
Critical Role of Estrogens on Bone Homeostasis in Both Male and Female: From Physiology to Medical Implications


Abstract
1. Introduction
1.1. Bone Physiology
1.2. Bone Cells
1.3. Bone Modeling and Remodeling
2. Skeletal Evolution throughout Life
2.1. Skeletal Development
2.2. Skeletal Maintenance
2.3. Pregnancy and Lactation
2.4. Skeletal Involution
2.5. Sex Steroid Deficiency
2.6. Aging
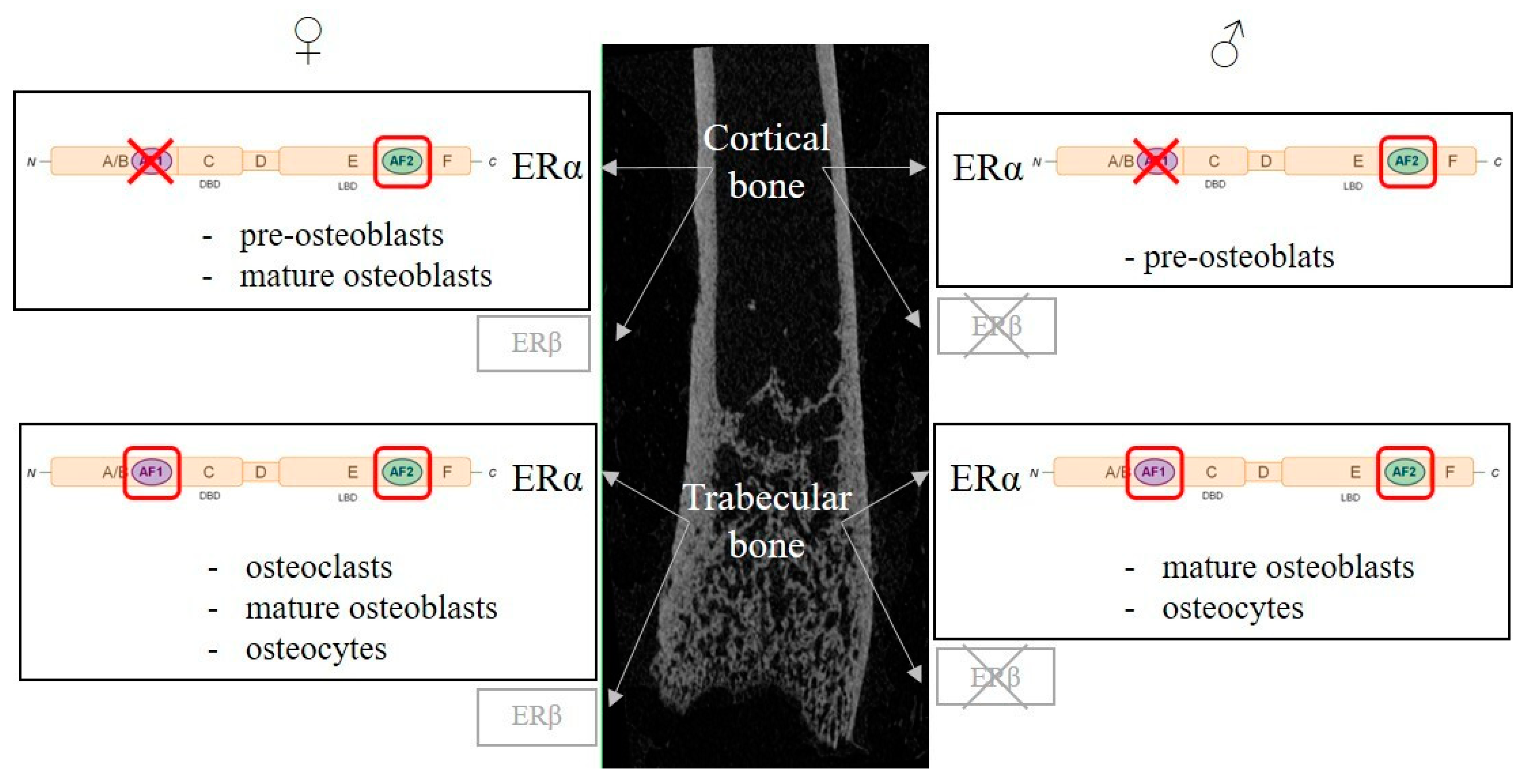
3. Roles of Estrogen Receptors
3.1. Estrogen Receptors
3.2. Estrogen Receptors in Bone Cells
3.3. Nuclear vs. Non-Nuclear Erα-Mediated Pathways
3.4. Selective Estrogen Receptor Modulators
3.5. Mechanical Loading
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Valéra, M.C.; Fontaine, C.; Dupuis, M.; Noirrit-Esclassan, E.; Vinel, A.; Guillaume, M.; Gourdy, P.; Lenfant, F.; Arnal, J.F. Towards optimization of estrogen receptor modulation in medicine. Pharmacol. Ther. 2018, 189, 123–129. [Google Scholar] [CrossRef]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R.A. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Bouillon, R.; Bex, M.; Vanderschueren, D.; Boonen, S. Estrogens Are Essential for Male Pubertal Periosteal Bone Expansion. J. Clin. Endocrinol. Metab. 2004, 89, 6025–6029. [Google Scholar] [CrossRef]
- Oldknow, K.J.; Macrae, V.E.; Farquharson, C. Endocrine role of bone: Recent and emerging perspectives beyond osteocalcin. J. Endocrinol. 2015, 225, R1–R19. [Google Scholar] [CrossRef] [PubMed]
- Alford, A.I.; Kozloff, K.M.; Hankenson, K.D. Extracellular matrix networks in bone remodeling. Int. J. Biochem. Cell Biol. 2015, 65, 20–31. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [PubMed]
- Ono, T.; Nakashima, T. Recent advances in osteoclast biology. Histochem. Cell Biol. 2018, 149, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of Proliferation, Differentiation and Functions of Osteoblasts by Runx2. Int. J. Mol. Sci. 2019, 20, 1694. [Google Scholar] [CrossRef]
- Florencio-Silva, R.; Sasso, G.R.D.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef]
- Ducy, P.; Schinke, T.; Karsenty, G. The Osteoblast: A Sophisticated Fibroblast under Central Surveillance. Science 2000, 289, 1501–1504. [Google Scholar] [CrossRef]
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and endocrine actions of bone—The functions of secretory proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res. 2018, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The Osteocyte: An Endocrine Cell … and More. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef]
- Tresguerres, F.; Torres, J.; López-Quiles, J.; Hernández, G.; Vega, J.; Tresguerres, I. The osteocyte: A multifunctional cell within the bone. Ann. Anat. Anat. Anz. 2020, 227, 151422. [Google Scholar] [CrossRef] [PubMed]
- Seeman, E.; Delmas, P. Bone Quality—The Material and Structural Basis of Bone Strenght and Fragility. N. Engl. J. Med. 2006, 354, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.; Martin, J. Coupling the Activities of Bone Formation and Resorption: A Multitude of Signals Within the Basic Multi-cellular Unit. BoneKEy Rep. 2014, 3, 1–10. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef]
- Golden, N.H. Bones and Birth Control in Adolescent Girls. J. Pediatr. Adolesc. Gynecol. 2020, 33, 249–254. [Google Scholar] [CrossRef]
- Jafari Nasabian, P.; Inglis, J.E.; Reilly, W.; Kelly, O.J.; Ilich, J.Z. Aging human body: Changes in bone, muscle and body fat with con-sequent changes in nutrient intake. J. Endocrinol. 2017, 234, R37–R51. [Google Scholar] [CrossRef]
- Ben Kahla, R.; Barkaoui, A.; Merzouki, T. Age-related mechanical strength evolution of trabecular bone under fatigue damage for both genders: Fracture risk evaluation. J. Mech. Behav. Biomed. Mater. 2018, 84, 64–73. [Google Scholar] [CrossRef]
- Weaver, C.M.; Gordon, C.M.; Janz, K.F.; Kalkwarf, H.J.; Lappe, J.M.; Lewis, R.; O’Karma, M.; Wallace, T.C.; Zemel, B.S. The National Osteoporosis Foundation’s position statement on peak bone mass development and lifestyle factors: A systematic review and implementation recommendations. Osteoporos. Int. 2016, 27, 1281–1386. [Google Scholar] [CrossRef]
- Bonjour, J.-P.; Chevalley, T. Pubertal Timing, Bone Acquisition, and Risk of Fracture Throughout Life. Endocr. Rev. 2014, 35, 820–847. [Google Scholar] [CrossRef] [PubMed]
- Agas, D.; Lacava, G.; Sabbieti, M.G. Bone and bone marrow disruption by endocrine-active substances. J. Cell. Physiol. 2019, 234, 192–213. [Google Scholar] [CrossRef]
- Kirmani, S.; Christen, D.; Van Lenthe, G.H.; Fischer, P.R.; Bouxsein, M.L.; McCready, L.K.; Melton, L.J.; Riggs, B.L.; Amin, S.; Müller, R.; et al. Bone Structure at the Distal Radius During Adolescent Growth. J. Bone Miner. Res. 2009, 24, 1033–1042. [Google Scholar] [CrossRef]
- Xue, S.; Kemal, O.; Lu, M.; Lix, L.M.; Leslie, W.D.; Yang, S. Age at attainment of peak bone mineral density and its associated fac-tors: The National Health and Nutrition Examination Survey 2005–2014. Bone 2020, 131, 115163. [Google Scholar] [CrossRef]
- Gabel, L.; Macdonald, H.M.; McKay, H.A. Sex Differences and Growth-Related Adaptations in Bone Microarchitecture, Geome-try, Density, and Strength from Childhood to Early Adulthood: A Mixed Longitudinal HR-pQCT Study. J. Bone Miner. Res. 2017, 32, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Laurent, M.; Antonio, L.; Sinnesael, M.; Dubois, V.; Gielen, E.; Classens, F.; Vanderschueren, D. Androgens and estrogens in skeletal sexual dimor-phism. Asian J. Androl. 2014, 16, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Connelly, K.J.; Larson, E.A.; Marks, D.L.; Klein, R.F. Neonatal estrogen exposure results in biphasic age-dependent effects on the skeletal development of male mice. Endocrinology 2015, 156, 193–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Almeida, M.; Laurent, M.R.; Dubois, V.; Claessens, F.; O’Brien, C.A.; Bouillon, R.; Vanderschueren, D.; Manolagas, S.C. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol. Rev. 2017, 97, 135–187. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.S.; Henry, Y.M.; Fatayerji, D.; Eastell, R. Lumbar spine peak bone mass and bone turnover in men and women: A longi-tudinal study. Osteoporos. Int. 2009, 20, 355–362. [Google Scholar] [CrossRef]
- Iravani, M.; Lagerquist, M.K.; Ohlsson, C.; Sävendahl, L. Regulation of bone growth via ligand-specific activation of estrogen receptor alpha. J. Endocrinol. 2017, 232, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Morishima, A.; Grumbach, M.M.; Simpson, E.R.; Fisher, C.; Qin, K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J. Clin. Endocrinol. Metab. 1995, 80, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, M.H.; Morishima, A.; Conte, F.; Grumbach, M.; Smith, E.P. Pediatric endocrinology update: An overview. The essen-tial roles of estrogens in pubertal growth, epiphyseal fusion and bone turnover: Lessons from mutations in the genes for aro-matase and the estrogen receptor. Horm. Res. 1998, 49 (Suppl. 1), 2–8. [Google Scholar] [CrossRef]
- Smith, E.P.; Boyd, J.; Frank, G.R.; Takahashi, H.; Cohen, R.M.; Specker, B.; Williams, T.C.; Lubahn, D.B.; Korach, K.S. Estrogen Resistance Caused by a Mutation in the Estrogen-Receptor Gene in a Man. N. Engl. J. Med. 1994, 331, 1056–1061. [Google Scholar] [CrossRef]
- Rochira, V.; Zirilli, L.; Madeo, B.; Aranda, C.; Caffagni, G.; Fabre, B.; Montangero, V.E.; Roldan, E.J.; Maffei, L.; Carani, C. Skeletal effects of long-term estrogen and testosterone replacement treatment in a man with congenital aromatase deficiency: Evidences of a priming effect of estrogen for sex ster-oids action on bone. Bone 2007, 40, 1662–1668. [Google Scholar] [CrossRef]
- Hadji, P.; Colli, E.; Regidor, P.-A. Bone health in estrogen-free contraception. Osteoporos. Int. 2019, 30, 2391–2400. [Google Scholar] [CrossRef]
- Manolagas, S.C. From Estrogen-Centric to Aging and Oxidative Stress: A Revised Perspective of the Pathogenesis of Osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef] [PubMed]
- Mosekilde, L.; Vestergaard, P.; Rejnmark, L. The Pathogenesis, Treatment and Prevention of Osteoporosis in Men. Drugs 2013, 73, 15–29. [Google Scholar] [CrossRef]
- Walsh, J.S.; Paggiosi, M.A.; Eastell, R. Cortical Consolidation of the Radius and Tibia in Young Men and Women. J. Clin. Endocrinol. Metab. 2012, 97, 3342–3348. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohlsson, C.; Darelid, A.; Nilsson, M.; Melin, J.; Mellström, D.; Lorentzon, M. Cortical consolidation due to increased mineraliza-tion and endosteal contraction in young adult men: A five-year longitudinal study. J. Clin. Endocrinol. Metab. 2011, 96, 2262–2269. [Google Scholar] [CrossRef]
- Bertelloni, S.; Meriggiola, M.C.; Dati, E.; Balsamo, A.; Baroncelli, G.I. Bone Mineral Density in Women Living with Complete An-drogen Insensitivity Syndrome and Intact Testes or Removed Gonads. Sex Dev. 2017, 11, 182–189. [Google Scholar] [CrossRef]
- Szeliga, A.; Maciejewska-Jeske, M.; Męczekalski, B. Bone health and evaluation of bone mineral density in patients with prema-ture ovarian insufficiency. Prz. Menop. 2018, 17, 112–116. [Google Scholar]
- Lauretani, F.; Bandinelli, S.; E Griswold, M.; Maggio, M.; Semba, R.; Guralnik, J.M.; Ferrucci, L. Longitudinal Changes in BMD and Bone Geometry in a Population-Based Study. J. Bone Miner. Res. 2007, 23, 400–408. [Google Scholar] [CrossRef]
- Kovacs, C.S. Maternal Mineral and Bone Metabolism During Pregnancy, Lactation, and Post-Weaning Recovery. Physiol. Rev. 2016, 96, 449–547. [Google Scholar] [CrossRef] [PubMed]
- Paton, L.M.; Alexander, J.L.; Nowson, C.; Margerison, C.; Frame, M.G.; Kaymakci, B.; Wark, J.D. Pregnancy and lactation have no long-term deleterious effect on measures of bone mineral in healthy women: A twin study. Am. J. Clin. Nutr. 2003, 77, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Anne, T.F.; Pouillès, J.-M.; Drewniak, N.; Laparra, J.; Ribot, C.A.; Dargent-Molina, P. Fracture risk prediction using BMD and clinical risk factors in early postmenopausal women: Sensitivity of the WHO FRAX tool. J. Bone Miner. Res. 2010, 25, 1002–1009. [Google Scholar] [CrossRef]
- Winter, E.M.; Ireland, A.; Butterfield, N.C.; Haffner-Luntzer, M.; Horcajada, M.N.; Veldhuis-Vlug, A.G.; Oei, L.; Colaianni, G.; Bonnet, N. Pregnancy and lactation, a challenge for the skeleton. Endocr. Connect. 2020, 9, R143–R157. [Google Scholar] [CrossRef]
- Xiao, H.; Zhou, Q.; Niu, G.; Han, G.; Zhang, Z.; Zhang, Q.; Bai, J.; Zhu, X. Association between breastfeeding and osteoporotic hip fracture in women: A dose-response meta-analysis. J. Orthop. Surg. Res. 2020, 15, 1–7. [Google Scholar] [CrossRef]
- Polat, S.B.; Evranos, B.; Aydin, C.; Cuhaci, N.; Ersoy, R.; Cakir, B. Effective treatment of severe pregnancy and lactation-related osteoporosis with teriparatide: Case report and review of the literature. Gynecol. Endocrinol. 2015, 31, 522–525. [Google Scholar] [CrossRef]
- Yun, K.Y.; Han, S.E.; Kim, S.C.; Kil Joo, J.; Lee, K.S. Pregnancy-related osteoporosis and spinal fractures. Obstet. Gynecol. Sci. 2017, 60, 133–137. [Google Scholar] [CrossRef]
- Miyamoto, T.; Miyakoshi, K.; Sato, Y.; Kasuga, Y.; Ikenoue, S.; Miyamoto, K.; Nishiwaki, Y.; Tanaka, M.; Nakamura, M.; Matsumoto, M. Changes in bone metabolic profile associated with pregnancy or lactation. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Aspray, T.J.; Hill, T.R. Osteoporosis and the Ageing Skeleton. Macromol. Protein Complexes III Struct. Funct. 2019, 91, 453–476. [Google Scholar] [CrossRef]
- Karlamangla, A.S.; Burnett-Bowie, S.-A.M.; Crandall, C.J. Bone Health During the Menopause Transition and Beyond. Obstet. Gynecol. Clin. N. Am. 2018, 45, 695–708. [Google Scholar] [CrossRef]
- Cauley, J.A.; Danielson, M.E.; Greendale, G.A.; Finkelstein, J.S.; Chang, Y.F.; Lo, J.C.; Crandall, C.J.; Neer, R.M.; Ruppert, K.; Meyn, L.; et al. Bone resorption and fracture across the menopausal transition: The Study of Women’s Health Across the Nation. Menopause 2012, 19, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.; Greendale, G.A.; Cauley, J.A.; Karlamangla, A.S. The Association between Fast Increase in Bone Turnover During the Menopause Transition and Subsequent Fracture. J. Clin. Endocrinol. Metab. 2019, 105, e1440–e1448. [Google Scholar] [CrossRef]
- Martin-Millan, M.; Almeida, M.; Ambrogini, E.; Han, L.; Zhao, H.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A.; Manolagas, S.C. The Estrogen Receptor-α in Osteoclasts Mediates the Protective Effects of Estrogens on Cancellous but Not Cortical Bone. Mol. Endocrinol. 2010, 24, 323–334. [Google Scholar] [CrossRef]
- Johnell, O.; Gullberg, B.; Kanis, J.A.; Allander, E.; Elffors, L.; Dequeker, J.; Dilsen, G.; Gennari, C.; Vaz, L.A.; Lyritis, G.; et al. Risk factors for hip fracture in european women: The MEDOS study. J. Bone Miner. Res. 2009, 10, 1802–1815. [Google Scholar] [CrossRef] [PubMed]
- Chevalley, T.; Bonjour, J.-P.; Ferrari, S.; Rizzoli, R. Deleterious Effect of Late Menarche on Distal Tibia Microstructure in Healthy 20-Year-Old and Premenopausal Middle-Aged Women. J. Bone Miner. Res. 2009, 24, 144–152. [Google Scholar] [CrossRef]
- Vandenput, L.; Kindblom, J.M.; Bygdell, M.; Nethander, M.; Ohlsson, C. Pubertal timing and adult fracture risk in men: A popula-tion-based cohort study. PLoS Med. 2019, 16, e1002986. [Google Scholar] [CrossRef] [PubMed]
- Cauley, J.A.; Danielson, M.E.; Jammy, G.R.; Bauer, D.C.; Jackson, R.; Wactawski-Wende, J.; Chlebowski, R.T.; Ensrud, K.E.; Boudreau, R. Sex Steroid Hormones and Fracture in a Multiethnic Cohort of Women: The Women’s Health Initiative Study (WHI). J. Clin. Endocrinol. Metab. 2017, 102, 1538–1547. [Google Scholar] [CrossRef]
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99, 11–15. [Google Scholar] [CrossRef]
- Khosla, S.; Monroe, D.G. Regulation of Bone Metabolism by Sex Steroids. Cold Spring Harb. Perspect. Med. 2018, 8, a031211. [Google Scholar] [CrossRef]
- Vlot, M.C.; Wiepjes, C.M.; de Jongh, R.T.; T’Sjoen, G.; Heijboer, A.C.; den Heijer, M. Gender-Affirming Hormone Treatment Decreas-es Bone Turnover in Transwomen and Older Transmen. J. Bone Miner. Res. 2019, 34, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, E.S.; Nielson, C.M.; Marshall, L.M.; Lapidus, J.A.; Barrett-Connor, E.; Ensrud, K.E.; Hoffman, A.R.; Laughlin, G.; Ohlsson, C.; Orwoll, E.S.; et al. The Effects of Serum Testosterone, Estradiol, and Sex Hormone Binding Globulin Levels on Fracture Risk in Older Men. J. Clin. Endocrinol. Metab. 2009, 94, 3337–3346. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, H.M.; Nishiyama, K.K.; Kang, J.; Hanley, D.A.; Boyd, S.K. Age-related patterns of trabecular and cortical bone loss differ between sexes and skeletal sites: A population-based HR-pQCT study. J. Bone Miner. Res. 2011, 26, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, A.J.; Kazakia, G.J.; Ramachandran, S.; Link, T.M.; Majumdar, S. Age and Gender Related Differences in the Geometric Properties and Biomechanical Significance of Intra-Cortical Porosity in the Distal Radius and Tibia. J. Bone Miner. Res. 2009, 25, 983–993. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.-M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to verb-A. Nat. Cell Biol. 1986, 320, 134–139. [Google Scholar] [CrossRef]
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor com-plementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef]
- Kuiper, G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.-A. Cloning of a Novel Estrogen Receptor Expressed in Rat Prostate and Ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ERβ: Identificatino and Characterization of a Novel Human Estrogen Receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Couse, J.F.; Lindzey, J.; Grandien, K.; Gustafsson, J.A.; Korach, K.S. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 1997, 138, 4613–4621. [Google Scholar] [CrossRef]
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Lindberg, M.K.; Alatalo, S.L.; Halleen, J.M.; Mohan, S.; A Gustafsson, J.; Ohlsson, C. Estrogen receptor specificity in the regulation of the skeleton in female mice. J. Endocrinol. 2001, 171, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Movérare, S.; Venken, K.; Eriksson, A.-L.; Andersson, N.; Skrtic, S.; Wergedal, J.; Mohan, S.; Salmon, P.; Bouillon, R.; Gustafsson, J.-A.; et al. Differencial Effects on Bone of Estrogen Receptor α and Androgen Receptor Activation in Orchidectomized Adult Male Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13573–13578. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Krust, A.; Gansmuller, A.; Dierich, A.; Chambon, P.; Mark, M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 2000, 127, 4277–4291. [Google Scholar] [PubMed]
- Sims, N.A.; Dupont, S.; Krust, A.; Clement-Lacroix, P.; Minet, D.; Resche-Rigon, M.; Gaillard-Kelly, M.; Baron, R. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone 2002, 30, 18–25. [Google Scholar] [CrossRef]
- Sims, N.A.; Clément-Lacroix, P.; Minet, D.; Fraslon-Vanhulle, C.; Gaillard-Kelly, M.; Resche-Rigon, M.; Baron, R. A functional androgen receptor is not sufficient to allow estradiol to protect bone after gonadectomy in estradiol receptor–deficient mice. J. Clin. Investig. 2003, 111, 1319–1327. [Google Scholar] [CrossRef]
- Vinel, A.; Coudert, A.E.; Buscato, M.; Valera, M.C.; Ostertag, A.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; Berdal, A.; Babajko, S.; Arnal, J.F.; et al. Respective role of membrane and nu-clear estrogen receptor (ER) α in the mandible of growing mice: Implications for ERα modulation. J. Bone Miner. Res. 2018, 33, 1520–1531. [Google Scholar] [CrossRef]
- Ohlsson, C.; Farman, H.H.; Gustafsson, K.L.; Wu, J.; Henning, P.; Windahl, S.H.; Sjögren, K.; Gustafsson, J.-Å.; Movérare-Skrtic, S.; Lagerquist, M.K. The effects of estradiol are modulated in a tissue-specific manner in mice with inducible inactivation of ERα after sexual maturation. Am. J. Physiol. Metab. 2020, 318, E646–E654. [Google Scholar] [CrossRef]
- Idelevich, A.; Baron, R. Brain to bone: What is the contribution of the brain to skeletal homeostasis? Bone 2018, 115, 31–42. [Google Scholar] [CrossRef]
- Farman, H.H.; Windahl, S.H.; Westberg, L.; Isaksson, H.; Egecioglu, E.; Schele, E.; Ryberg, H.; Jansson, J.O.; Tuukkanen, J.; Koskela, A.; et al. Female Mice Lacking Estrogen Receptor-α in Hypothalamic Proopiomelanocortin (POMC) Neurons Display Enhanced Estrogenic Response on Cortical Bone Mass. Endocrinology 2016, 157, 3242–3252. [Google Scholar] [CrossRef]
- Melville, K.; Kelly, N.; Surita, G.; Buchalter, D.; Schimenti, J.; Main, R.; Ross, F.P.; van der Meulen, M.C. Effects of Deletion of ER α in Osteoblast-Lineage Cells on Bone Mass and Adptation to Mechanical Loading Differ in Female and Male Mice. J. Bone Miner. Res. 2015, 30, 1468–1480. [Google Scholar] [CrossRef] [PubMed]
- Määttä, J.A.; Büki, K.G.; Gu, G.; Alanne, M.H.; Vääräniemi, J.; Liljenbäck, H.; Poutanen, M.; Härkönen, P.; Väänänen, K. Inactivation of estrogen receptor α in bone-forming cells induces bone loss in female mice. FASEB J. 2013, 27, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Windahl, S.H.; Börjesson, A.E.; Farman, H.H.; Engdahl, C.; Movérare-Skrtic, S.; Sjögren, K.; Lagerquist, M.K.; Kindblom, J.M.; Koskela, A.; Tuukkanen, J.; et al. Estrogen receptor-α in osteocytes is important for trabecular bone formation in male mice. Proc. Natl. Acad. Sci. USA 2013, 110, 2294–2299. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, S.; Inoue, K.; Igarashi, K.; Sugizaki, H.; Shirode-Fukuda, Y.; Inoue, E.; Yu, T.; Takeuchi, J.K.; Kanno, J.; Bonewald, L.F.; et al. Estrogen receptor α in osteocytes regulates trabecular bone formation in female mice. Bone 2014, 60, 68–77. [Google Scholar] [CrossRef]
- Ucer, S.; Iyer, S.; Bartell, S.M.; Martin-Millan, M.; Han, L.; Kim, H.-N.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A.; Almeida, M.; et al. The Effects of Androgens on Murine Cortical Bone Do Not Require AR or ERα Signaling in Osteoblasts and Osteoclasts. J. Bone Miner. Res. 2015, 30, 1138–1149. [Google Scholar] [CrossRef]
- Nakamura, T.; Imai, Y.; Matsumoto, T.; Sato, S.; Takeuchi, K.; Igarashi, K.; Harada, Y.; Azuma, Y.; Krust, A.; Yamamoto, Y.; et al. Estrogen Prevents Bone Loss via Estrogen Receptor α and Induction of Fas Ligand in Osteoclasts. Cell 2007, 130, 811–823. [Google Scholar] [CrossRef]
- Almeida, M.; Iyer, S.; Martin-Millan, M.; Bartell, S.; Han, L.; Ambrogini, E.; Onal, M.; Xiong, J.; Weinstein, R.; Jilka, R.; et al. Estrogen Receptor-α Signaling in Osteoblast Pro-genitors Stimulates Cortical Bone Accrual. J. Clin. Investig. 2013, 123, 394–404. [Google Scholar] [CrossRef]
- Melville, K.M.; Kelly, N.H.; Khan, S.A.; Schimenti, J.C.; Ross, F.P.; Main, R.P.; Van Der Meulen, M.C.H. Female Mice Lacking Estrogen Receptor-Alpha in Osteoblasts Have Compromised Bone Mass and Strength. J. Bone Miner. Res. 2014, 29, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Nicks, K.M.; Fujita, K.; Fraser, D.G.; McGregor, U.; Drake, M.T.; McGee-Lawrence, M.E.; Westendorf, J.J.; Monroe, D.G.; Khosla, S. Deletion of Estrogen Receptor Beta in Osteoprogenitor Cells Increases Trabecular but Not Cortical Bone Mass in Female Mice. J. Bone Miner. Res. 2016, 31, 606–614. [Google Scholar] [CrossRef]
- Zhang, J.; Link, D.C. Targeting of Mesenchymal Stromal Cells by Cre-Recombinase Transgenes Commonly Used to Target Osteoblast Lineage Cells. J. Bone Miner. Res. 2016, 31, 2001–2007. [Google Scholar] [CrossRef]
- Henning, P.; Ohlsson, C.; Engdahl, C.; Farman, H.; Windahl, S.H.; Carlsten, H.; Lagerquist, M.K. The effect of estrogen on bone requires ERα in nonhematopoietic cells but is enhanced by ERα in hematopoietic cells. Am. J. Physiol. Metab. 2014, 307, E589–E595. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R. Role of T cells in ovariectomy induced bone loss-revisited. J. Bone Miner. Res. 2012, 27, 231–239. [Google Scholar] [CrossRef]
- Horowitz, M.C.; Fretz, J.A.; Lorenzo, J.A. How B cells influence bone biology in health and disease. Bone 2010, 47, 472–479. [Google Scholar] [CrossRef]
- Gustafsson, K.L.; Nilsson, K.H.; Farman, H.H.; Andersson, A.; Lionikaite, V.; Henning, P.; Wu, J.; Windahl, S.H.; Islander, U.; Movérare-Skrtic, S.; et al. ERα expression in T lymphocytes is dispensable for estrogenic effects in bone. J. Endocrinol. 2018, 238, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Piemontese, M.; Liu, Y.; Thostenson, J.D.; Xiong, J.; O’Brien, C.A. RANKL (Receptor Activator of NFκB Ligand) Pro-duced by Osteocytes Is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J. Biol. Chem. 2016, 291, 24838–24850. [Google Scholar] [CrossRef] [PubMed]
- Billon-Galés, A.; Fontaine, C.; Filipe, C.; Douin-Echinard, V.; Fouque, M.-J.; Flouriot, G.; Gourdy, P.; Lenfant, F.; Laurell, H.; Krust, A.; et al. The transactivating function 1 of estrogen receptor α is dispensable for the vasculoprotective actions of 17β-estradiol. Proc. Natl. Acad. Sci. USA 2009, 106, 2053–2058. [Google Scholar] [CrossRef]
- Billon-Galés, A.; Krust, A.; Fontaine, C.; Abot, A.; Flouriot, G.; Toutain, C.; Berges, H.; Gadeau, A.-P.; Lenfant, F.; Gourdy, P.; et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. USA 2011, 108, 13311–13316. [Google Scholar] [CrossRef]
- Börjesson, A.E.; Farman, H.H.; Engdahl, C.; Koskela, A.; Sjögren, K.; Kindblom, J.M.; Stubelius, A.; Islander, U.; Carlsten, H.; Antal, M.C.; et al. The role of activation functions 1 and 2 of estrogen receptor-α for the effects of estradiol and selective estrogen receptor modulators in male mice. J. Bone Miner. Res. 2013, 28, 1117–1126. [Google Scholar] [CrossRef]
- Börjesson, A.E.; Windahl, S.H.; Lagerquist, M.K.; Engdahl, C.; Frenkel, B.; Movérare-Skrtic, S.; Sjögren, K.; Kindblom, J.M.; Stubelius, A.; Islander, U.; et al. Roles of transactivating func-tions 1 and 2 of estrogen receptor-alpha in bone. Proc. Natl. Acad. Sci. USA 2011, 108, 6288–6293. [Google Scholar] [CrossRef]
- Fontaine, C.; Buscato, M.; Vinel, A.; Giton, F.; Raymond-Letron, I.; Kim, S.H.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Gourdy, P.; Milon, A.; et al. The tissue-specific effects of different 17β-estradiol doses reveal the key sensitizing role of AF1 domain in ERα activity. Mol. Cell. Endocrinol. 2020, 505, 110741. [Google Scholar] [CrossRef]
- Bartell, S.M.; Han, L.; Kim, H.-N.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; Chambliss, K.L.; Shaul, P.W.; Roberson, P.K.; Weinstein, R.S.; et al. Non-Nuclear–Initiated Actions of the Estrogen Receptor Protect Cortical Bone Mass. Mol. Endocrinol. 2013, 27, 649–656. [Google Scholar] [CrossRef]
- Vinel, A.; Hay, E.; Valera, M.C.; Buscato, M.; Adlanmerini, M.; Guillaume, M.; Cohen-Solal, M.; Ohlsson, C.; Lenfant, F.; Arnal, J.F.; et al. Role of ERα in the Effect of Estradiol on Cancel-lous and Cortical Femoral Bone in Growing Female Mice. Endocrinology 2016, 157, 2533–2544. [Google Scholar] [CrossRef]
- Gustafsson, K.L.; Farman, H.; Henning, P.; Lionikaite, V.; Movérare-Skrtic, S.; Wu, J.; Ryberg, H.; Koskela, A.; Gustafsson, J.-Å.; Tuukkanen, J.; et al. The role of membrane ERα signaling in bone and other major estrogen responsive tissues. Sci. Rep. 2016, 6, 29473. [Google Scholar] [CrossRef]
- Banerjee, S.; Chambliss, K.L.; Mineo, C.; Shaul, P.W. Recent insights into non-nuclear actions of estrogen receptor alpha. Steroids 2014, 81, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.L.; Boudou, F.; Sautier, L.; Vessieres, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [PubMed]
- Pedram, A.; Razandi, M.; Lewis, M.; Hammes, S.; Levin, E.R. Membrane-localized estrogen receptor α is required for normal or-gan development and function. Dev. Cell 2014, 29, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Adlanmerini, M.; Fébrissy, C.; Zahreddine, R.; Vessières, E.; Buscato, M.; Solinhac, R.; Favre, J.; Anquetil, T.; Guilhot, A.L.; Boudou, F.; et al. Mutation of Arginine 264 on ERα (Estrogen Receptor Alpha) Selectively Abrogates the Rapid Signaling of Estradiol in the Endothelium Without Altering Fertili-ty. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2143–2158. [Google Scholar] [CrossRef]
- Gustafsson, K.L.; Farman, H.H.; Nilsson, K.H.; Henning, P.; Movérare-Skrtic, S.; Lionikaite, V.; Lawenius, L.; Engdahl, C.; Ohlsson, C.; Lagerquist, M.K. Arginine site 264 in murine estrogen receptor alpha is dispensable for the regulation of the skeleton. Am. J. Physiol. Metab. 2020. [Google Scholar] [CrossRef]
- Abot, A.; Fontaine, C.; Buscato, M.; Solinhac, R.; Flouriot, G.; Fabre, A.; Drougard, A.; Rajan, S.; Laine, M.; Milon, A.; et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation. EMBO Mol. Med. 2014, 6, 1328–1346. [Google Scholar] [CrossRef]
- Benoit, T.; Valera, M.-C.; Fontaine, C.; Buscato, M.; Lenfant, F.; Raymond-Letron, I.; Tremollieres, F.; Soulie, M.; Foidart, J.-M.; Gamé, X.; et al. Estetrol, a Fetal Selective Estrogen Receptor Modulator, Acts on the Vagina of Mice through Nuclear Estrogen Receptor α Activation. Am. J. Pathol. 2017, 187, 2499–2507. [Google Scholar] [CrossRef]
- Valéra, M.-C.; Noirrit, E.; Dupuis, M.; Fontaine, C.; Lenfant, F.; Briaux, A.; Cabou, C.; Garcia, C.; Lairez, O.; Foidart, J.-M.; et al. Effect of estetrol, a selective nuclear estrogen receptor modulator, in mouse models of arterial and venous thrombosis. Mol. Cell. Endocrinol. 2018, 477, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Coelingh Bennink, H.J.; Holinka, C.F.; Diczfalusy, E. Estetrol review: Profile and potential clinical applications. Climacteric 2008, 11 (Suppl. 1), 47–58. [Google Scholar] [CrossRef]
- Coelingh Bennink, H.J.; Heegaard, A.M.; Visser, M.; Holinka, C.F.; Christiansen, C. Oral bioavailability and bone-sparing effects of estetrol in an osteoporosis model. Climacteric 2008, 11 (Suppl. 1), 2–14. [Google Scholar] [CrossRef]
- Coelingh Bennink, H.J.T.; Verhoeven, C.; Zimmerman, Y.; Visser, M.; Foidart, J.M.; Gemzell-Danielsson, K. Pharmacodynamic effects of the fetal estrogen estetrol in postmenopausal women: Results from a multiple-rising-dose study. Menopause 2017, 24, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Coelingh Bennink, H.J.; Verhoeven, C.; Zimmerman, Y.; Visser, M.; Foidart, J.M.; Gemzell-Danielsson, K. Clinical effects of the fetal estrogen estetrol in a multiple-rising-dose study in postmenopausal women. Maturitas 2016, 91, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Coelingh Bennink, H.J.T.; Zimmerman, Y.; Verhoeven, C.; Dutman, A.E.; Mensinga, T.; Kluft, C.; Reisman, Y.; Debruyne, F.M.J. A Dose-Escalating Study with the Fetal Estrogen Estetrol in Healthy Men. J. Clin. Endocrinol. Metab. 2018, 103, 3239–3249. [Google Scholar] [CrossRef] [PubMed]
- Harrington, W.; Sung Hoon, K.; Funk, C.; Madak-Erdogan, Z.; Schiff, R.; Katzenellenbogen, J.; Katzenellenbogen, B. Estrogen Dendrimer Conju-gates that Preferentially Activate Extranuclear, Nongenomic Versus Genomic Pathways of Estrogen Action. Mol. Endo-Crinol. 2006, 20, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Chambliss, K.L.; Wu, Q.; Oltmann, S.; Konaniah, E.S.; Umetani, M.; Korach, K.S.; Thomas, G.; Minea, C.; Yyhanna, I.; Sung Hoon, K.; et al. Non-nuclear estrogen receptor alpha signal-ing promotes cardiovascular protection but not uterine or breast cancer growth in mice. J. Clin. Investig. 2010, 120, 2319–3230. [Google Scholar] [CrossRef]
- Farman, H.H.; Wu, J.; Gustafsson, K.L.; Windahl, S.H.; Kim, S.H.; A Katzenellenbogen, J.; Ohlsson, C.; Lagerquist, M.K. Extra-nuclear effects of estrogen on cortical bone in males require ERαAF-1. J. Mol. Endocrinol. 2017, 58, 105–111. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Kim, S.H.; Gong, P.; Zhao, Y.C.; Zhang, H.; Chambliss, K.L.; Carlson, K.E.; Mayne, C.G.; Shaul, P.W.; Korach, K.S.; et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci. Signal. 2016, 9, ra53. [Google Scholar] [CrossRef]
- Farman, H.H.; Gustafsson, K.L.; Henning, P.; Grahnemo, L.; Lionikaite, V.; Movérare-Skrtic, S.; Wu, J.; Ryberg, H.; Koskela, A.; Tuukkanen, J.; et al. Membrane estrogen receptor α is essential for estrogen signaling in the male skeleton. J. Endocrinol. 2018, 239, 303–312. [Google Scholar] [CrossRef]
- Riggs, L.; Hartmann, L. Selective Estrogen-Receptor Modulators: Mechanisms of Action and Application to Clinical Practice. N. Engl. J. Med. 2003, 348, 618–629. [Google Scholar] [CrossRef]
- Pinkerton, J.V.; Conner, E.A. Beyond estrogen: Advances in tissue selective estrogen complexes and selective estrogen receptor modulators. Climacteric 2019, 22, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.A.; Sun, W.; Chen, H.; Zhang, H.; Lay, Y.-A.E.; Lane, N.E.; Yao, W. Optimizing tamoxifen-inducible Cre/loxp system to reduce tamoxifen effect on bone turnover in long bones of young mice. Bone 2015, 81, 614–619. [Google Scholar] [CrossRef]
- Starnes, L.M.; Downey, C.M.; Boyd, S.K.; Jirik, F.R. Increased bone mass in male and female mice following tamoxifen administra-tion. Genesis 2007, 45, 229–235. [Google Scholar] [CrossRef]
- Jardí, F.; Laurent, M.R.; Dubois, V.; Khalil, R.; Deboel, L.; Schollaert, D.; Bosch, L.V.D.; Decallonne, B.; Carmeliet, G.; Claessens, F.; et al. A shortened tamoxifen induction scheme to induce CreER recombinase without side effects on the male mouse skeleton. Mol. Cell. Endocrinol. 2017, 452, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Tando, T.; Morita, M.; Miyamoto, K.; Kobayashi, T.; Watanabe, R.; Oike, T.; Matsumoto, M.; Nakamura, M.; Miyamoto, T. Selective estrogen receptor modulators and the vitamin D analogue eldecalcitol block bone loss in male osteoporosis. Biochem. Biophys. Res. Commun. 2017, 482, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Kanis, J.A.; Cooper, C.; Rizzoli, R.; Reginster, J.Y. (IOF) SABotESfCaEAoOEatCoSAaNSotIOF. European guidance for the diagno-sis and management of osteoporosis in postmenopausal women. Osteoporos. Int. 2019, 30, 3–44. [Google Scholar] [CrossRef]
- Börjesson, A.E.; Farman, H.H.; Movérare-Skrtic, S.; Engdahl, C.; Antal, M.C.; Koskela, A.; Tuukkanen, J.; Carlsten, H.; Krust, A.; Chambon, P.; et al. SERMs have substance-specific ef-fects on bone, and these effects are mediated via ErαAF-1 in female mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E912–E918. [Google Scholar] [CrossRef]
- Palacios, S.; Silverman, S.L.; de Villiers, T.J.; Levine, A.B.; Goemaere, S.; Brown, J.P.; De Cicco Nardone, F.; Williams, R.; Hines, T.L.; Mirkin, S.; et al. A 7-year randomized, placebo-controlled trial assessing the long-term efficacy and safety of bazedoxifene in postmenopausal women with osteoporosis: Effects on bone density and fracture. Menopause 2015, 22, 806–813. [Google Scholar] [CrossRef]
- Cummings, S.R.; Ensrud, K.; Delmas, P.D.; LaCroix, A.Z.; Vukicevic, S.; Reid, D.M.; Goldstein, S.; Sriram, U.; Lee, A.; Thompson, J.; et al. Lasofoxifene in postmenopausal women with osteoporosis. N. Engl. J. Med. 2010, 362, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Vico, L.; Collet, P.; Guignandon, A.; Lafage-Proust, M.-H.; Thomas, T.; Rehailia, M.; Alexandre, C. Effects of long-term microgravity exposure on cancellous and cortical weight-bearing bones of cosmonauts. Lancet 2000, 355, 1607–1611. [Google Scholar] [CrossRef]
- Klentrou, P. Influence of Exercise and Training on Critical Stages of Bone Growth and Development. Pediatr. Exerc. Sci. 2016, 28, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.I.; Plotkin, L.I.; Gortazar, A.R.; Millan, M.M.; O’Brien, C.A.; Manolagas, S.C.; Bellido, T. A Novel Ligand-independent Function of the Estrogen Receptor Is Essential for Osteocyte and Osteoblast Mechanotransduction. J. Biol. Chem. 2007, 282, 25501–25508. [Google Scholar] [CrossRef] [PubMed]
- Galea, G.L.; Meakin, L.B.; Sugiyama, T.; Zebda, N.; Sunters, A.; Taipaleenmaki, H.; Stein, G.S.; Van Wijnen, A.J.; Lanyon, L.E.; Price, J.S. Estrogen Receptor α Mediates Proliferation of Osteoblastic Cells Stimulated by Estrogen and Mechanical Strain, but Their Acute Down-regulation of the Wnt Antagonist Sost Is Mediated by Estrogen Receptor β. J. Biol. Chem. 2013, 288, 9035–9048. [Google Scholar] [CrossRef]
- Suuriniemi, M.; Mahonen, A.; Kovanen, V.; Alén, M.; Lyytikäinen, A.; Wang, Q.; Kröger, H.; Cheng, S. Association Between Exercise and Pubertal BMD Is Modulated by Estrogen Receptor α Genotype. J. Bone Miner. Res. 2004, 19, 1758–1765. [Google Scholar] [CrossRef] [PubMed]
- Saxon, L.K.; Galea, G.; Meakin, L.; Price, J.; Lanyon, L.E. Estrogen Receptors α and β Have Different Gender-Dependent Effects on the Adaptive Responses to Load Bearing in Cancellous and Cortical Bone. Endocrinology 2012, 153, 2254–2266. [Google Scholar] [CrossRef]
- Windahl, S.; Saxon, L.; Börjesson, A.; Lagerquist, M.; Frenkel, B.; Henning, P.; Lerner, U.; Galea, G.; Meakin, L.; Engdahl, C.; et al. Estrogen Receptor-α Is Required of the Osteo-genic Response to Mechanical Loading in a Ligand-Independant Manner Involving Its Activatin Function 1 but Not 2. J. Bone Miner. Res. 2013, 28, 291–301. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Targeted Cells | Osteoclasts | Osteoblasts | Osteocytes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Differentiation Stage | Myeloid Progenitors | Mature Osteoclasts | Pluripotent Mesenchymal Progenitors | Osteoblastic Progenitors | Mature Matrix Maturation | Mature Mineralization | NA | |||||||
| gene promotor | LysM | CtsK | Prx1 | Osx1 | Col1a1 | Ocn | Dmp1 | |||||||
| gender | female | male | female | male | female | male | female | male | female | male | female | male | female | male |
| trabecular bone | ↘ | ↔ | ↘ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↔ | ↘ | ↔ [81] ↘ [82] | ↔ [83] ↘ [84] | ↘ |
| cortical bone | ↔ | ↔ | ↔ | ↔ | ↘ | ↘ * | ↘ | ↘ * | ↔ | ↔ | ↘ | ↔ | ↔ | ↔ |
| references | [55] | [85] | [86] | [86] | [87] | [87] | [87] | [85] | [87] | [87] | [81,82,88] | [81,82] | [83,84] | [83] |
| Trabecular Bone | Cortical Bone | Alveolar Bone | ||||||
|---|---|---|---|---|---|---|---|---|
| Mouse Model | Treatment | Female | Male | Female | Male | Female | Male | References |
| WT | E2 | ↗↗ | ↗↗ | ↗↗ | ↗↗ | ↗↗ | NT | [76,77] |
| EDC | ↔ | NT | ↗ | NT | ↗ | NT | [77,101] | |
| PaPEs | NT | NT | NT | NT | ↗ | NT | [77] | |
| ERα−/− | E2 | ↔ | ↔ | ↔ | ↔ | ↔ | NT | [76,77] |
| ERβ−/− | E2 | ↗ | ↗↗ | ↗ | ↗↗ | ↗↗ | NT | [76,77] |
| ERα AF1° | E2 | ↔ | ↔ | ↗↗ | ↗↗ | NT | NT | [98,99] |
| ERα AF2° | E2 | ↔ | ↔ | ↔ | ↔ | ↔ | NT | [77,98,99] |
| C451A-ERα | E2 | ↗ | ↗ | ↗ | ↗ | ↗ | NT | [77,102,103] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noirrit-Esclassan, E.; Valera, M.-C.; Tremollieres, F.; Arnal, J.-F.; Lenfant, F.; Fontaine, C.; Vinel, A. Critical Role of Estrogens on Bone Homeostasis in Both Male and Female: From Physiology to Medical Implications. Int. J. Mol. Sci. 2021, 22, 1568. https://doi.org/10.3390/ijms22041568
Noirrit-Esclassan E, Valera M-C, Tremollieres F, Arnal J-F, Lenfant F, Fontaine C, Vinel A. Critical Role of Estrogens on Bone Homeostasis in Both Male and Female: From Physiology to Medical Implications. International Journal of Molecular Sciences. 2021; 22(4):1568. https://doi.org/10.3390/ijms22041568
Chicago/Turabian StyleNoirrit-Esclassan, Emmanuelle, Marie-Cécile Valera, Florence Tremollieres, Jean-Francois Arnal, Françoise Lenfant, Coralie Fontaine, and Alexia Vinel. 2021. "Critical Role of Estrogens on Bone Homeostasis in Both Male and Female: From Physiology to Medical Implications" International Journal of Molecular Sciences 22, no. 4: 1568. https://doi.org/10.3390/ijms22041568
APA StyleNoirrit-Esclassan, E., Valera, M.-C., Tremollieres, F., Arnal, J.-F., Lenfant, F., Fontaine, C., & Vinel, A. (2021). Critical Role of Estrogens on Bone Homeostasis in Both Male and Female: From Physiology to Medical Implications. International Journal of Molecular Sciences, 22(4), 1568. https://doi.org/10.3390/ijms22041568