Hendricks, R.L. Stratification of Antigen-presenting Cells within the Normal Cornea. Ophthalmol. Eye Dis. 2009, 1, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Takahashi, H.; Dana, R. Novel Insights Into the Immunoregulatory Function and Localization of Dendritic Cells. Cornea 2016, 35 (Suppl. S1), S49–S54. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Yin, J.; Yoon, G.S.; Mi, Q.S.; Yu, F.S. Dendritic cell-epithelium interplay is a determinant factor for corneal epithelial wound repair. Am. J. Pathol. 2011, 179, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, C.; Lee, I.X.Y.; Lin, M.T.Y.; Liu, Y.C. Corneal dendritic cells in diabetes mellitus: A narrative review. Front. Endocrinol. 2023, 14, 1078660. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.J.; Liu, J. Human Microbiota and Ophthalmic Disease. Yale J. Biol. Med. 2016, 89, 325–330. [Google Scholar]
- Dong, Q.; Brulc, J.M.; Iovieno, A.; Bates, B.; Garoutte, A.; Miller, D.; Revanna, K.V.; Gao, X.; Antonopoulos, D.A.; Slepak, V.Z.; et al. Diversity of bacteria at healthy human conjunctiva. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5408–5413. [Google Scholar] [CrossRef]
- Miller, D.; Iovieno, A. The role of microbial flora on the ocular surface. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 466–470. [Google Scholar] [CrossRef]
- van der Meulen, I.J.; van Rooij, J.; Nieuwendaal, C.P.; Van Cleijnenbreugel, H.; Geerards, A.J.; Remeijer, L. Age-related risk factors, culture outcomes, and prognosis in patients admitted with infectious keratitis to two Dutch tertiary referral centers. Cornea 2008, 27, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Cogen, A.L.; Nizet, V.; Gallo, R.L. Skin microbiota: A source of disease or defence? Br. J. Dermatol. 2008, 158, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Kooragayala, K.; Gotoh, N.; Cogliati, T.; Nellissery, J.; Kaden, T.R.; French, S.; Balaban, R.; Li, W.; Covian, R.; Swaroop, A. Quantification of Oxygen Consumption in Retina Ex Vivo Demonstrates Limited Reserve Capacity of Photoreceptor Mitochondria. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8428–8436. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef]
- Dando, S.J.; Naranjo Golborne, C.; Chinnery, H.R.; Ruitenberg, M.J.; McMenamin, P.G. A case of mistaken identity: CD11c-eYFP(+) cells in the normal mouse brain parenchyma and neural retina display the phenotype of microglia, not dendritic cells. Glia 2016, 64, 1331–1349. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Foulds, W.S.; Ling, E.A.; Kaur, C. Retinal microglia—A key player in healthy and diseased retina. Prog. Neurobiol. 2019, 173, 18–40. [Google Scholar] [CrossRef]
- Uckermann, O.; Wolf, A.; Kutzera, F.; Kalisch, F.; Beck-Sickinger, A.G.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Glutamate release by neurons evokes a purinergic inhibitory mechanism of osmotic glial cell swelling in the rat retina: Activation by neuropeptide Y. J. Neurosci. Res. 2006, 83, 538–550. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Wosik, K.; Antel, J.P. Glial cell influence on the human blood-brain barrier. Glia 2001, 36, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Bu, Y.; Shih, K.C.; Tong, L. The ocular surface and diabetes, the other 21st Century epidemic. Exp. Eye Res. 2022, 220, 109099. [Google Scholar] [CrossRef] [PubMed]
- Amorim, M.; Martins, B.; Caramelo, F.; Goncalves, C.; Trindade, G.; Simao, J.; Barreto, P.; Marques, I.; Leal, E.C.; Carvalho, E.; et al. Putative Biomarkers in Tears for Diabetic Retinopathy Diagnosis. Front. Med. 2022, 9, 873483. [Google Scholar] [CrossRef] [PubMed]
- Naik, K.; Magdum, R.; Ahuja, A.; Kaul, S.; Johnson, S.; Mishra, A.; Patil, M.; Dhore, D.N.; Alapati, A. Ocular Surface Diseases in Patients With Diabetes. Cureus 2022, 14, e23401. [Google Scholar] [CrossRef] [PubMed]
- Han, J.X.; Wang, H.; Liang, H.H.; Guo, J.X. Correlation of the retinopathy degree with the change of ocular surface and corneal nerve in patients with type 2 diabetes mellitus. Int. J. Ophthalmol. 2021, 14, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Markoulli, M.; Flanagan, J.; Tummanapalli, S.S.; Wu, J.; Willcox, M. The impact of diabetes on corneal nerve morphology and ocular surface integrity. Ocul. Surf. 2018, 16, 45–57. [Google Scholar] [CrossRef]
- Zhou, T.; Lee, A.; Lo, A.C.Y.; Kwok, J. Diabetic Corneal Neuropathy: Pathogenic Mechanisms and Therapeutic Strategies. Front. Pharmacol. 2022, 13, 816062. [Google Scholar] [CrossRef]
- Nadeem, H.; Malik, T.G.; Mazhar, A.; Ali, A. Association of Dry Eye Disease with Diabetic Retinopathy. J. Coll. Physicians Surg. Pak. 2020, 30, 493–497. [Google Scholar] [CrossRef]
- Csosz, E.; Deak, E.; Kallo, G.; Csutak, A.; Tozser, J. Diabetic retinopathy: Proteomic approaches to help the differential diagnosis and to understand the underlying molecular mechanisms. J. Proteom. 2017, 150, 351–358. [Google Scholar] [CrossRef]
- Amil-Bangsa, N.H.; Mohd-Ali, B.; Ishak, B.; Abdul-Aziz, C.N.N.; Ngah, N.F.; Hashim, H.; Ghazali, A.R. Total Protein Concentration and Tumor Necrosis Factor alpha in Tears of Nonproliferative Diabetic Retinopathy. Optom. Vis. Sci. 2019, 96, 934–939. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Ru, Y.S.; Wang, X.W.; Yang, J.Z.; Li, C.H.; Wang, H.X.; Li, X.R.; Li, B. Ocular surface changes in type II diabetic patients with proliferative diabetic retinopathy. Int. J. Ophthalmol. 2015, 8, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.C.; Lam, K.S.L.; Tong, L. A systematic review on the impact of diabetes mellitus on the ocular surface. Nutr. Diabetes 2017, 7, e251. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.P.; Parrish, S.T.; Vernon, S.A. Corneal epithelial oxygen uptake rate in diabetes mellitus. Eye 1990, 4 Pt 5, 757–759. [Google Scholar] [CrossRef]
- Niimi, N.; Yako, H.; Takaku, S.; Chung, S.K.; Sango, K. Aldose Reductase and the Polyol Pathway in Schwann Cells: Old and New Problems. Int. J. Mol. Sci. 2021, 22, 1031. [Google Scholar] [CrossRef] [PubMed]
- Hager, A.; Wegscheider, K.; Wiegand, W. Changes of extracellular matrix of the cornea in diabetes mellitus. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Perez-Rico, C.; Gutierrez-Ortiz, C.; Gonzalez-Mesa, A.; Zandueta, A.M.; Moreno-Salgueiro, A.; Germain, F. Effect of diabetes mellitus on Corvis ST measurement process. Acta Ophthalmol. 2015, 93, e193–e198. [Google Scholar] [CrossRef]
- Kotecha, A.; Oddone, F.; Sinapis, C.; Elsheikh, A.; Sinapis, D.; Sinapis, A.; Garway-Heath, D.F. Corneal biomechanical characteristics in patients with diabetes mellitus. J. Cataract Refract. Surg. 2010, 36, 1822–1828. [Google Scholar] [CrossRef]
- Goldich, Y.; Barkana, Y.; Gerber, Y.; Rasko, A.; Morad, Y.; Harstein, M.; Avni, I.; Zadok, D. Effect of diabetes mellitus on biomechanical parameters of the cornea. J. Cataract Refract. Surg. 2009, 35, 715–719. [Google Scholar] [CrossRef]
- Scheler, A.; Spoerl, E.; Boehm, A.G. Effect of diabetes mellitus on corneal biomechanics and measurement of intraocular pressure. Acta Ophthalmol. 2012, 90, e447–e451. [Google Scholar] [CrossRef]
- Luo, X.Y.; Dai, W.; Chee, M.L.; Tao, Y.; Chua, J.; Tan, N.Y.Q.; Tham, Y.C.; Aung, T.; Wong, T.Y.; Cheng, C.Y. Association of Diabetes With Central Corneal Thickness Among a Multiethnic Asian Population. JAMA Netw. Open 2019, 2, e186647. [Google Scholar] [CrossRef]
- El-Agamy, A.; Alsubaie, S. Corneal endothelium and central corneal thickness changes in type 2 diabetes mellitus. Clin. Ophthalmol. 2017, 11, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Bhadra, S.; Mittal, P.; Shyam, K. Corneal endothelial morphology and central corneal thickness in type 2 diabetes mellitus patients. Indian J. Ophthalmol. 2021, 69, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- GM, I. Ocular problems in diabetes mellitus. Sudan. J. Ophthalmol. 2014, 6, 43–48. [Google Scholar]
- He, F.; Zhao, Z.; Liu, Y.; Lu, L.; Fu, Y. Assessment of Ocular Surface Damage during the Course of Type 2 Diabetes Mellitus. J. Ophthalmol. 2018, 2018, 1206808. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Qin, G.; Xie, H.; Lv, P. Tear film function in type 2 diabetic patients with retinopathy. Ophthalmologica 2008, 222, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, J.; Shi, B.; He, S.; Yao, X.; Willcox, M.D. Advanced glycation end product (AGE) modified proteins in tears of diabetic patients. Mol. Vis. 2010, 16, 1576–1584. [Google Scholar]
- Sandra Johanna, G.P.; Antonio, L.A.; Andres, G.S. Correlation between type 2 diabetes, dry eye and Meibomian glands dysfunction. J. Optom. 2019, 12, 256–262. [Google Scholar] [CrossRef]
- Kern, T.S. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 95103. [Google Scholar] [CrossRef]
- Messmer, E.M.; Schmid-Tannwald, C.; Zapp, D.; Kampik, A. In vivo confocal microscopy of corneal small fiber damage in diabetes mellitus. Graefes Arch. Clin. Exp. Ophthalmol. 2010, 248, 1307–1312. [Google Scholar] [CrossRef]
- Beckman, K.A. Characterization of dry eye disease in diabetic patients versus nondiabetic patients. Cornea 2014, 33, 851–854. [Google Scholar] [CrossRef]
- Alves Mde, C.; Carvalheira, J.B.; Modulo, C.M.; Rocha, E.M. Tear film and ocular surface changes in diabetes mellitus. Arq. Bras. Oftalmol. 2008, 71, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Herber, S.; Grus, F.H.; Sabuncuo, P.; Augustin, A.J. Two-dimensional analysis of tear protein patterns of diabetic patients. Electrophoresis 2001, 22, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Khuong, T.; Everest-Dass, A.V.; Kautto, L.; Zhao, Z.; Willcox, M.D.; Packer, N.H. Glycomic characterization of basal tears and changes with diabetes and diabetic retinopathy. Glycobiology 2015, 25, 269–283. [Google Scholar] [CrossRef]
- Negre-Salvayre, A.; Salvayre, R.; Auge, N.; Pamplona, R.; Portero-Otin, M. Hyperglycemia and glycation in diabetic complications. Antioxid. Redox Signal. 2009, 11, 3071–3109. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ghosh, S.; Azharuddin, M.; Bera, S.; Datta, H.; Dasgupta, A. Change in tear protein profile in diabetic retinopathy with duration of diabetes. Diabetes Metab. Syndr. 2014, 8, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.R.; Kim, Y.H.; Ha, S.J.; Byeon, H.E.; Cho, C.H.; Kim, J.H.; Lee, K. Role of Inflammation in Classification of Diabetic Macular Edema by Optical Coherence Tomography. J. Diabetes Res. 2019, 2019, 8164250. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.Z.; Guo, X.; Klein, R.; Klein, B.E.; Cui, J.; Rotter, J.I.; Ipp, E.; Chen, Y.D. Systemic soluble tumor necrosis factor receptors 1 and 2 are associated with severity of diabetic retinopathy in Hispanics. Ophthalmology 2012, 119, 1041–1046. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, S.S.; Kim, J.C.; Kim, H.C.; Im, Y.S.; Ahn, C.W.; Lee, H.K. Serum and tear levels of nerve growth factor in diabetic retinopathy patients. Am. J. Ophthalmol. 2008, 145, 432–437. [Google Scholar] [CrossRef]
- Csosz, E.; Boross, P.; Csutak, A.; Berta, A.; Toth, F.; Poliska, S.; Torok, Z.; Tozser, J. Quantitative analysis of proteins in the tear fluid of patients with diabetic retinopathy. J. Proteom. 2012, 75, 2196–2204. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, P.K.; Yoo, H.S.; Kim, C.W. Comparison of tear proteins between healthy and early diabetic retinopathy patients. Clin. Biochem. 2012, 45, 60–67. [Google Scholar] [CrossRef]
- Xue, W.; Li, J.J.; Zou, Y.; Zou, B.; Wei, L. Microbiota and Ocular Diseases. Front. Cell. Infect. Microbiol. 2021, 11, 759333. [Google Scholar] [CrossRef]
- Berbudi, A.; Rahmadika, N.; Tjahjadi, A.I.; Ruslami, R. Type 2 Diabetes and its Impact on the Immune System. Curr. Diabetes Rev. 2020, 16, 442–449. [Google Scholar] [CrossRef]
- Moutschen, M.P.; Scheen, A.J.; Lefebvre, P.J. Impaired immune responses in diabetes mellitus: Analysis of the factors and mechanisms involved. Relevance to the increased susceptibility of diabetic patients to specific infections. Diabete Metab. 1992, 18, 187–201. [Google Scholar] [PubMed]
- Daoud, A.K.; Tayyar, M.A.; Fouda, I.M.; Harfeil, N.A. Effects of diabetes mellitus vs. in vitro hyperglycemia on select immune cell functions. J. Immunotoxicol. 2009, 6, 36–41. [Google Scholar] [CrossRef]
- Xu, K.; Yu, F.S. Impaired epithelial wound healing and EGFR signaling pathways in the corneas of diabetic rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3301–3308. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Liu, Y.; Sullivan, D.A. Effects of Insulin and High Glucose on Human Meibomian Gland Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7814–7820. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, H.; Zhao, Z.; Luo, X.; Zhang, M.; Bu, J.; Liang, M.; Wu, H.; Yu, J.; He, H.; et al. Hyperglycemia Induces Meibomian Gland Dysfunction. Investig. Ophthalmol. Vis. Sci. 2022, 63, 30. [Google Scholar] [CrossRef]
- Yin, J.; Huang, J.; Chen, C.; Gao, N.; Wang, F.; Yu, F.S. Corneal complications in streptozocin-induced type I diabetic rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6589–6596. [Google Scholar] [CrossRef]
- Dota, A.; Sakamoto, A.; Nagano, T.; Murakami, T.; Matsugi, T. Effect of Diquafosol Ophthalmic Solution on Airflow-Induced Ocular Surface Disorder in Diabetic Rats. Clin. Ophthalmol. 2020, 14, 1019–1024. [Google Scholar] [CrossRef]
- Liu, H.; Sheng, M.; Liu, Y.; Wang, P.; Chen, Y.; Chen, L.; Wang, W.; Li, B. Expression of SIRT1 and oxidative stress in diabetic dry eye. Int. J. Clin. Exp. Pathol. 2015, 8, 7644–7653. [Google Scholar]
- Bettahi, I.; Sun, H.; Gao, N.; Wang, F.; Mi, X.; Chen, W.; Liu, Z.; Yu, F.S. Genome-wide transcriptional analysis of differentially expressed genes in diabetic, healing corneal epithelial cells: Hyperglycemia-suppressed TGFbeta3 expression contributes to the delay of epithelial wound healing in diabetic corneas. Diabetes 2014, 63, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Gao, N.; Sun, H.; Yin, J.; Lee, P.; Zhou, L.; Fan, X.; Yu, F.S. Targeting Imbalance between IL-1beta and IL-1 Receptor Antagonist Ameliorates Delayed Epithelium Wound Healing in Diabetic Mouse Corneas. Am. J. Pathol. 2016, 186, 1466–1480. [Google Scholar] [CrossRef]
- Alba-Loureiro, T.C.; Hirabara, S.M.; Mendonca, J.R.; Curi, R.; Pithon-Curi, T.C. Diabetes causes marked changes in function and metabolism of rat neutrophils. J. Endocrinol. 2006, 188, 295–303. [Google Scholar] [CrossRef]
- Gao, N.; Yan, C.; Lee, P.; Sun, H.; Yu, F.S. Dendritic cell dysfunction and diabetic sensory neuropathy in the cornea. J. Clin. Investig. 2016, 126, 1998–2011. [Google Scholar] [CrossRef]
- Yu, F.X.; Lee, P.S.Y.; Yang, L.; Gao, N.; Zhang, Y.; Ljubimov, A.V.; Yang, E.; Zhou, Q.; Xie, L. The impact of sensory neuropathy and inflammation on epithelial wound healing in diabetic corneas. Prog. Retin. Eye Res. 2022, 89, 101039. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Shi, D.; Chen, P.; Yu, Y.; Yang, L.; Xie, L. Overexpression of SIRT1 promotes high glucose-attenuated corneal epithelial wound healing via p53 regulation of the IGFBP3/IGF-1R/AKT pathway. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3806–3814. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Salimian, M.; Konduru, S.R.; Choi, J.; Kumar, P.; Long, A.; Klimova, N.; Ho, C.Y.; Kristian, T.; Russell, J.W. Overexpression of Sirtuin 1 protein in neurons prevents and reverses experimental diabetic neuropathy. Brain 2019, 142, 3737–3752. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Bai, X.; Zhou, Q.; Chen, C.; Wang, H.; Liu, T.; Xue, J.; Wei, C.; Xie, L. The advanced glycation end-products (AGEs)/ROS/NLRP3 inflammasome axis contributes to delayed diabetic corneal wound healing and nerve regeneration. Int. J. Biol. Sci. 2022, 18, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch. Pharm. Res. 2021, 44, 16–35. [Google Scholar] [CrossRef]
- Youm, Y.H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef]
- Zhang, X.; Fu, Y.; Li, H.; Shen, L.; Chang, Q.; Pan, L.; Hong, S.; Yin, X. H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. J. Cell. Mol. Med. 2018, 22, 1816–1825. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, X.; Zong, B.; Yuan, H.; Wang, Z.; Wei, Y.; Wang, X.; Liu, G.; Zhang, J.; Li, S.; et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J. Cell. Mol. Med. 2018, 22, 4437–4448. [Google Scholar] [CrossRef]
- Yu, Z.W.; Zhang, J.; Li, X.; Wang, Y.; Fu, Y.H.; Gao, X.Y. A new research hot spot: The role of NLRP3 inflammasome activation, a key step in pyroptosis, in diabetes and diabetic complications. Life Sci. 2020, 240, 117138. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, H.; Lu, X.; Zhao, S. N-acetylcysteine alleviates ocular surface damage in STZ-induced diabetic mice by inhibiting the ROS/NLRP3/Caspase-1/IL-1beta signaling pathway. Exp. Eye Res. 2021, 209, 108654. [Google Scholar] [CrossRef]
- Yang, C.; Fei, Y.; Qin, Y.; Luo, D.; Yang, S.; Kou, X.; Zi, Y.; Deng, T.; Jin, M. Bacterial Flora Changes in Conjunctiva of Rats with Streptozotocin-Induced Type I Diabetes. PLoS ONE 2015, 10, e0133021. [Google Scholar] [CrossRef] [PubMed]
- Hara, N.; Alkanani, A.K.; Ir, D.; Robertson, C.E.; Wagner, B.D.; Frank, D.N.; Zipris, D. The role of the intestinal microbiota in type 1 diabetes. Clin. Immunol. 2013, 146, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Sambhav, K.; Mathai, A.; Reddy, A.K.; Reddy, B.V.; Bhatia, K.; Balne, P.K. Endogenous endophthalmitis caused by Enterococcus casseliflavus. J. Med. Microbiol. 2011, 60, 670–672. [Google Scholar] [CrossRef]
- Woo, P.C.; Tam, D.M.; Lau, S.K.; Fung, A.M.; Yuen, K.Y. Enterococcus cecorum empyema thoracis successfully treated with cefotaxime. J. Clin. Microbiol. 2004, 42, 919–922. [Google Scholar] [CrossRef]
- Tewari, R.; Dudeja, M.; Das, A.K.; Nandy, S. Kocuria kristinae in catheter associated urinary tract infection: A case report. J. Clin. Diagn. Res. 2013, 7, 1692–1693. [Google Scholar] [CrossRef]
- Citro, R.; Prota, C.; Greco, L.; Mirra, M.; Masullo, A.; Silverio, A.; Bossone, E.; Piscione, F. Kocuria kristinae endocarditis related to diabetic foot infection. J. Med. Microbiol. 2013, 62, 932–934. [Google Scholar] [CrossRef]
- Fernandes, R.; Girao, H.; Pereira, P. High glucose down-regulates intercellular communication in retinal endothelial cells by enhancing degradation of connexin 43 by a proteasome-dependent mechanism. J. Biol. Chem. 2004, 279, 27219–27224. [Google Scholar] [CrossRef]
- Fernandes, R.; Carvalho, A.L.; Kumagai, A.; Seica, R.; Hosoya, K.; Terasaki, T.; Murta, J.; Pereira, P.; Faro, C. Downregulation of retinal GLUT1 in diabetes by ubiquitinylation. Mol. Vis. 2004, 10, 618–628. [Google Scholar] [PubMed]
- Bento, C.F.; Fernandes, R.; Matafome, P.; Sena, C.; Seica, R.; Pereira, P. Methylglyoxal-induced imbalance in the ratio of vascular endothelial growth factor to angiopoietin 2 secreted by retinal pigment epithelial cells leads to endothelial dysfunction. Exp. Physiol. 2010, 95, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, R.; Hosoya, K.; Pereira, P. Reactive oxygen species downregulate glucose transport system in retinal endothelial cells. Am. J. Physiol. Cell Physiol. 2011, 300, C927–C936. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Marques, C.; Leal, E.; Ribeiro, C.F.; Reis, F.; Ambrosio, A.F.; Fernandes, R. Dipeptidyl peptidase-IV inhibition prevents blood-retinal barrier breakdown, inflammation and neuronal cell death in the retina of type 1 diabetic rats. Biochim. Biophys. Acta 2014, 1842, 1454–1463. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, C.; Lu, L.; Tian, H.; Liu, K.; Luo, D.; Qiu, Q.; Xu, G.T.; Zhang, J. Melatonin Maintains Inner Blood-Retinal Barrier by Regulating Microglia via Inhibition of PI3K/Akt/Stat3/NF-kappaB Signaling Pathways in Experimental Diabetic Retinopathy. Front. Immunol. 2022, 13, 831660. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xie, H.; Zhang, C.; Wang, T.; Tian, H.; Lu, L.; Xu, J.Y.; Xu, G.T.; Liu, L.; Zhang, J. Enhancing fractalkine/CX3CR1 signalling pathway can reduce neuroinflammation by attenuating microglia activation in experimental diabetic retinopathy. J. Cell. Mol. Med. 2022, 26, 1229–1244. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Barber, A.J. Visual dysfunction associated with diabetic retinopathy. Curr. Diabetes Rep. 2010, 10, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Trento, M.; Durando, O.; Lavecchia, S.; Charrier, L.; Cavallo, F.; Costa, M.A.; Hernandez, C.; Simo, R.; Porta, M.; EUROCONDOR Trial Investigators. Vision related quality of life in patients with type 2 diabetes in the EUROCONDOR trial. Endocrine 2017, 57, 83–88. [Google Scholar] [CrossRef]
- Wolff, B.E.; Bearse, M.A., Jr.; Schneck, M.E.; Dhamdhere, K.; Harrison, W.W.; Barez, S.; Adams, A.J. Color vision and neuroretinal function in diabetes. Doc. Ophthalmol. 2015, 130, 131–139. [Google Scholar] [CrossRef]
- Chhablani, J.; Sharma, A.; Goud, A.; Peguda, H.K.; Rao, H.L.; Begum, V.U.; Barteselli, G. Neurodegeneration in Type 2 Diabetes: Evidence From Spectral-Domain Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6333–6338. [Google Scholar] [CrossRef]
- Di Leo, M.A.; Caputo, S.; Falsini, B.; Porciatti, V.; Greco, A.V.; Ghirlanda, G. Presence and further development of retinal dysfunction after 3-year follow up in IDDM patients without angiographically documented vasculopathy. Diabetologia 1994, 37, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Juen, S.; Kieselbach, G.F. Electrophysiological changes in juvenile diabetics without retinopathy. Arch. Ophthalmol. 1990, 108, 372–375. [Google Scholar] [CrossRef]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef]
- Tyrberg, M.; Lindblad, U.; Melander, A.; Lovestam-Adrian, M.; Ponjavic, V.; Andreasson, S. Electrophysiological studies in newly onset type 2 diabetes without visible vascular retinopathy. Doc. Ophthalmol. 2011, 123, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.G.; Lee, J.Y.; Kim, C.; Park, Y.H. Early Microglial Changes Associated with Diabetic Retinopathy in Rats with Streptozotocin-Induced Diabetes. J. Diabetes Res. 2021, 2021, 4920937. [Google Scholar] [CrossRef]
- Harrison, W.W.; Bearse, M.A., Jr.; Ng, J.S.; Jewell, N.P.; Barez, S.; Burger, D.; Schneck, M.E.; Adams, A.J. Multifocal electroretinograms predict onset of diabetic retinopathy in adult patients with diabetes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Dodo, Y.; Murakami, T.; Uji, A.; Yoshitake, S.; Yoshimura, N. Disorganized retinal lamellar structures in nonperfused areas of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2012–2020. [Google Scholar] [CrossRef]
- Sorrentino, F.S.; Matteini, S.; Bonifazzi, C.; Sebastiani, A.; Parmeggiani, F. Diabetic retinopathy and endothelin system: Microangiopathy versus endothelial dysfunction. Eye 2018, 32, 1157–1163. [Google Scholar] [CrossRef]
- Rodriguez, M.L.; Perez, S.; Mena-Molla, S.; Desco, M.C.; Ortega, A.L. Oxidative Stress and Microvascular Alterations in Diabetic Retinopathy: Future Therapies. Oxid. Med. Cell. Longev. 2019, 2019, 4940825. [Google Scholar] [CrossRef]
- Santiago, A.R.; Boia, R.; Aires, I.D.; Ambrosio, A.F.; Fernandes, R. Sweet Stress: Coping With Vascular Dysfunction in Diabetic Retinopathy. Front. Physiol. 2018, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Ahmad, A.; Siddiquei, M.M.; De Zutter, A.; Allegaert, E.; Gikandi, P.W.; De Hertogh, G.; Van Damme, J.; Opdenakker, G.; Struyf, S. The Proinflammatory and Proangiogenic Macrophage Migration Inhibitory Factor Is a Potential Regulator in Proliferative Diabetic Retinopathy. Front. Immunol. 2019, 10, 2752. [Google Scholar] [CrossRef] [PubMed]
- Martins, B.; Amorim, M.; Reis, F.; Ambrosio, A.F.; Fernandes, R. Extracellular Vesicles and MicroRNA: Putative Role in Diagnosis and Treatment of Diabetic Retinopathy. Antioxidants 2020, 9, 705. [Google Scholar] [CrossRef] [PubMed]
- Aouiss, A.; Anka Idrissi, D.; Kabine, M.; Zaid, Y. Update of inflammatory proliferative retinopathy: Ischemia, hypoxia and angiogenesis. Curr. Res. Transl. Med. 2019, 67, 62–71. [Google Scholar] [CrossRef]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef]
- Aboualizadeh, E.; Ranji, M.; Sorenson, C.M.; Sepehr, R.; Sheibani, N.; Hirschmugl, C.J. Retinal oxidative stress at the onset of diabetes determined by synchrotron FTIR widefield imaging: Towards diabetes pathogenesis. Analyst 2017, 142, 1061–1072. [Google Scholar] [CrossRef]
- Liao, P.L.; Lin, C.H.; Li, C.H.; Tsai, C.H.; Ho, J.D.; Chiou, G.C.; Kang, J.J.; Cheng, Y.W. Anti-inflammatory properties of shikonin contribute to improved early-stage diabetic retinopathy. Sci. Rep. 2017, 7, 44985. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Lai, T.T.; Li, C.J. The Oxidative Stress and Mitochondrial Dysfunction during the Pathogenesis of Diabetic Retinopathy. Oxid. Med. Cell. Longev. 2018, 2018, 3420187. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Zhang, W.; McDonald, D. Diabetic retinopathy: Mechanism, diagnosis, prevention, and treatment. BioMed Res. Int. 2015, 2015, 854593. [Google Scholar] [CrossRef]
- Al-Kharashi, A.S. Role of oxidative stress, inflammation, hypoxia and angiogenesis in the development of diabetic retinopathy. Saudi J. Ophthalmol. 2018, 32, 318–323. [Google Scholar] [CrossRef]
- Yue, T.; Shi, Y.; Luo, S.; Weng, J.; Wu, Y.; Zheng, X. The role of inflammation in immune system of diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Front. Immunol. 2022, 13, 1055087. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ashander, L.M.; Appukuttan, B.; Ryan, F.J.; Tan, A.C.R.; Matthews, J.M.; Michael, M.Z.; Lynn, D.J.; Smith, J.R. Selective Transcription Factor Blockade Reduces Human Retinal Endothelial Cell Expression of Intercellular Adhesion Molecule-1 and Leukocyte Binding. Int. J. Mol. Sci. 2023, 24, 3304. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A.; et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci. Rep. 2018, 8, 2847. [Google Scholar] [CrossRef] [PubMed]
- Yumnamcha, T.; Devi, T.S.; Singh, L.P. Auranofin Mediates Mitochondrial Dysregulation and Inflammatory Cell Death in Human Retinal Pigment Epithelial Cells: Implications of Retinal Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1065. [Google Scholar] [CrossRef]
- Adhya, P.; Sharma, S.S. Redox TRPs in diabetes and diabetic complications: Mechanisms and pharmacological modulation. Pharmacol. Res. 2019, 146, 104271. [Google Scholar] [CrossRef]
- Rossino, M.G.; Casini, G. Nutraceuticals for the Treatment of Diabetic Retinopathy. Nutrients 2019, 11, 771. [Google Scholar] [CrossRef]
- Xu, H.; Chen, M. Diabetic retinopathy and dysregulated innate immunity. Vis. Res. 2017, 139, 39–46. [Google Scholar] [CrossRef]
- Lu, K.; Cho, C.L.; Liang, C.L.; Chen, S.D.; Liliang, P.C.; Wang, S.Y.; Chen, H.J. Inhibition of the MEK/ERK pathway reduces microglial activation and interleukin-1-beta expression in spinal cord ischemia/reperfusion injury in rats. J. Thorac. Cardiovasc. Surg. 2007, 133, 934–941. [Google Scholar] [CrossRef]
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. [Google Scholar] [CrossRef]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.; Vaucher, E.; Couture, R. Bradykinin Type 1 Receptor–Inducible Nitric Oxide Synthase: A New Axis Implicated in Diabetic Retinopathy. Front. Pharmacol. 2019, 10, 300. [Google Scholar] [CrossRef]
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023. [Google Scholar] [CrossRef] [PubMed]
- Cecilia, O.M.; Jose Alberto, C.G.; Jose, N.P.; Ernesto German, C.M.; Ana Karen, L.C.; Luis Miguel, R.P.; Ricardo Raul, R.R.; Adolfo Daniel, R.C. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J. Diabetes Res. 2019, 2019, 8562408. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Long, P.; Guo, L.; Zhang, M.; Wang, S.; He, H. Fushiming Capsule Attenuates Diabetic Rat Retina Damage via Antioxidation and Anti-Inflammation. Evid. Based Complement. Altern. Med. 2019, 2019, 5376439. [Google Scholar] [CrossRef]
- Liu, L.; Jiang, Y.; Steinle, J.J. Glycyrrhizin Protects the Diabetic Retina against Permeability, Neuronal, and Vascular Damage through Anti-Inflammatory Mechanisms. J. Clin. Med. 2019, 8, 957. [Google Scholar] [CrossRef]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A New Human Blood-Retinal Barrier Model Based on Endothelial Cells, Pericytes, and Astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef]
- Kitamura, H. Effects of Propolis Extract and Propolis-Derived Compounds on Obesity and Diabetes: Knowledge from Cellular and Animal Models. Molecules 2019, 24, 4394. [Google Scholar] [CrossRef]
- Picconi, F.; Parravano, M.; Ylli, D.; Pasqualetti, P.; Coluzzi, S.; Giordani, I.; Malandrucco, I.; Lauro, D.; Scarinci, F.; Giorno, P.; et al. Retinal neurodegeneration in patients with type 1 diabetes mellitus: The role of glycemic variability. Acta Diabetol. 2017, 54, 489–497. [Google Scholar] [CrossRef]
- Rasmussen, K.L.; Nordestgaard, B.G.; Nielsen, S.F. Complement C3 and Risk of Diabetic Microvascular Disease: A Cohort Study of 95202 Individuals from the General Population. Clin. Chem. 2018, 64, 1113–1124. [Google Scholar] [CrossRef]
- Zhang, J.; Gerhardinger, C.; Lorenzi, M. Early complement activation and decreased levels of glycosylphosphatidylinositol-anchored complement inhibitors in human and experimental diabetic retinopathy. Diabetes 2002, 51, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Amadi-Obi, A.; Yu, C.R.; Dambuza, I.; Kim, S.H.; Marrero, B.; Egwuagu, C.E. Interleukin 27 induces the expression of complement factor H (CFH) in the retina. PLoS ONE 2012, 7, e45801. [Google Scholar] [CrossRef] [PubMed]
- Ciecko, A.E.; Foda, B.; Barr, J.Y.; Ramanathan, S.; Atkinson, M.A.; Serreze, D.V.; Geurts, A.M.; Lieberman, S.M.; Chen, Y.G. Interleukin-27 Is Essential for Type 1 Diabetes Development and Sjogren Syndrome-like Inflammation. Cell Rep. 2019, 29, 3073–3086 e3075. [Google Scholar] [CrossRef]
- Houssen, M.E.; El-Hussiny, M.A.B.; El-Kannishy, A.; Sabry, D.; El Mahdy, R.; Shaker, M.E. Serum and aqueous humor concentrations of interleukin-27 in diabetic retinopathy patients. Int. Ophthalmol. 2018, 38, 1817–1823. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.R.P.; Boia, R.; Campos, E.J.; Martins, J.; Nunes, S.; Madeira, M.H.; Santiago, A.R.; Pereira, F.C.; Reis, F.; Ambrosio, A.F.; et al. Subtle thinning of retinal layers without overt vascular and inflammatory alterations in a rat model of prediabetes. Mol. Vis. 2018, 24, 353–366. [Google Scholar] [PubMed]
- van Dijk, H.W.; Verbraak, F.D.; Stehouwer, M.; Kok, P.H.; Garvin, M.K.; Sonka, M.; DeVries, J.H.; Schlingemann, R.O.; Abramoff, M.D. Association of visual function and ganglion cell layer thickness in patients with diabetes mellitus type 1 and no or minimal diabetic retinopathy. Vis. Res. 2011, 51, 224–228. [Google Scholar] [CrossRef]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Prasad, R.; Asare-Bediko, B.; Harbour, A.; Floyd, J.L.; Chakraborty, D.; Duan, Y.; Lamendella, R.; Wright, J.; Grant, M.B. Microbial Signatures in The Rodent Eyes With Retinal Dysfunction and Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2022, 63, 5. [Google Scholar] [CrossRef]
- Taylor, A.W.; Kaplan, H.J. Ocular immune privilege in the year 2010: Ocular immune privilege and uveitis. Ocul. Immunol. Inflamm. 2010, 18, 488–492. [Google Scholar] [CrossRef]
- Fernandes, R.; Viana, S.D.; Nunes, S.; Reis, F. Diabetic gut microbiota dysbiosis as an inflammaging and immunosenescence condition that fosters progression of retinopathy and nephropathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1876–1897. [Google Scholar] [CrossRef] [PubMed]
- Madaan, A.; Chaudhari, P.; Nadeau-Vallee, M.; Hamel, D.; Zhu, T.; Mitchell, G.; Samuels, M.; Pundir, S.; Dabouz, R.; Howe Cheng, C.W.; et al. Muller Cell-Localized G-Protein-Coupled Receptor 81 (Hydroxycarboxylic Acid Receptor 1) Regulates Inner Retinal Vasculature via Norrin/Wnt Pathways. Am. J. Pathol. 2019, 189, 1878–1896. [Google Scholar] [CrossRef]
- Coburn, P.S.; Wiskur, B.J.; Astley, R.A.; Callegan, M.C. Blood-Retinal Barrier Compromise and Endogenous Staphylococcus aureus Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7303–7311. [Google Scholar] [CrossRef] [PubMed]
- Manfredo Vieira, S.; Hiltensperger, M.; Kumar, V.; Zegarra-Ruiz, D.; Dehner, C.; Khan, N.; Costa, F.R.C.; Tiniakou, E.; Greiling, T.; Ruff, W.; et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science 2018, 359, 1156–1161. [Google Scholar] [CrossRef]
- Talreja, D.; Singh, P.K.; Kumar, A. In Vivo Role of TLR2 and MyD88 Signaling in Eliciting Innate Immune Responses in Staphylococcal Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Yu, F.S.; Kumar, A. Targeting toll-like receptor signaling as a novel approach to prevent ocular infectious diseases. Indian J. Med. Res. 2013, 138, 609–619. [Google Scholar]
- Singh, P.K.; Shiha, M.J.; Kumar, A. Antibacterial responses of retinal Muller glia: Production of antimicrobial peptides, oxidative burst and phagocytosis. J. Neuroinflamm. 2014, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Shamsuddin, N.; Kumar, A. TLR2 mediates the innate response of retinal Muller glia to Staphylococcus aureus. J. Immunol. 2011, 186, 7089–7097. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amorim, M.; Martins, B.; Fernandes, R. Immune Fingerprint in Diabetes: Ocular Surface and Retinal Inflammation. Int. J. Mol. Sci. 2023, 24, 9821. https://doi.org/10.3390/ijms24129821
Amorim M, Martins B, Fernandes R. Immune Fingerprint in Diabetes: Ocular Surface and Retinal Inflammation. International Journal of Molecular Sciences. 2023; 24(12):9821. https://doi.org/10.3390/ijms24129821
Chicago/Turabian StyleAmorim, Madania, Beatriz Martins, and Rosa Fernandes. 2023. "Immune Fingerprint in Diabetes: Ocular Surface and Retinal Inflammation" International Journal of Molecular Sciences 24, no. 12: 9821. https://doi.org/10.3390/ijms24129821
APA StyleAmorim, M., Martins, B., & Fernandes, R. (2023). Immune Fingerprint in Diabetes: Ocular Surface and Retinal Inflammation. International Journal of Molecular Sciences, 24(12), 9821. https://doi.org/10.3390/ijms24129821