Abstract
Despite extensive preclinical research on immunotherapeutic approaches, malignant glioma remains a devastating disease of the central nervous system for which standard of care treatment is still confined to resection and radiochemotherapy. For peripheral solid tumors, immune checkpoint inhibition has shown substantial clinical benefit, while promising preclinical results have yet failed to translate into clinical efficacy for brain tumor patients. With the advent of high-throughput sequencing technologies, tumor antigens and corresponding T cell receptors (TCR) and antibodies have been identified, leading to the development of chimeric antigen receptors (CAR), which are comprised of an extracellular antibody part and an intracellular T cell receptor signaling part, to genetically engineer T cells for antigen recognition. Due to efficacy in other tumor entities, a plethora of CARs has been designed and tested for glioma, with promising signs of biological activity. In this review, we describe glioma antigens that have been targeted using CAR T cells preclinically and clinically, review their drawbacks and benefits, and illustrate how the emerging field of transgenic TCR therapy can be used as a potent alternative for cell therapy of glioma overcoming antigenic limitations.
1. Introduction
Despite intensive research over the last decades, standard of care (SOC) treatment for malignant gliomas is still restricted to resection and radiochemotherapy. The tremendous clinical effects of immune checkpoint inhibition (ICI) have revolutionized therapy for many cancer entities such as melanoma but have not conferred clinical benefit to brain tumor patients, yet despite promising preclinical results [1,2]. However, none of the phase 3 clinical trials using checkpoint-inhibiting molecules in gliomas met their primary clinical endpoints for patients with newly diagnosed or relapsed glioblastoma (GBM) (Checkmate 143, 498) [3,4,5]. Conversely, more recently, two independent phase 2 trials showed response of neoadjuvant programmed cell death protein 1 (PD-1) therapy in recurrent and operable GBM with response-associated distinct immunogenomic features [6,7,8].
Cellular therapies have become an emerging field in preclinical and clinical cancer research. The first cellular therapies in solid tumors were conducted in 1980 by Rosenberg and colleagues using expanded tumor-infiltrating leukocytes (TIL) and high dose interleukin (IL) 2 [9,10,11]. For brain tumors, TIL therapy in patients with GBM and melanoma brain metastases has been investigated [12,13,14]. However, although promising in some post-hoc analyzed subgroups, the overall outcome of these trials was unsatisfactory even though ex vivo TIL cultures from GBM patients have been shown to exert tumor reactivity [15]. As the usage of endogenous T cells comes with a variety of caveats, such as potential incomplete in vitro reinvigoration of exhausted TIL, limited capacity of TIL to expand in vivo after a strong preceding in vitro stimulation, and potential predominant expansion of bystander T cells, the use of genetically modified T cells could circumvent these obstacles. In recent years, there has been remarkable effort in identifying suitable targets for cellular glioma immunotherapy [16,17]. Chimeric antigen receptor (CAR) T cells have shown tremendous effects in non-solid tumors such as multiple myeloma and leukemia and have recently been approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA). For solid tumors, a plethora of early CAR T cell clinical trials has recently been initiated [18]. CARs are designed by using an antibody-derived extracellular recognition domain, a hinging transmembrane domain, and an intracellular T cell receptor (TCR)-derived signaling domain. The antibody-derived variable regions are able to recognize extracellular domains and proteins and bypass major histocompatibility complex (MHC) expression and presentation by tumor cells or professional antigen presenting cells (APC). Alternatively, modified natural ligands of surface receptors may be used as extracellular recognition domains. Modifying the intracellular signaling domain and the addition of co-stimulatory signals has led to the development of second, third, and fourth-generation CARs [19]. In preclinical studies, several CARs against glioma-associated target structures have been developed. In this review, we focus on CAR T cell therapies, highlighting targets for such therapy (Figure 1), we then discuss early phase clinical trials (Table 1), and elaborate on the benefits and drawbacks of CAR T cell therapy, especially in comparison to TCR-engineered T cell therapy. Attention will be turned to the consideration of application routes.
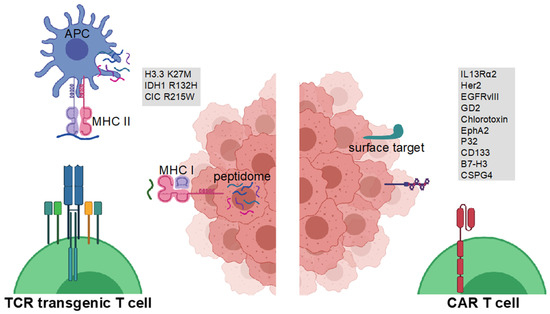
Figure 1.
Glioma antigens for CAR- and TCR-engineered T cell therapy. TCR-engineered T cells target MHC class I-bound short peptides or MHC class II-bound long glioma-specific peptides on glioma cells or glioma-associated myeloid professional antigen-presenting cells (APC), respectively (left). CAR T cells target cell surface proteins on glioma cells (right). Figure created with BioRender.com.

Table 1.
Clinical trials investigating genetically modified cellular therapies in brain tumors.
2. Glioma Antigens for CAR-Engineered T Cell Therapy
2.1. IL13Ra2
Interleukin-13 receptor subunit alpha-2 (IL13Ra2) was the first target in GBM to have been exploited for CAR T cell therapy. As required for tumor-associated targets, IL13Ra2 is highly expressed in a high frequency on tumor cells of GBM patients [20,21,22]. In contrast, healthy tissue, except the testis, express low levels of IL13Ra2, making it an adequate target for targeted therapy. The ubiquitously expressed IL13Ra1 binds IL13 with lower affinity than IL13Ra2, allowing for predominant targeting with modified IL13 variants [23,24,25]. The first CAR targeting IL13Ra2 was developed in 2004 and included a so-called zetakine, which was composed of an extracellular altered IL13 domain tethered to an immunoglobulin transmembrane domain and a CD3-zeta cytoplasmic part [26]. In vitro and in vivo studies in human xenografts showed specific and effective tumor lysis. Second-generation CARs, which include a co-stimulatory 4-1BB domain, outperformed the first-generation CARs while not leading to cytotoxicity in non-obese diabetic (NOD) scid gamma (NSG) mice [27]. Further improvements, such as the addition of an activation-induced expression of IL15 in CAR T cells, led to increased CAR proliferation, cytokine production and persistence [28]. The first in-human clinical trial evaluating IL13Ra2-specific CARs was completed in 2015 and enrolled three patients with recurrent GBM being treated with an IL13-zetakine CAR product [29]. The CAR product was injected directly into the resection cavity in 12 doses over 5 weeks. Overall, the treatment was well tolerated in these patients with only temporary brain inflammation events, indicating promising tolerability of T cell products. Of note, a decrease in tumoral IL13Ra2 expression after CAR therapy was reported in one patient, suggesting therapy-driven antigen loss. On basis of an outstanding casuistic report on a patient with recurrent GBM treated with a 4-1BB-modified CAR product [30], intraventricular IL13Ra2 CAR T cell therapy is currently assessed in a phase 1 clinical trial for ependymoma, GBM, and medulloblastoma (NCT04661384), while intratumoral delivery is being tested in recurrent or refractory malignant glioma (NCT02208362).
2.2. Her2
The human epidermal growth factor receptor 2 (Her2) constitutes a GBM-associated antigen, being expressed in approximately 80% of all GBM patients [31,32]. Her2-specific CAR T cells demonstrated preclinical efficacy in several tumor models, were shown to effectively target Her2-positive glioma cells and glioma stem cells, and to lead to regression of GBM xenografts [31,33,34,35,36]. Interestingly, the xenograft study used syngeneic T cells and GBM cells in a patient-derived xenograft (PDX) model, avoiding allogenic immune responses. In another study, Ahmed et al. showed sustained regression of medulloblastoma xenografts in immunodeficient mice using intratumoral adoptive transfer of Her2-specific CAR T cells. As 40% of medulloblastomas overexpress Her2, CAR T cell therapy might represent a promising therapeutic option [37]. Recently, Her2-specific CAR NK cells, derived from the human NK cell line NK-92, have been reported to specifically lyse GBM-derived cell lines and to show in vivo anti-tumor activity in xenografts and immunocompetent mouse models [38]. This NK cell-line-based concept is currently investigated in a phase 1 clinical trial (NCT03383978). Overall, the clinical usage of HER2-specific CARs has been challenged by a case report in 2010 where the administration of a HER2-directed CAR product in a patient with metastatic colon cancer led to a severe and lethal cytokine storm [39]. More recently, subsequent clinical studies reported no severe systemic toxicities [40,41]. In GBM, a phase 1 dose escalation study using HER2-specific CAR T cells derived from virus-specific T cells (VST) showed tolerability with no dose-limiting toxic effect in 17 patients [42]. However, the HER2-VST CARs did not expand in the peripheral blood and clinical efficacy was limited with a median OS of 11.1 months. An interim analysis of a phase 1 clinical trial using locoregional delivery of HER2-specific CAR T cells recently reported no dose-limiting toxicity of multiple CAR T cell infusions and demonstrated highly elevated interferon-induced C-X-C motif chemokine ligand 10 (CXCL10) and CC-chemokine ligand 2 (CCL2) levels in the cerebrospinal fluid (CSF) after CAR T cell infusion [43]. Magnetic resonance (MR) imaging showed vasogenic edema and local intensified contrast enhancement, indicating an inflammatory response called pseudoprogression. The associated clinical trial is composed of two arms, delivering the CAR T cell product into the tumor cavity or intraventricularly, respectively (NCT03500991).
2.3. EGFRvIII
The mutated epidermal growth factor receptor variant III (EGFRvIII) results from an amplification of the wildtype EGFR and is expressed in approximately 30% of all GBM patients [44,45,46]. Vaccination or antibody treatment against EGFRvIII has been shown to induce increased survival and long-term immunological memory in various preclinical models [47,48,49], indicating that EGFRvIII can serve as a potent target for cellular therapies. The feasibility of using EGFRvIII-directed CARs has been extensively studied. In a comprehensive study, Morgan et al. screened seven different antibodies for the ability to be used in a CAR T cell product [50]. CAR T cells based on three of these seven antibodies were shown to produce effector cytokines in response to EGFRvIII-expressing glioma cells. In a follow-up study, the authors could show preclinical efficacy of a murine third generation EGFRvIII-directed CAR product in a syngeneic mouse model [51]. Since this study was conducted in immunocompetent mice, the authors reported two important key findings in their preclinical glioma model: first, it was shown that lymphodepletion is needed for a response to systemically injected CAR T cells. Second, CAR T cell therapy was able to induce endogenous long-term immunity against the tumor, even when mice were rechallenged with tumor cells not expressing EGFRvIII. Another study using EGFRvIII CAR T cells showed that overexpression of the mircoRNA miR-17-92, a microRNA that has been reported to enhance T cell survival and interferon (IFN)-γ production and to be downregulated in GBM-infiltrating T cells, increases EGFRvIII CAR T cell function [52]. Interestingly, EGFR806-CAR T cells, recognizing not only EGFRvIII but also full-length gene amplified wildtype EGFR, retained their specificity due to specific steric accessibility of tumor-expressed EGFR [53]. A first in-human study using EGFRvIII-directed CARs was published in 2017 by O’Rourke and colleagues [54]. The CAR design used for this study was selected based on a comprehensive preclinical study [55]. Ten patients were treated with a single peripherally infused dose of the CAR T cell product. Upon recurrence, tissue analysis post CAR T cell transfer revealed that CAR T cells were effectively trafficking to the brain. Reduced EGFRvIII expression in infiltrated regions was rather representing the natural cause of the disease than a sign of immunological escape [56]. However, CAR T cell infiltration led to an increase in T regulatory cell abundance and increased expression of inhibitory molecules such as programmed death-ligand 1 (PD-L1), transforming growth factor ß (TGF-ß), and IL10. More recently, EGFRvIII-specific CAR T cell clinical trials for intracranial tumors, including GBM, have been closed prior to completion due to lack of funding, observed toxicity, shift towards next CAR T cell iteration or combinatorial treatments, or missing objective clinical responses (NCT01454596, NCT02664363, NCT02209376, NCT03283631).
2.4. GD2
The disialoganglioside GD2 is frequently overexpressed in neuroblastoma with only restricted expression in healthy tissue, classifying it a tumor-associated antigen. CAR T cell therapy was able to abrogate tumor progression in a xenograft model [57]. CAR T cells against GD2 have also shown tremendous preclinical efficacy in PDX models of H3.3.K27M-mutated midline gliomas [58]. Using second-generation 4-1BB-overexpressing CAR T cells, tumors were almost cleared from different localizations (pons, thalamus, and spinal cord) with a small amount of GD2-negative tumor cells remaining. However, accompanying the strong anti-tumor effect, the authors also observed severe neuroinflammation in immunodeficient mice. GD2 CAR T cell therapy is currently tested in a phase 1 trial in diffuse midline gliomas with retroviral vectors manufactured in the closed CliniMACS Prodigy system (NCT04196413), in combination with constitutive active IL7 receptor (NCT04099797), or in recurrent gliomas (NCT03423992). In the terminated above-mentioned phase 1 clinical trial (NCT04196413) using a retro-viral (14g2a-CD8.BB.z.iCasp9) expressing GD2-chimeric antigen receptor, three of four patients exhibited marked improvement or resolution of neurological deficits as well as radiographic improvements. Moreover, no on-target off-tumor toxicity was observed [59].
2.5. Chlorotoxin
Chlorotoxin, a venom-derived peptide, has been described to specifically bind to GBM cells [60,61]. Recently, researchers developed a chlorotoxin-based CAR with the peptide as targeting domain that efficiently targeted tumors with heterogenous expression of GBM-associated antigens such as IL13Rα2, HER2, and EGFR [62]. Of note, the chlorotoxin-based CAR required matrix metalloproteinase 2 (MMP2) expression on the tumor cells for efficient binding. Antitumor capacity was assessed in orthotopic GBM PDX models without severe off-target effects after systemic intravenous or regional intraventricular or intratumoral CAR T cell delivery. A phase 1 clinical trial assessing CAR T cells with a chlorotoxin tumor-targeting domain in MMP2-positive recurrent or progressive GBM is currently recruiting patients (NCT04214392).
2.6. EphA2
Ephrin type-A receptor 2 (EphA2) is a receptor tyrosine kinase that binds ephrin-A family ligands and its downstream signaling participates in migration, proliferation, differentiation, and integrin-mediated adhesion [63,64]. It has been described as a glioma-associated antigen with limited expression in healthy tissue with the exception of some epithelial cells [65]. Its overexpression has been reported in several tumor types and linked to decreased overall survival in patients with GBM [66]. Several preclinical studies used EphA2-directed CAR T cells to treat GBM xenografts and showed potent anti-tumor activity against glioma-initiating cells [67,68]. Preclinical locoregional delivery of CAR T cells was validated as effective treatment in medulloblastoma mouse models [69]. However, to date, to our knowledge, clinical studies evaluating EphA2-directed CAR T cells have not yet been initiated.
2.7. P32
P32, also known as complement component 1 Q subcomponent-binding protein (C1QBP), has previously been reported to be expressed in tumor cells and tumor-associated endothelial cells [70]. More recently, Rousso-Noori et al. reported P32 to be specifically expressed on murine and human glioma cells [71]. In their study, CAR T cell therapy was able to reduce tumor growth in xenograft and syngeneic mouse models. The authors used a combination of intratumoral and intraventricular injection of the CAR T cell product, leading to sustained infiltration of CAR T cells into the tumor. Conceptualization of clinical trials investigating P32 as CAR T cell target can be expected.
2.8. CD133
CD133 is a pentaspan transmembrane glycoprotein reported to be predominantly expressed on cancer, hematopoietic, and neural stem cells [72,73,74]. Several studies have reported CD133 to be involved in tumor initiation and resistance to radio- and chemotherapy [75,76,77]. Upregulation is considered to be prognostically unfavorable. In a side-by-side comparison, while different modalities against CD133 showed efficacy in orthotopic GBM xenografts, CD133-specific CAR-T cells represented the most efficacious [62]. Interestingly, in hematopoietic stem-cell-humanized NOD scid gamma (NSG) mice, intraventricular injection of CD133-specific CAR T cells was effective and did not lead to reduced frequencies of CD34 CD133 double positive hematopoietic cells [73]. As of now, no phase 1 clinical trial using CD133-specific CAR T cells has been initiated.
2.9. CSPG4
Chondroitin sulfate proteoglycan 4 (CSPG4) is a type I transmembrane protein that is overexpressed in 67% of GBM [78]. GBM with high CSPG4 expression are considered to be more aggressive than their low expressing counterparts. Specific and cell ratio-dependent killing was observed when CSPG4-specific CAR T cells were co-cultured with CSPG4-expressing primary GBM cell lines or when injected intratumorally in human GBM-bearing nude mice. In these PDX model, no relevant post-treatment antigen loss was observed, which is suggestive for homogeneous target expression in primary GBM cell lines and throughout in vivo growth. To our knowledge, thus far, no clinical trials have been initiated targeting CSPG4 in gliomas.
2.10. B7-H3
B7 Homolog 3 (B7-H3) is a type I transmembrane protein that is overexpressed in 76% of GBM [79]. Using established human glioma cell lines such as U87 in nude mice, intratumorally injected B7-H3-specific CAR T cells lead to durable responses independent of applied co-stimulatory domains. Similarly to observations by Nehama et al., Tang et al. reported increased preclinical survival after intravenous injection of B7-H3-specific CAR T cells in U87-bearing NOD SCID mice [80]. More recently, B7-H3 has been successfully co-targeted by B7-H3-CD70 tandem CARs (Tan-CAR) [81]. Compared to single targeting of CD70 or B7-H3, respectively, improved preclinical survival was observed when Tan-CAR T cells were adoptively transferred in non-glioma PDX. Interestingly, B7-H3 co-expression was also observed in glioma. Currently, three clinical trials investigating B7-H3-specific CAR T therapy in recurrent or refractory GBM or various other central nervous system (CNS) tumors are recruiting patients (Table 1).
3. Preclinical Strategies to Overcome Off-Target and On-Target Off-Tumor Side Effects
Beyond potential genotoxicity (due to the risk of oncogenic insertional mutagenesis by viral expression vectors), immunogenicity (of CAR epitopes), and off-target side effects, (i.e., interaction of CARs with Fc receptors expressed on myeloid cells inducing antigen-independent T cell activation), severe on-target off-tumor side effects can occur, because CAR T cells usually target tumor-associated rather than tumor-specific antigens. For clinical translation in brain tumors, it is essential to carefully assess potential on-target off-tumor side effects that can lead to severe neuroinflammation and neurotoxicity. Recently, in an observational study, Parker and colleagues identified CD19-expressing brain mural cells as a potential on-target off-site in CD19-specific CAR T cell therapy [82]. Interestingly, on-target off-site-relevant CD19 expression was found in human brain mural cells but not in murine systems, highlighting the limitations of studies on off- and on-target side effects in mouse model systems. One option to overcome on-target off-tumor side effects is the insertion of additional targetable epitopes in CAR constructs themselves [83]. In a preclinical study using an acute myeloid leukemia xenograft mouse model, Bonifant et al. introduced a CD20 domain into their CAR construct, enabling elimination of CAR T cells via rituximab, a CD20-specific therapeutic antibody, if required [84]. To our knowledge, such strategy thus far has not been pursued in the context of preclinical brain tumors. The design of dual CAR constructs could potentially prevent CAR activation distant from the tumor site. By using a synNotch receptor in a bilateral K562-bearing xenograft model, Roybal and colleagues could show specific CAR expression on T cells only after delivery of a first expression-inducing signal [85]. After antigenic ligand encounter, synNotch receptor activation leads to cleavage of a transcription factor within its intracellular signaling domain, which enables expression of the CAR specific for a second antigen. Recently, synNotch CAR T cells that are activated by local and tumor-specific antigen encounters have shown to lead to a specific and locally restricted anti-tumor response [86]. The authors used the CNS- or tumor-specific antigens myelin oligodendrocyte glycoprotein (MOG) or EGFRvIII, respectively, as activating signal for the expression of anti-EphA2 or anti-IL13Ra2 CARs. Using this system, the authors showed tumor microenvironment-restricted CAR expression, T cell priming, and effective killing of tumor cells in PDX models.
4. Overcoming Resistance to CAR T Cell Therapy
Shared features of resistance to CAR T cell therapy in non-solid tumors and concepts to overcome these obstacles have comprehensively been discussed elsewhere [87,88,89,90]. In particular, gliomas are signified by a profoundly immunosuppressive microenvironment. Therefore, CAR T cell combinatorial therapies with immunomodulatory agents seem obligatory and have been comprehensively discussed elsewhere [91]. In brief, Agliardi et al. reported that the intratumoral application of IL12 increases response to EGFRvIII CAR T cell therapy by boosting cytotoxicity and remodeling the tumor microenvironment to a more proinflammatory state [92]. Anti-GD2 CARs have been modified with an IL15 cytokine domain and CAR-expressing T cells showed prolonged anti-tumor activity upon repetitive antigen exposure [93], overcoming exhaustion. In GBM xenografts, temozolomide-induced lymphodepletion enhanced CAR T cell expansion and persistence and prolonged survival of tumor-bearing mice [94]. Antigen loss is considered a frequent tumor escape mechanism when only single tumor antigens are targeted. Several reports have shown improved CAR product efficacy and reduced resistance to CAR therapy when multiple antigens were targeted in one therapeutic approach [69,95,96,97]. However, directing CAR T cells towards multiple targets also increases the risk for off-target effects, and clinical trials targeting multiple antigens have not yet been initiated. Loss of antigen as underlying mechanism of CAR T cell resistance is not strictly linked to mutational events; frequently, downregulation of tumor antigens is mediated by epigenetic silencing. Thus, it is tempting to modulate epigenetic silencing of CAR T cell targets using epigenetic drugs. In several entities, blocking DNA methyltransferases in combination with CAR T cell therapy showed signs of improved efficacy in preclinical models [98,99,100,101]. In medulloblastoma xenograft models, the demethylating agent azacytidine led to increased target expression and prolonged survival upon CAR T cell therapy [69].
5. Application Routes for Genetically Modified T Cells in Preclinical Models
First preclinical studies for other tumor entities used a systemic intravenous injection of CAR T cell products, resulting in potent anti-tumor effects. Using Her2 CAR T cells in an intracranial breast cancer metastasis model, Priceman and colleagues reported improved tumor eradication rates after intratumoral or intraventricular adoptive transfer [102], and Brown et al. showed no therapeutic effect at all for systemic intravenous injection [27]. Recently, two studies from Theruvath and colleagues and Donovan and colleagues reported enhanced anti-tumor efficacy of locally transferred CAR T cells in several xenograft models of brain tumors. Donovan et al. showed increased survival of intraventricularly injected mice compared to intravenously injected mice in medulloblastoma and ependymoma models using CAR T cells against EPHA2, HER2, and IL13a2 [69]. Theruvath et al. used previously described B7-H3-targeting CAR T cells to show improved survival and reduced systemic inflammatory cytokine levels upon locoregional delivery [103,104]. Particularly preclinically, the literature implies a superior performance of locally administered CAR T cells over systemic application. Nonetheless, all studies were performed in fully immunodeficient mice, while effective trafficking of intravenously injected genetically manipulated T cells to the parenchyma requires cytokine gradients that will not be established in immunodeficient mice. Therefore, more investigations of how transferred T cells efficiently migrate to the brain are required in immunocompetent mice in order to draw definitive conclusions. Likewise, clinical response to CAR T cell therapy is associated with an induction of endogenous tumor-specific T cell responses and microenvironmental reprogramming [105]. Immunocompetent model systems displaying syngeneic MHC-proficient microenvironments represent a pre-requisite to assess therapeutic efficacy, particularly for T cell receptor-engineered T cell therapy targeting T helper cell epitopes, as discussed in the following section.
6. T Cell Receptor-Engineered T Cell Therapy
In contrast to CAR T cells, which are able to target only extracellular, mostly non-tumor-specific targets, TCR-transgenic T cells provide the possibility to target intracellular antigens that are presented on MHC class I and II. These targets can be glioma-associated antigens that have been reported to be overexpressed in brain tumors such as SART1 or MAGE1 [106,107,108]. Initial clinical trials in several tumor entities have reported promising results with TCR-engineered T cell therapy [109,110,111]. Most reports to date are based on tumor-associated antigens that are shared across a broad patient population [112,113,114]. Although, the use of tumor-associated antigens led to severe off-target and on-target off-tumor side effects in many early trials [82,113,115], however, the true strength in using transgenic TCR therapy lies in the ability to target tumor-specific mutations, without targeting heathy tissue. Yet, GBMs harbor only 30 to 50 mutations, leading to a limited repertoire of potential targets [116,117,118]. Recently, two clinical trials could show that vaccination against actively personalized or warehouse patient-specific neoepitopes can trigger T cell-driven immune responses [119,120]. Tumor-specific mutations are usually patient-individual mutations, leading to a prolonged time for target identification and therapy manufacturing. This could be circumvented by using frequently mutated targets. We and others have shown T cell immunogenicity for several common mutations in brain tumors, as reported for isocitrate dehydrogenase I (IDH1) mutation R132H, histone H3.3 mutation K27M, and more recently capicua transcriptional repressor (CIC) R215W/Q [121,122,123,124,125,126,127]. As these targets are also known to be driver mutations, the risk of antigen loss is reduced [128,129,130,131,132]. However, specific driver mutations such as mutant IDH1 have been reported to have immunosuppressive capacities themselves by, for example, tumor-genotype dependent education of myeloid cells [133,134,135] or suppression of antitumor T cell responses [123,136,137] in the tumor microenvironment and in peripheral immune compartments [138]. These observations have important implications for the future development of locoregional or systemic cellular therapies for IDH mutant gliomas.
In summary, an ideal target for T cellular therapy would be tumor-specific, shared by a significant cohort of patients, and a driver mutation to reduce the risk of immune escape by antigen loss. The first glioma-specific TCR that has been reported targets the K27M mutation in histone H3.3 [139]. Despite being able to detect vaccine-induced T cell clones in the periphery of diffuse midline gliomas (DIPG) patients, the authors could retrieve an H3.3K27M-reactive TCR sequence from peptide-pulsed healthy donor peripheral mononuclear cells. In in vitro and in vivo assays against an H3.3K27M-transfected human GBM cell line, H3.3K27M-specific TCR-transduced primary T cells specifically lysed tumor cells and inhibited tumor progression. More recently, we described a TCR targeting the IDH1 R132H mutation [122]. Using single cell transcriptomic and TCR sequencing, we were able to retrieve an IDH1 R132H-specific TCR from a post peptide vaccine inflamed CNS lesion. This example demonstrates that, with the emerging accessibility of single cell sequencing, identification of tumor-reactive and patient-specific TCRs will be increasingly achievable, hence exploitable for therapeutic approaches [140,141].
7. Perspective
Main hurdles for efficacious cell therapies for brain tumors will remain sufficient infiltration, persistence, and resilience of genetically modified T cells into or within the hostile brain tumor microenvironment. More recent studies suggest an IDH mutation status-associated reduced antigen presentation capacity and particularly profound exclusion of T cells within or from the IDH mutant tumor microenvironment. Despite proneural to mesenchymal transitions, mesenchymal subtype glioblastomas are considered an immunologically more active glioblastoma subtype [142]. These glioma entity- or glioblastoma subtype-specificities are suggestive for potentially required appropriate adjustments of cellular therapeutics.
Current concepts to overcome the main hurdles include the utilization of alternative application routes and combinatorial treatments in preclinical and in early clinical trials. Some evidence of relevant peripheral antigen presentation in brain tumors exists [143,144]. As of now, however, it remains unclear if antigen presentation has an impact on efficacy of local versus systemically applied TCR- or CAR-engineered T cells in gliomas. If systemic antigen presentation proves to be relevant, synthetic vaccine-based in vivo CAR T cell boosting concepts, which have already been designed [145], will gain importance. Some trials are even terminated prior to completion to pursue combination therapies with i.e., checkpoint inhibition instead. CAR- and TCR-engineered T cell therapy have both unique advantages and disadvantages, and combinations of both cellular concepts should be explored preclinically in the near future. At the same time, inducible cellular systems are in preclinical development to reduce on-target off-tumor side effects. Whereas CAR T cells target predominantly tumor-associated cell surface antigens, TCR-engineered T cell therapy will strictly depend on intratumoral MHC class I or class II expression and antigen presentation by glioma cells and associated professional antigen-presenting cells, respectively (Figure 1). This prerequisite should be considered when conceptualizing clinical trials investigating TCR-engineered T cell therapy. In this regard, for biomarker assessment, preceding biopsies or resected tumor tissues should be representative for the current tumor disease at time of treatment start.
8. Conclusions
In this review we discuss emerging targets and recent preclinical and clinical developments in the field of CAR- and TCR-engineered T cell therapy for malignant gliomas. In comparison to other immunotherapeutic modalities for gliomas, such as dendritic cell vaccination, peptide vaccination, or TIL therapy, CAR- and TCR-engineered T cell therapy offers several advantages: (i) independence of the patient’s immune system to mount meaningful T cell responses, which is, in general, limited in vaccination approaches; (ii) immune receptor engineered T cells target known and defined antigens; (iii) T cells can be additionally genetically modified ex vivo to enhance T cell responses; (iv) in contrast to TIL therapy, the re-infusion of an engineered T cell product allows the exact enumeration of truly tumor-reactive T cells; and (v) defined TCR as well as CAR sequences or epitopes facilitate the analysis of T cell fate and dynamics during monitoring. Up to now, however, these advantages have been inseparable of great financial and regulatory challenges in the processes of manufacturing and engineering cellular products. At the same time, CAR- and TCR-engineered T cell therapy bears the potential—as in other tumor entities–to cure gliomas.
Author Contributions
M.K., T.B. and L.B. wrote the manuscript. W.W., M.P. and L.B. conceptualized and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
M.K. was supported by the Helmholtz International Graduate School. L.B. is supported by the Else Kröner Fresenius Foundation (2019_EKMS.49).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
T.B., W.W. and M.P. are inventors and patent-holders on ‘Peptides for use in treating or diagnosing IDH1R132H positive cancers’ (EP2800580B1). T.B., W.W., M.P. and L.B. are patent holders of ‘Histone Anti-Cancer Vaccines’ (WO/2017/009349). M.K. declares no conflict of interest.
References
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Nunez, N.G.; De Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; de Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: CheckMate 143. Neuro Oncol. 2017, 19, iii21. [Google Scholar] [CrossRef]
- Maxwell, R.; Jackson, C.M.; Lim, M. Clinical Trials Investigating Immune Checkpoint Blockade in Glioblastoma. Curr. Treat. Options Oncol. 2017, 18, 51. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Topalian, S.L.; Solomon, D.; Avis, F.P.; Chang, A.E.; Freerksen, D.L.; Linehan, W.M.; Lotze, M.T.; Robertson, C.N.; Seipp, C.A.; Simon, P.; et al. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: A pilot study. J. Clin. Oncol. 1988, 6, 839–853. [Google Scholar] [CrossRef]
- Dillman, R.O.; Duma, C.M.; Ellis, R.A.; Cornforth, A.N.; Schiltz, P.M.; Sharp, S.L.; DePriest, M.C. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J. Immunother. 2009, 32, 914–919. [Google Scholar] [CrossRef]
- Dillman, R.O.; Duma, C.M.; Schiltz, P.M.; DePriest, C.; Ellis, R.A.; Okamoto, K.; Beutel, L.D.; De Leon, C.; Chico, S. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J. Immunother. 2004, 27, 398–404. [Google Scholar] [CrossRef]
- Mehta, G.U.; Malekzadeh, P.; Shelton, T.; White, D.E.; Butman, J.A.; Yang, J.C.; Kammula, U.S.; Goff, S.L.; Rosenberg, S.A.; Sherry, R.M. Outcomes of Adoptive Cell Transfer with Tumor-infiltrating Lymphocytes for Metastatic Melanoma Patients with and Without Brain Metastases. J. Immunother. 2018, 41, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Meng, Q.; Bartek, J., Jr.; Poiret, T.; Persson, O.; Rane, L.; Rangelova, E.; Illies, C.; Peredo, I.H.; Luo, X.; et al. Tumor-infiltrating lymphocytes (TILs) from patients with glioma. Oncoimmunology 2017, 6, e1252894. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wucherpfennig, K.W.; Freeman, G.; Wu, C.J.; Chiocca, E.A.; Wen, P.Y.; Curry, W.T., Jr.; Mitchell, D.A.; Fecci, P.E.; Sampson, J.H.; et al. An update on vaccine therapy and other immunotherapeutic approaches for glioblastoma. Expert Rev. Vaccines 2013, 12, 597–615. [Google Scholar] [CrossRef]
- Okada, H.; Kohanbash, G.; Zhu, X.; Kastenhuber, E.R.; Hoji, A.; Ueda, R.; Fujita, M. Immunotherapeutic approaches for glioma. Crit. Rev. Immunol. 2009, 29, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M.; Slagle, B.; Powers, S.K.; Gillespie, G.Y. Receptor for interleukin 13 is abundantly and specifically over-expressed in patients with glioblastoma multiforme. Int. J. Oncol. 1999, 15, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.H.; Plautz, G.E.; Puri, R.K. Interleukin-13 receptor α chain: A novel tumor-associated transmembrane protein in primary explants of human malignant gliomas. Cancer Res. 2000, 60, 1168–1172. [Google Scholar]
- Debinski, W.; Obiri, N.I.; Powers, S.K.; Pastan, I.; Puri, R.K. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. 1995, 1, 1253–1258. [Google Scholar] [PubMed]
- Krebs, S.; Chow, K.K.; Yi, Z.; Rodriguez-Cruz, T.; Hegde, M.; Gerken, C.; Ahmed, N.; Gottschalk, S. T cells redirected to interleukin-13Ralpha2 with interleukin-13 mutein-chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Ralpha1. Cytotherapy 2014, 16, 1121–1131. [Google Scholar] [CrossRef]
- Debinski, W.; Gibo, D.M.; Obiri, N.I.; Kealiher, A.; Puri, R.K. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat. Biotechnol. 1998, 16, 449–453. [Google Scholar] [CrossRef]
- Thaci, B.; Brown, C.E.; Binello, E.; Werbaneth, K.; Sampath, P.; Sengupta, S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014, 16, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Ralpha2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Ralpha2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Ahmed, N.; Salsman, V.S.; Kew, Y.; Shaffer, D.; Powell, S.; Zhang, Y.J.; Grossman, R.G.; Heslop, H.E.; Gottschalk, S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin. Cancer Res. 2010, 16, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ying, H.; Zeng, G.; Wheeler, C.J.; Black, K.L.; Yu, J.S. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004, 64, 4980–4986. [Google Scholar] [CrossRef] [PubMed]
- Turatti, F.; Figini, M.; Alberti, P.; Willemsen, R.A.; Canevari, S.; Mezzanzanica, D. Highly efficient redirected anti-tumor activity of human lymphocytes transduced with a completely human chimeric immune receptor. J. Gene Med. 2005, 7, 158–170. [Google Scholar] [CrossRef]
- Toth, G.; Szollosi, J.; Abken, H.; Vereb, G.; Szoor, A. A Small Number of HER2 Redirected CAR T Cells Significantly Improves Immune Response of Adoptively Transferred Mouse Lymphocytes against Human Breast Cancer Xenografts. Int. J. Mol. Sci. 2020, 21, 1039. [Google Scholar] [CrossRef]
- Stancovski, I.; Schindler, D.G.; Waks, T.; Yarden, Y.; Sela, M.; Eshhar, Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J. Immunol. 1993, 151, 6577–6582. [Google Scholar]
- Li, S.; Yang, J.; Urban, F.A.; MacGregor, J.N.; Hughes, D.P.; Chang, A.E.; McDonagh, K.T.; Li, Q. Genetically engineered T cells expressing a HER2-specific chimeric receptor mediate antigen-specific tumor regression. Cancer Gene Ther. 2008, 15, 382–392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gajjar, A.; Hernan, R.; Kocak, M.; Fuller, C.; Lee, Y.; McKinnon, P.J.; Wallace, D.; Lau, C.; Chintagumpala, M.; Ashley, D.M.; et al. Clinical, histopathologic, and molecular markers of prognosis: Toward a new disease risk stratification system for medulloblastoma. J. Clin. Oncol. 2004, 22, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Hegde, M.; Joseph, S.K.; Pashankar, F.; DeRenzo, C.; Sanber, K.; Navai, S.; Byrd, T.T.; Hicks, J.; Xu, M.L.; Gerken, C.; et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat. Commun. 2020, 11, 3549. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Vitanza, N.A.; Johnson, A.J.; Wilson, A.L.; Brown, C.; Yokoyama, J.K.; Kunkele, A.; Chang, C.A.; Rawlings-Rhea, S.; Huang, W.; Seidel, K.; et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: An interim analysis. Nat. Med. 2021, 27, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; Longo, N.; Hamid, M.L.; Olson, J.J.; Liu, L.; Collins, V.P.; James, C.D. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene 1994, 9, 2313–2320. [Google Scholar]
- Wikstrand, C.J.; McLendon, R.E.; Friedman, A.H.; Bigner, D.D. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997, 57, 4130–4140. [Google Scholar]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Crotty, L.E.; Archer, G.E.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar]
- Heimberger, A.B.; Archer, G.E.; Crotty, L.E.; McLendon, R.E.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Dendritic cells pulsed with a tumor-specific peptide induce long-lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery 2002, 50, 158–164; discussion 156–164. [Google Scholar] [CrossRef]
- Choi, B.D.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; McLendon, R.E.; Bigner, D.D.; Sampson, J.H. EGFRvIII-targeted vaccination therapy of malignant glioma. Brain Pathol. 2009, 19, 713–723. [Google Scholar] [CrossRef]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef]
- Sampson, J.H.; Choi, B.D.; Sanchez-Perez, L.; Suryadevara, C.M.; Snyder, D.J.; Flores, C.T.; Schmittling, R.J.; Nair, S.K.; Reap, E.A.; Norberg, P.K.; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 2014, 20, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Ohkuri, T.; Kosaka, A.; Tanahashi, K.; June, C.H.; Natsume, A.; Okada, H. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 2013, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Ravanpay, A.C.; Gust, J.; Johnson, A.J.; Rolczynski, L.S.; Cecchini, M.; Chang, C.A.; Hoglund, V.J.; Mukherjee, R.; Vitanza, N.A.; Orentas, R.J.; et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget 2019, 10, 7080–7095. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Prapa, M.; Caldrer, S.; Spano, C.; Bestagno, M.; Golinelli, G.; Grisendi, G.; Petrachi, T.; Conte, P.; Horwitz, E.M.; Campana, D.; et al. A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget 2015, 6, 24884–24894. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Ramakrishna, S.; Mochizuki, A.; Patel, S.; Chinnasamy, H.; Yeom, K.; Schultz, L.; Richards, R.; Campen, C.; Reschke, A.; et al. Abstract CT031: GD2 CAR T cells mediate clinical activity and manageable toxicity in children and young adults with DIPG and H3K27M-mutated diffuse midline gliomas. Cancer Res. 2021, 81, CT031. [Google Scholar] [CrossRef]
- Soroceanu, L.; Gillespie, Y.; Khazaeli, M.B.; Sontheimer, H. Use of chlorotoxin for targeting of primary brain tumors. Cancer Res. 1998, 58, 4871–4879. [Google Scholar] [PubMed]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Starr, R.; Chang, W.C.; Aguilar, B.; Alizadeh, D.; Wright, S.L.; Yang, X.; Brito, A.; Sarkissian, A.; Ostberg, J.R.; et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci. Transl. Med. 2020, 12, eaaw2672. [Google Scholar] [CrossRef]
- Ieguchi, K.; Maru, Y. Roles of EphA1/A2 and ephrin-A1 in cancer. Cancer Sci. 2019, 110, 841–848. [Google Scholar] [CrossRef]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2000, 2, 62–69. [Google Scholar] [CrossRef]
- Kang, B.H.; Jensen, K.J.; Hatch, J.A.; Janes, K.A. Simultaneous profiling of 194 distinct receptor transcripts in human cells. Sci. Signal. 2013, 6, rs13. [Google Scholar] [CrossRef]
- Wang, L.F.; Fokas, E.; Bieker, M.; Rose, F.; Rexin, P.; Zhu, Y.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.X. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol. Rep. 2008, 19, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.K.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Prinzing, B.L.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Agemy, L.; Kotamraju, V.R.; Friedmann-Morvinski, D.; Sharma, S.; Sugahara, K.N.; Ruoslahti, E. Proapoptotic peptide-mediated cancer therapy targeted to cell surface p32. Mol. Ther. 2013, 21, 2195–2204. [Google Scholar] [CrossRef]
- Rousso-Noori, L.; Mastandrea, I.; Talmor, S.; Waks, T.; Globerson Levin, A.; Haugas, M.; Teesalu, T.; Alvarez-Vallina, L.; Eshhar, Z.; Friedmann-Morvinski, D. P32-specific CAR T cells with dual antitumor and antiangiogenic therapeutic potential in gliomas. Nat. Commun. 2021, 12, 3615. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef]
- Venugopal, C.; Hallett, R.; Vora, P.; Manoranjan, B.; Mahendram, S.; Qazi, M.A.; McFarlane, N.; Subapanditha, M.; Nolte, S.M.; Singh, M.; et al. Pyrvinium Targets CD133 in Human Glioblastoma Brain Tumor-Initiating Cells. Clin. Cancer Res. 2015, 21, 5324–5337. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Shibahara, I.; Sonoda, Y.; Saito, R.; Kanamori, M.; Yamashita, Y.; Kumabe, T.; Watanabe, M.; Suzuki, H.; Watanabe, T.; Ishioka, C.; et al. The expression status of CD133 is associated with the pattern and timing of primary glioblastoma recurrence. Neuro Oncol. 2013, 15, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Pellegatta, S.; Savoldo, B.; Di Ianni, N.; Corbetta, C.; Chen, Y.; Patane, M.; Sun, C.; Pollo, B.; Ferrone, S.; DiMeco, F.; et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 2018, 10, eaao2731. [Google Scholar] [CrossRef]
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patane, M.; Pollo, B.; Finocchiaro, G.; Park, J.J.H.; Dunn, D.E.; Edwards, D.S.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine 2019, 47, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhao, S.; Zhang, Y.; Wang, Y.; Zhang, Z.; Yang, M.; Zhu, Y.; Zhang, G.; Guo, G.; Tong, A.; et al. B7-H3 as a Novel CAR-T Therapeutic Target for Glioblastoma. Mol. Ther. Oncolytics 2019, 14, 279–287. [Google Scholar] [CrossRef]
- Yang, M.; Tang, X.; Zhang, Z.; Gu, L.; Wei, H.; Zhao, S.; Zhong, K.; Mu, M.; Huang, C.; Jiang, C.; et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics 2020, 10, 7622–7634. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Szoor, A.; Torres, D.; Joseph, N.; Velasquez, M.P.; Iwahori, K.; Gaikwad, A.; Nguyen, P.; Arber, C.; Song, X.T.; et al. CD123-Engager T Cells as a Novel Immunotherapeutic for Acute Myeloid Leukemia. Mol. Ther. 2016, 24, 1615–1626. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.; Themeli, M.; Usmani, S.Z. Determinants of response and mechanisms of resistance of CAR T-cell therapy in multiple myeloma. Blood Cancer Discov. 2021, 2, 302–318. [Google Scholar] [CrossRef] [PubMed]
- Song, M.K.; Park, B.B.; Uhm, J.E. Resistance Mechanisms to CAR T-Cell Therapy and Overcoming Strategy in B-Cell Hematologic Malignancies. Int. J. Mol. Sci. 2019, 20, 5010. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; De Feo, D.; Nunez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef]
- Suryadevara, C.M.; Desai, R.; Abel, M.L.; Riccione, K.A.; Batich, K.A.; Shen, S.H.; Chongsathidkiet, P.; Gedeon, P.C.; Elsamadicy, A.A.; Snyder, D.J.; et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology 2018, 7, e1434464. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- El Khawanky, N.; Hughes, A.; Yu, W.; Taromi, S.; Clarson, J.; Lopez, A.F.; Brown, M.P.; Duyster, J.; Hughes, T.P.; White, D.L.; et al. Azacytidine Sensitizes AML Cells for Effective Elimination by CD123 CAR T-Cells. Blood 2019, 134, 3904. [Google Scholar] [CrossRef]
- Terracina, K.P.; Graham, L.J.; Payne, K.K.; Manjili, M.H.; Baek, A.; Damle, S.R.; Bear, H.D. DNA methyltransferase inhibition increases efficacy of adoptive cellular immunotherapy of murine breast cancer. Cancer Immunol. Immunother. 2016, 65, 1061–1073. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-Dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef]
- You, L.; Han, Q.; Zhu, L.; Zhu, Y.; Bao, C.; Yang, C.; Lei, W.; Qian, W. Decitabine-Mediated Epigenetic Reprograming Enhances Anti-leukemia Efficacy of CD123-Targeted Chimeric Antigen Receptor T-Cells. Front. Immunol. 2020, 11, 1787. [Google Scholar] [CrossRef]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Hirabayashi, K.; Ahn, S.; Kren, N.P.; Montgomery, S.A.; Wang, X.; Tiruthani, K.; Mirlekar, B.; Michaud, D.; Greene, K.; et al. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell 2019, 35, 221–237.e8. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Jonsson, V.; Hibbard, J.; Yahn, S.; Wong, R.A.; Yang, X.; Ng, R.; Dullerud, N.; Maker, M.; et al. Abstract 59: CAR T cell therapy reshapes the tumor microenvironment to promote host antitumor immune repsonses in glioblastoma. Cancer Res. 2021, 81, 59. [Google Scholar] [CrossRef]
- Skog, J. Glioma-Specific antigens for immune tumor therapy. Expert Rev. Vaccines 2006, 5, 793–802. [Google Scholar] [CrossRef]
- Rimoldi, D.; Romero, P.; Carrel, S. The human melanoma antigen-encoding gene, MAGE-1, is expressed by other tumour cells of neuroectodermal origin such as glioblastomas and neuroblastomas. Int. J. Cancer 1993, 54, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Kuramoto, T.; Matsunaga, K.; Shichijo, S.; Yutani, S.; Shigemori, M.; Oizumi, K.; Itoh, K. Expression of the tumor-rejection antigen SART1 in brain tumors. Int. J. Cancer 1999, 83, 760–764. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal. Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Kilian, M.; Friedrich, M.; Sanghvi, K.; Green, E.; Pusch, S.; Kawauchi, D.; Löwer, M.; Sonner, J.K.; Krämer, C.; Zaman, J.; et al. T cell receptor therapy targeting mutant capicua transcriptional repressor in experimental gliomas. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Bunse, L.; Schumacher, T.; Sahm, F.; Pusch, S.; Oezen, I.; Rauschenbach, K.; Gonzalez, M.; Solecki, G.; Osswald, M.; Capper, D.; et al. Proximity ligation assay evaluates IDH1R132H presentation in gliomas. J. Clin. Investig. 2015, 125, 593–606. [Google Scholar] [CrossRef]
- Ochs, K.; Ott, M.; Bunse, T.; Sahm, F.; Bunse, L.; Deumelandt, K.; Sonner, J.K.; Keil, M.; von Deimling, A.; Wick, W.; et al. K27M-mutant histone-3 as a novel target for glioma immunotherapy. Oncoimmunology 2017, 6, e1328340. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Wick, W.; Platten, M. Mutant IDH1: An immunotherapeutic target in tumors. Oncoimmunology 2014, 3, e974392. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patane, M.; Zucca, I.; Riccardi Sirtori, F.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Friedrich, M.; Sankowski, R.; Bunse, L.; Kilian, M.; Green, E.; Ramallo Guevara, C.; Pusch, S.; Poschet, G.; Sanghvi, K.; Hahn, M.; et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat. Cancer 2021, 2, 723–740. [Google Scholar] [CrossRef]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Nunez, F.J.; Liu, Y.; Yu, M.; Taher, A.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131, e139542. [Google Scholar] [CrossRef] [PubMed]
- Kohanbash, G.; Carrera, D.A.; Shrivastav, S.; Ahn, B.J.; Jahan, N.; Mazor, T.; Chheda, Z.S.; Downey, K.M.; Watchmaker, P.B.; Beppler, C.; et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Investig. 2017, 127, 1425–1437. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Rao, A.; Sandlesh, P.; Yerneni, S.S.; Swain, A.D.; Bullock, K.M.; Hansen, K.M.; Zhang, X.; Jaman, E.; Allen, J.; et al. Characterization of systemic immunosuppression by IDH mutant glioma small extracellular vesicles. Neuro Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Green, E.W.; Bunse, L.; Bozza, M.; Sanghvi, K.; Platten, M. TCR validation toward gene therapy for cancer. Methods Enzym. 2019, 629, 419–441. [Google Scholar] [CrossRef]
- Bunse, L.; Green, E.W.; Platten, M. High-throughput discovery of cancer-targeting TCRs. Methods Enzym. 2019, 629, 401–417. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Louveau, A.; Herz, J.; Alme, M.N.; Salvador, A.F.; Dong, M.Q.; Viar, K.E.; Herod, S.G.; Knopp, J.; Setliff, J.C.; Lupi, A.L.; et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci 2018, 21, 1380–1391. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dichwalkar, T.; Chang, J.Y.H.; Cossette, B.; Garafola, D.; Zhang, A.Q.; Fichter, M.; Wang, C.; Liang, S.; Silva, M.; et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019, 365, 162–168. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).