
Functional In Vitro Assessment of VEGFA/NOTCH2 Signaling Pathway and pRB Proteasomal Degradation and the Clinical Relevance of Mucolipin TRPML2 Overexpression in Glioblastoma Patients

 ,
,  ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Silenced or Enforced TRPML2 Expression and TRPML2 Activation in T98 and U251 Cells
2.2. Gene Expression Profile in TRPML2 Silenced T98 and U251 Cells
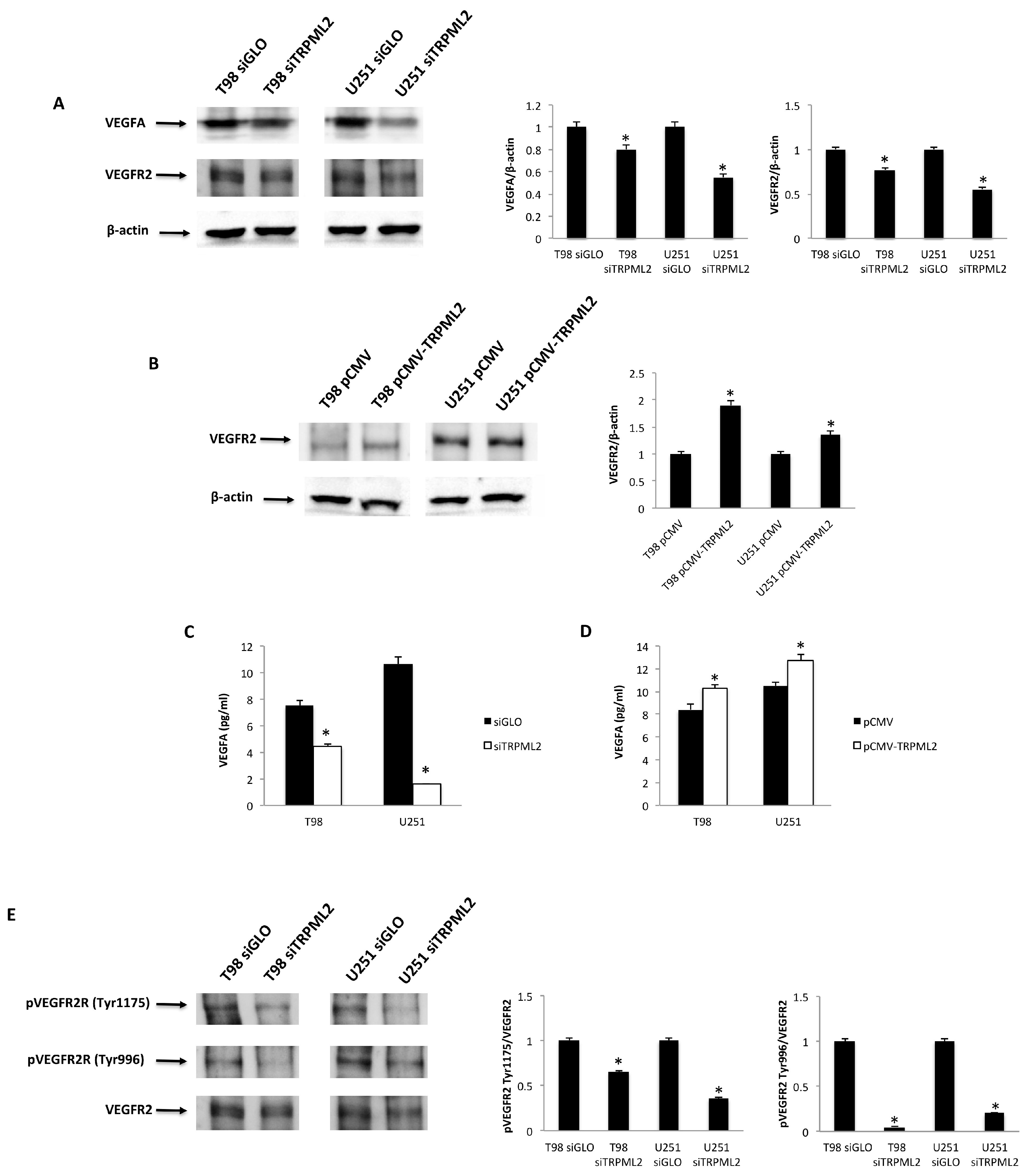
2.3. The VEGFA/VEGFR2 Signaling Pathway in TRPML2-Silenced, -Enforced and ML2-SA1-Activated T98 and U251 Cells
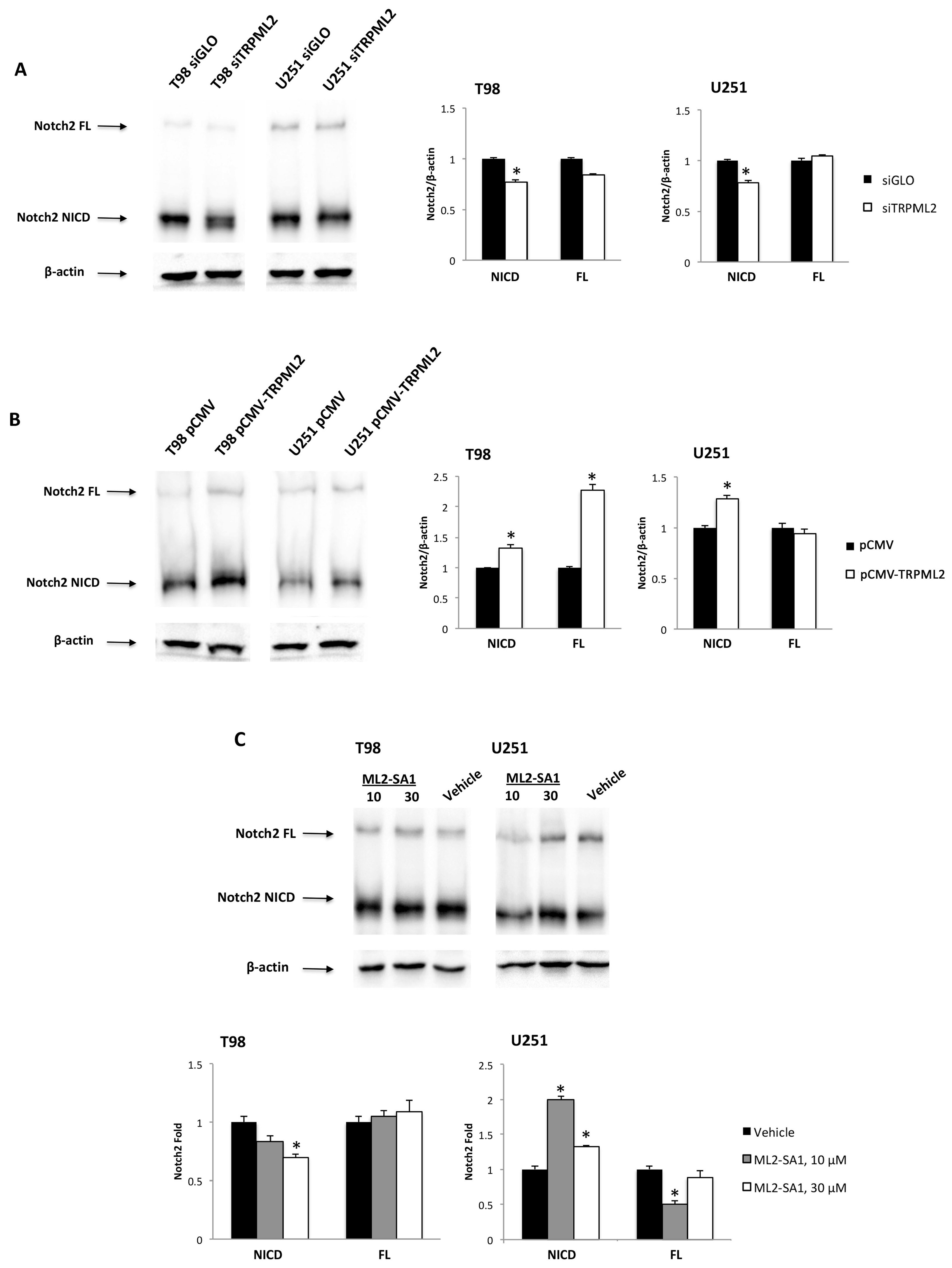
2.4. Crosstalk between Notch2 Signaling and the TRPML2 Channels in GBM Cell Lines
2.5. TRPML2 Silencing Triggers the Migration/Invasion in T98 and U251 Cells
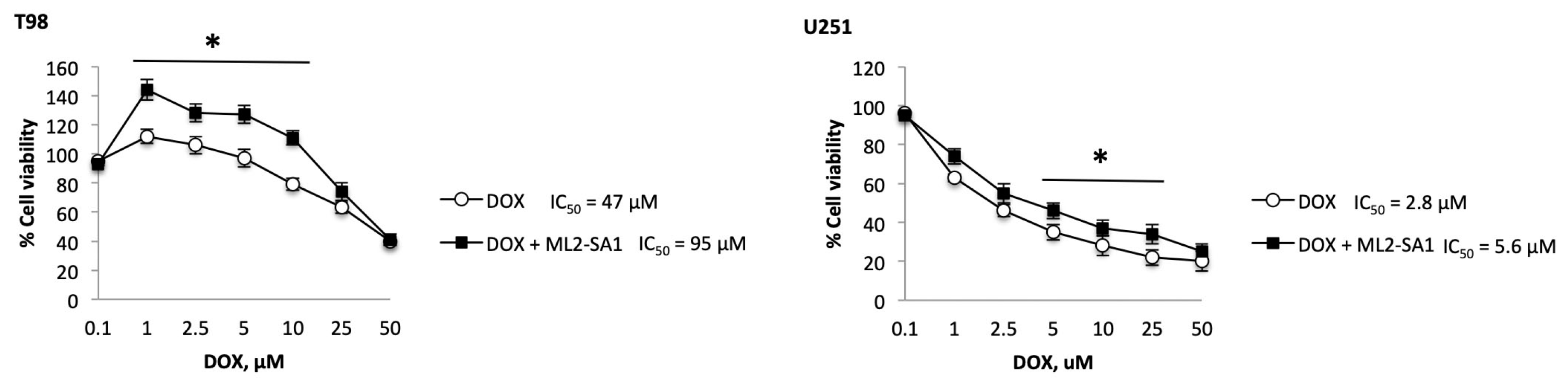
2.6. TRPML2 Silencing or ML2-SA1 Treatment Modulates the Doxorubicin Resistance in T98 and U251 Cells
2.7. Silencing of TRPML2 Triggers pRB Degradation in T98 and U251 Cells
2.8. TRPML2 Overexpression as a Negative Prognostic Factor in GBM Patients
3. Discussion
4. Materials and Methods
4.1. Cells and Tissues
4.2. Chemical and Reagents
4.3. MTT Assay
4.4. Immuno-Quantitative Enzyme-Linked Immunosorbent Assay (ELISA) for VEGFA
4.5. Western Blot Analysis
4.6. Invasion Assay
4.7. TRPML2 Transfection Models
4.8. Gene Expression Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maher, E.A.; Furnari, F.B.; Bachoo, R.M.; Rowitch, D.H.; Louis, D.N.; Cavenee, W.K.; DePinho, R.A. Malignant Glioma: Genetics and Biology of a Grave Matter. Genes Dev. 2001, 15, 1311–1333. [Google Scholar] [CrossRef]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of Transient Receptor Potential Mucolipin-2 Ion Channels in Gliomas: Role in Tumor Growth and Progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Tomassoni, D.; Nabissi, M.; Arcella, A.; Santoni, G. Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers 2019, 11, 525. [Google Scholar] [CrossRef]
- Santoni, G.; Santoni, M.; Nabissi, M.; Marinelli, O.; Morelli, M.B. Emerging Role of TRPML1 Mucolipin Endolysosomal Channel in Cancer. J. Cell. Signal. 2020, 1, 4–7. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Owsianik, G. The Transient Receptor Potential Family of Ion Channels. Genome Biol. 2011, 12, 218. [Google Scholar] [CrossRef]
- Samie, M.A.; Grimm, C.; Evans, J.A.; Curcio-Morelli, C.; Heller, S.; Slaugenhaupt, S.A.; Cuajungco, M.P. The Tissue-Specific Expression of TRPML2 (MCOLN-2) Gene Is Influenced by the Presence of TRPML1. Pflügers Arch.Eur. J. Physiol. 2009, 459, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.N.; García-Añoveros, J. TRPML2 and the Evolution of Mucolipins. Adv. Exp. Med. Biol. 2011, 704, 221–228. [Google Scholar] [CrossRef]
- Sun, L.; Hua, Y.; Vergarajauregui, S.; Diab, H.I.; Puertollano, R. Novel Role of TRPML2 in the Regulation of the Innate Immune Response. J. Immunol. 2015, 195, 4922–4932. [Google Scholar] [CrossRef]
- Santoni, G.; Morelli, M.B.; Amantini, C.; Nabissi, M.; Santoni, M.; Santoni, A. Involvement of the TRPML Mucolipin Channels in Viral Infections and Anti-Viral Innate Immune Responses. Front. Immunol. 2020, 11, 739. [Google Scholar] [CrossRef]
- Dong, X.P.; Wang, X.; Xu, H. TRP Channels of Intracellular Membranes. J. Neurochem. 2010, 113, 313–328. [Google Scholar] [CrossRef]
- Grimm, C.; Jors, S.; Guo, Z.; Obukhov, A.G.; Heller, S. Constitutive Activity of TRPML2 and TRPML3 Channels versus Activation by Low Extracellular Sodium and Small Molecules. J. Biol. Chem. 2012, 287, 22701–22708. [Google Scholar] [CrossRef]
- Karacsonyi, C.; Miguel, A.S.; Puertollano, R. Mucolipin-2 Localizes to the Arf6-Associated Pathway and Regulates Recycling of GPI-APs. Traffic 2007, 8, 1404–1414. [Google Scholar] [CrossRef]
- Zeevi, D.A.; Lev, S.; Frumkin, A.; Minke, B.; Bach, G. Heteromultimeric TRPML Channel Assemblies Play a Crucial Role in the Regulation of Cell Viability Models and Starvation-Induced Autophagy. J. Cell Sci. 2010, 123, 3112–3124. [Google Scholar] [CrossRef]
- Plesch, E.; Chen, C.-C.; Butz, E.; Scotto Rosato, A.; Krogsaeter, E.K.; Yinan, H.; Bartel, K.; Keller, M.; Robaa, D.; Teupser, D.; et al. Selective Agonist of TRPML2 Reveals Direct Role in Chemokine Release from Innate Immune Cells. eLife 2018, 7, e39720. [Google Scholar] [CrossRef]
- Almamun, M.; Levinson, B.T.; van Swaay, A.C.; Johnson, N.T.; McKay, S.D.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Integrated Methylome and Transcriptome Analysis Reveals Novel Regulatory Elements in Pediatric Acute Lymphoblastic Leukemia. Epigenetics 2015, 10, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Valadez, J.A.; Cuajungco, M.P. PAX5 Is the Transcriptional Activator of Mucolipin-2 (MCOLN2) Gene. Gene 2015, 555, 194–202. [Google Scholar] [CrossRef]
- Pérez-Riesgo, E.; Gutiérrez, L.G.; Ubierna, D.; Acedo, A.; Moyer, M.P.; Núñez, L.; Villalobos, C. Transcriptomic Analysis of Calcium Remodeling in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 922. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Cho, K.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven Cancer Cells Are Vulnerable to TRPML1 Inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef]
- Huang, C.C.; Tu, S.H.; Lien, H.H.; Jeng, J.Y.; Huang, C.S.; Huang, C.J.; Lai, L.C.; Chuang, E.Y. Concurrent Gene Signatures for Han Chinese Breast Cancers. PLoS ONE 2013, 8, e76421. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xie, M.; Meng, Z.; Lo, C.-Y.; Chan, F.L.; Jiang, L.; Meng, X.; Yao, X. Endolysosomal Ion Channel MCOLN2 (Mucolipin-2) Promotes Prostate Cancer Progression via IL-1β/NF-ΚB Pathway. Br. J. Cancer 2021, 125, 1420–1431. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor Angiogenesis: Causes, Consequences, Challenges and Opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. αv Integrins in Angiogenesis and Cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Yeager, K.; Morrison, S.J. Physiological Notch Signaling Promotes Gliogenesis in the Developing Peripheral and Central Nervous Systems. Development 2007, 134, 2435–2447. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Miele, L. Notch Signaling. Clin. Cancer Res. 2006, 12, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Miao, H.; Nickoloff, B. NOTCH Signaling as a Novel Cancer Therapeutic Target. Curr. Cancer Drug Targets 2006, 6, 313–323. [Google Scholar] [CrossRef]
- Teodorczyk, M.; Schmidt, M.H.H. Notching on Cancer’s Door: Notch Signaling in Brain Tumors. Front. Oncol. 2015, 4, 341. [Google Scholar] [CrossRef]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; van der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of Notch Signaling in Glioblastoma Targets Cancer Stem Cells via an Endothelial Cell Intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef]
- Qiang, L.; Wu, T.; Zhang, H.-W.; Lu, N.; Hu, R.; Wang, Y.-J.; Zhao, L.; Chen, F.-H.; Wang, X.-T.; You, Q.-D.; et al. HIF-1α Is Critical for Hypoxia-Mediated Maintenance of Glioblastoma Stem Cells by Activating Notch Signaling Pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef]
- Stockhausen, M.-T.; Kristoffersen, K.; Poulsen, H.S. Notch Signaling and Brain Tumors. Adv. Exp. Med. Biol. 2012, 727, 289–304. [Google Scholar] [CrossRef]
- Ferreira, W.A.S.; Araújo, M.D.; Anselmo, N.P.; de Oliveira, E.H.C.; Brito, J.R.N.; Burbano, R.R.; Harada, M.L.; do Nascimento Borges, B. Expression Analysis of Genes Involved in the RB/E2F Pathway in Astrocytic Tumors. PLoS ONE 2015, 10, e0137259. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Pei, Q.; Li, J.; Wan, X.; Ye, T. Emerging Role of E2F Family in Cancer Stem Cells. Front. Oncol. 2021, 11, 3164. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Olsson, A.-K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF Receptor Signalling-in Control of Vascular Function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Differential Roles of Vascular Endothelial Growth Factor Receptor-1 and Receptor-2 in Angiogenesis. J. Biochem. Mol. Biol. 2006, 39, 469–478. [Google Scholar] [CrossRef]
- Lu, Y.; Xiao, L.; Liu, Y.; Wang, H.; Li, H.; Zhou, Q.; Pan, J.; Lei, B.; Huang, A.; Qi, S. MIR517C Inhibits Autophagy and the Epithelial-to-Mesenchymal (-like) Transition Phenotype in Human Glioblastoma through KPNA2-Dependent Disruption of TP53 Nuclear Translocation. Autophagy 2015, 11, 2213–2232. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef]
- Niklasson, M.; Bergström, T.; Jarvius, M.; Sundström, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal Transition and Increased Therapy Resistance of Glioblastoma Cells Is Related to Astrocyte Reactivity. J. Pathol. 2019, 249, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 1314. [Google Scholar] [CrossRef]
- Huang, X.; Masselli, A.; Frisch, S.M.; Hunton, I.C.; Jiang, Y.; Wang, J.Y.J. Blockade of Tumor Necrosis Factor-Induced Bid Cleavage by Caspase-Resistant Rb. J. Biol. Chem. 2007, 282, 29401–29413. [Google Scholar] [CrossRef] [PubMed]
- Biasoli, D.; Kahn, S.A.; Cornélio, T.A.; Furtado, M.; Campanati, L.; Chneiweiss, H.; Moura-Neto, V.; Borges, H.L. Retinoblastoma Protein Regulates the Crosstalk between Autophagy and Apoptosis, and Favors Glioblastoma Resistance to Etoposide. Cell Death Dis. 2013, 4, e767. [Google Scholar] [CrossRef]
- Colletti, G.A.; Miedel, M.T.; Quinn, J.; Andharia, N.; Weisz, O.A.; Kiselyov, K. Loss of Lysosomal Ion Channel Transient Receptor Potential Channel Mucolipin-1 (TRPML1) Leads to Cathepsin B-Dependent Apoptosis. J. Biol. Chem. 2012, 287, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Nishinaka, T.; Yokoyama, K.; Chiu, R. A Retinoblastoma Susceptibility Gene Product, RB, Targeting Protease Is Regulated through the Cell Cycle. FEBS Lett. 1998, 421, 89–93. [Google Scholar] [CrossRef]
- Sever, N.; Filipic, M.; Brzin, J.; Lah, T.T. Effect of Cysteine Proteinase Inhibitors on Murine B16 Melanoma Cell Invasion in Vitro. Biol. Chem. 2002, 383, 839–842. [Google Scholar] [CrossRef]
- Karkkainen, M.J.; Ferrell, R.E.; Lawrence, E.C.; Kimak, M.A.; Levinson, K.L.; McTigue, M.A.; Alitalo, K.; Finegold, D.N. Missense Mutations Interfere with VEGFR-3 Signalling in Primary Lymphoedema. Nat. Genet. 2000, 25, 153–159. [Google Scholar] [CrossRef]
- Rahimi, N.; Dayanir, V.; Lashkari, K. Receptor Chimeras Indicate That the Vascular Endothelial Growth Factor Receptor-1 (VEGFR-1) Modulates Mitogenic Activity of VEGFR-2 in Endothelial Cells. J. Biol. Chem. 2000, 275, 16986–16992. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. Signal Transduction by Vascular Endothelial Growth Factor Receptors. Biochem. Soc. Trans. 2003, 31, 20–24. [Google Scholar] [CrossRef]
- Meyer, M.; Clauss, M.; Lepple-Wienhues, A.; Waltenberger, J.; Augustin, H.G.; Ziche, M.; Lanz, C.; Büttner, M.; Rziha, H.J.; Dehio, C. A Novel Vascular Endothelial Growth Factor Encoded by Orf Virus, VEGF-E, Mediates Angiogenesis via Signalling through VEGFR-2 (KDR) but Not VEGFR-1 (Flt-1) Receptor Tyrosine Kinases. EMBO J. 1999, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.; Waltenberger, J. The Vascular Endothelial Growth Factor Receptor KDR Activates Multiple Signal Transduction Pathways in Porcine Aortic Endothelial Cells. J. Biol. Chem. 1997, 272, 32521–32527. [Google Scholar] [CrossRef]
- Doughervermazen, M.; Hulmes, J.D.; Bohlen, P.; Terman, B.I. Biological Activity and Phosphorylation Sites of the Bacterially Expressed Cytosolic Domain of the KDR VEGF-Receptor. Biochem. Biophys. Res. Commun. 1994, 205, 728–738. [Google Scholar] [CrossRef]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A Single Autophosphorylation Site on KDR/Flk-1 Is Essential for VEGF-A-Dependent Activation of PLC-Gamma and DNA Synthesis in Vascular Endothelial Cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef]
- Holmqvist, K.; Cross, M.J.; Rolny, C.; Hägerkvist, R.; Rahimi, N.; Matsumoto, T.; Claesson-Welsh, L.; Welsh, M. The Adaptor Protein Shb Binds to Tyrosine 1175 in Vascular Endothelial Growth Factor (VEGF) Receptor-2 and Regulates VEGF-Dependent Cellular Migration. J. Biol. Chem. 2004, 279, 22267–22275. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Valapala, M.; Hose, S.; Gongora, C.; Dong, L.; Wawrousek, E.F.; Samuel Zigler, J.; Sinha, D. Impaired Endolysosomal Function Disrupts Notch Signalling in Optic Nerve Astrocytes. Nat. Commun. 2013, 4, 1629. [Google Scholar] [CrossRef] [PubMed]
- Rittierodt, M.; Harada, K. Repetitive Doxorubicin Treatment of Glioblastoma Enhances the PGP Expression–a Special Role for Endothelial Cells. Exp. Toxicol. Pathol. 2003, 55, 39–44. [Google Scholar] [CrossRef]
- da Ros, M.; de Gregorio, V.; Iorio, A.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-N.; Yang, J.-T.; Hsieh, M.-J.; Huang, C.; Huang, H.-C.; Ku, Y.-J.; Wu, Y.-P.; Huang, K.-C.; Chen, J.-C. Knockdown of Amphiregulin Triggers Doxorubicin-Induced Autophagic and Apoptotic Death by Regulating Endoplasmic Reticulum Stress in Glioblastoma Cells. J. Mol. Neurosci. 2020, 70, 1461–1470. [Google Scholar] [CrossRef]
- Chen, C.-C.; Krogsaeter, E.; Butz, E.S.; Li, Y.; Puertollano, R.; Wahl-Schott, C.; Biel, M.; Grimm, C. TRPML2 Is an Osmo/Mechanosensitive Cation Channel in Endolysosomal Organelles. Sci. Adv. 2020, 6, eabb5064. [Google Scholar] [CrossRef]
- Armento, A.; Ehlers, J.; Schötterl, S.; Naumann, U.; De Vleeschouwer, S. Molecular Mechanisms of Glioma Cell Motility. In Glioblastoma; Chapter 5; Codon Publications: Brisbane, Australia, 2017; pp. 73–93. ISBN 978-0-9944381-2-6. [Google Scholar] [CrossRef]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF Inhibits Tumor Cell Invasion and Mesenchymal Transition through a MET/VEGFR2 Complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef]
- Bien-Möller, S.; Balz, E.; Herzog, S.; Plantera, L.; Vogelgesang, S.; Weitmann, K.; Seifert, C.; Fink, M.A.; Marx, S.; Bialke, A.; et al. Association of Glioblastoma Multiforme Stem Cell Characteristics, Differentiation, and Microglia Marker Genes with Patient Survival. Stem Cells Int. 2018, 2018, 9628289. [Google Scholar] [CrossRef]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and Its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the Lysosome in Cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Dick, L.R.; Fleming, P.E. Building on Bortezomib: Second-Generation Proteasome Inhibitors as Anti-Cancer Therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Amantini, C.; Nabissi, M.; Maggi, F.; Arcella, A.; Marinelli, O.; Eleuteri, A.M.; Santoni, M.; Morelli, M.B. Knock-Down of Mucolipin 1 Channel Promotes Tumor Progression and Invasion in Human Glioblastoma Cell Lines. Front. Oncol. 2021, 11, 578928. [Google Scholar] [CrossRef]
- Kleihues, P.; Cavenee, W.K. World Health Organisation Classification of Tumours. In Pathology and Genetics of Tumours of the Nervous System; IARC Press: Lyon, France, 2000; p. 314. ISBN 9789283224099. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | T98 siGLO | T98 siTRPML2 |
|---|---|---|
| ALCAM | 826 ± 12 | 854 ± 9 |
| CD44 | 4510 ± 17 | 7800 ± 23 * |
| VIMENTIN | 6421 ± 19 | 7270 ± 7 * |
| ZEB1 | 566 ± 8 | 540 ± 12 |
| ZEB2 | 44 ± 2 | 50 ± 3 |
| VEGFA | 1506 ± 4 | 1273 ± 5 * |
| VEGFB | 1492 ± 7 | 1634 ± 10 * |
| NOTCH1 | 34 ± 4 | 30 ± 2 |
| NOTCH2 | 1492 ± 12 | 988 ± 9 * |
| STAT3 | 1090 ± 14 | 1368 ± 17 * |
| SPARC | 1835 ± 15 | 2810 ± 15 * |
| EPCAM | 5 ± 0 | 3 ± 0 |
| SHH | 0 ± 0 | 0 ± 0 |
| DHH | 7 ± 1 | 0 ± 0 |
| IHH | 0 ± 0 | 0 ± 0 |
| PTCH1 | 83 ± 3 | 79 ± 2 |
| PTCH2 | 2 ± 0 | 2 ± 0 |
| SMO | 257 ± 3 | 246 ± 6 |
| POU5F1B | 0 ± 0 | 0 ± 0 |
| ACTB | 7400 ± 37 | 7500 ± 53 |
| Target Gene | U251 siGLO | U251 siTRPML2 |
|---|---|---|
| ALCAM | 1153 ± 13 | 1218 ± 9 |
| CD44 | 10,200 ± 21 | 10,300 ± 16 |
| VIMENTIN | 6740 ± 12 | 7900 ± 10 * |
| ZEB1 | 110 ± 3 | 143 ± 6 |
| ZEB2 | 167 ± 5 | 130 ± 4 |
| VEGFA | 3440 ± 16 | 1617 ± 13 * |
| VEGFB | 537 ± 7 | 698 ± 10 * |
| NOTCH1 | 381 ± 7 | 132 ± 6 * |
| NOTCH2 | 2020 ± 12 | 1476 ± 14 * |
| STAT3 | 984 ± 13 | 1192 ± 6 * |
| SPARC | 1964 ± 9 | 2254 ± 6 * |
| EPCAM | 0 ± 0 | 2 ± 0 |
| SHH | 5 ± 0 | 1 ± 0 |
| DHH | 3 ± 1 | 1 ± 0 |
| IHH | 4 ± 0 | 1 ± 0 |
| PTCH1 | 27 ± 2 | 32 ± 1 |
| PTCH2 | 102 ± 3 | 59 ± 3 |
| SMO | 37 ± 2 | 54 ± 2 |
| POU5F1B | 1 ± 0 | 2 ± 0 |
| ACTB | 11,300 ± 32 | 11,300 ± 46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoni, G.; Amantini, C.; Nabissi, M.; Arcella, A.; Maggi, F.; Santoni, M.; Morelli, M.B. Functional In Vitro Assessment of VEGFA/NOTCH2 Signaling Pathway and pRB Proteasomal Degradation and the Clinical Relevance of Mucolipin TRPML2 Overexpression in Glioblastoma Patients. Int. J. Mol. Sci. 2022, 23, 688. https://doi.org/10.3390/ijms23020688
Santoni G, Amantini C, Nabissi M, Arcella A, Maggi F, Santoni M, Morelli MB. Functional In Vitro Assessment of VEGFA/NOTCH2 Signaling Pathway and pRB Proteasomal Degradation and the Clinical Relevance of Mucolipin TRPML2 Overexpression in Glioblastoma Patients. International Journal of Molecular Sciences. 2022; 23(2):688. https://doi.org/10.3390/ijms23020688
Chicago/Turabian StyleSantoni, Giorgio, Consuelo Amantini, Massimo Nabissi, Antonietta Arcella, Federica Maggi, Matteo Santoni, and Maria Beatrice Morelli. 2022. "Functional In Vitro Assessment of VEGFA/NOTCH2 Signaling Pathway and pRB Proteasomal Degradation and the Clinical Relevance of Mucolipin TRPML2 Overexpression in Glioblastoma Patients" International Journal of Molecular Sciences 23, no. 2: 688. https://doi.org/10.3390/ijms23020688
APA StyleSantoni, G., Amantini, C., Nabissi, M., Arcella, A., Maggi, F., Santoni, M., & Morelli, M. B. (2022). Functional In Vitro Assessment of VEGFA/NOTCH2 Signaling Pathway and pRB Proteasomal Degradation and the Clinical Relevance of Mucolipin TRPML2 Overexpression in Glioblastoma Patients. International Journal of Molecular Sciences, 23(2), 688. https://doi.org/10.3390/ijms23020688