Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Age-Related SOB Is the Result of a Vicious Cycle between Aging, Sarcopenia, and Obesity
3. Organokines Such as Myokines, Adipokines, and Hepatokines Are Involved in Modulating AMPK and PGC-1α Signaling Pathways, Which Are Important for the Induction of SOB
4. Dysregulated Autophagy Aggravates Sarcopenic Obesity through Dysfunctional AMPK and PGC-1α Signaling Pathway-Mediated Inflammation and Insulin Resistance
4.1. Role of Autophagy in the Induction of Sarcopenic Obesity
4.2. Dysregulated AMPK and PGC-1α Signaling Pathways Contribute to Inflammation and Insulin Resistance
4.3. Endoplasmic Reticulum Stress Disturbs the Function of Autophagy and Dysfunctional Autophagy Also Inhibits ER Function
5. Sarcopenic Obesity Increases the Risk of Various Diseases
5.1. Association of SOB with Metabolic Disease and Cardiovascular Disease
5.2. Association of SOB with Liver Disease
5.3. Association of SOB with Falls and Fractures and Osteoarthritis
5.4. Association of SOB with Pulmonary Disease and Mental Health
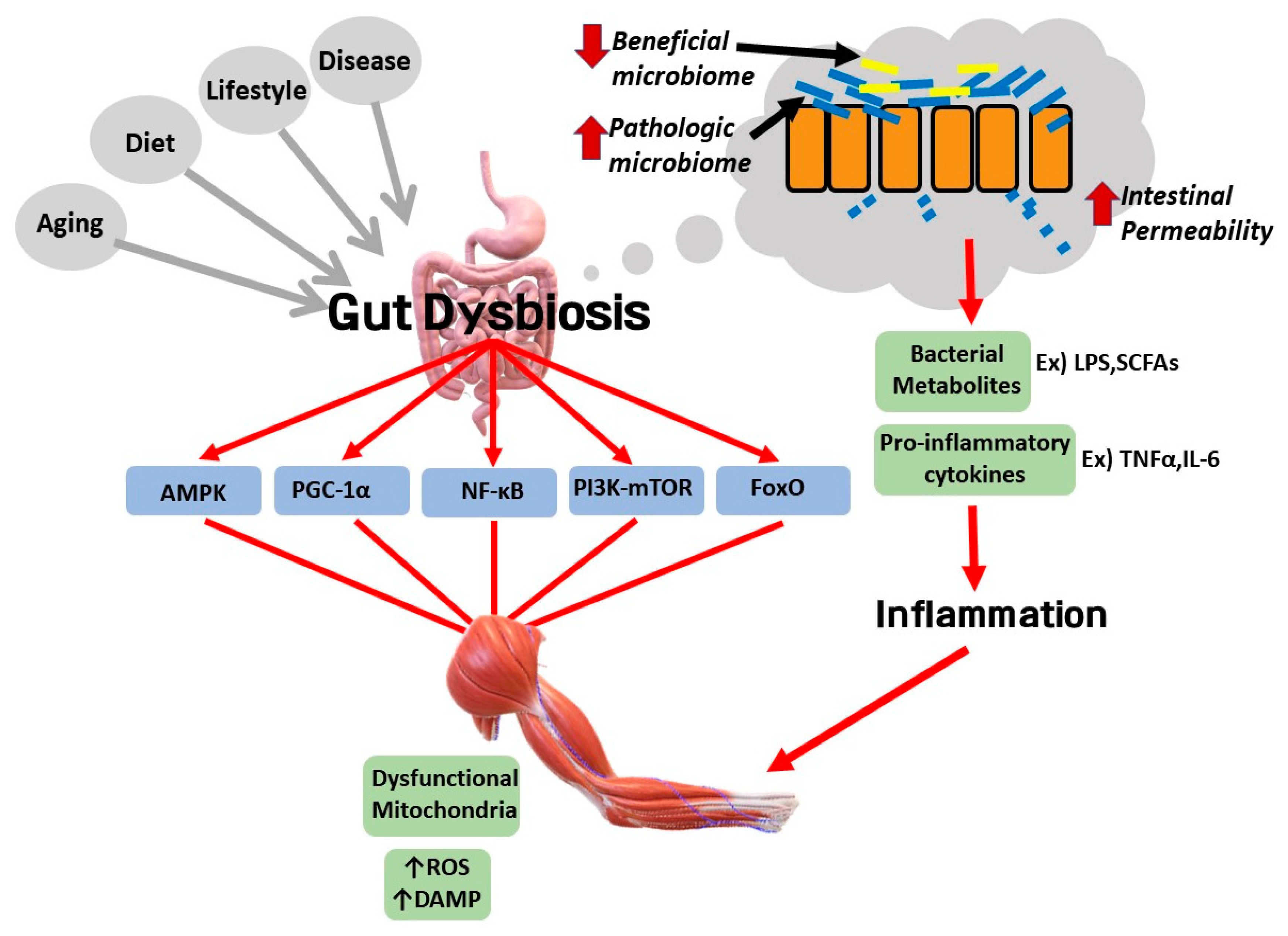
6. Potential Involvement of Gut Dysbiosis in the Pathological Link between Sarcopenic Obesity and Various Diseases
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SOB | Sarcopenic obesity |
| IR | Insulin resistance |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| ROS | Reactive oxygen species |
| AMPK | AMP-activated protein kinase |
| PGC1α | Peroxisome proliferator-activated receptor γ coactivator 1α |
| IRS | Insulin receptor substrate |
| ACC | Acetyl CoA carboxylase |
| SCFA | Single chain fatty acid |
| NLRP3 | Nod-like receptor 3 |
| ER | Endoplasmic reticulum |
| TAG | Triacylglycerol |
References
- Abarca-Gómez, L.; Abdeen, Z.A.; Hamid, Z.A.; Abu-Rmeileh, N.M.; Acosta-Cazares, B.; Acuin, C.; Adams, R.J.; Aekplakorn, W.; Afsana, K.; Agyemang, C.; et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- Jin, K.; Simpkins, J.W.; Ji, X.; Leis, M.; Stambler, I. The Critical Need to Promote Research of Aging and Aging-related Diseases to Improve Health and Longevity of the Elderly Population. Aging Dis. 2015, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.N.; Choi, K.M. Sarcopenic obesity. J. Korean Diabetes 2013, 14, 166–173. [Google Scholar] [CrossRef][Green Version]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The role of inflammation in age-related sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef]
- Jang, H.C. Recent Progression in Sarcopenia and Sarcopenic Obesity. J. Korean Geriatr. Soc. 2011, 15, 1–7. [Google Scholar] [CrossRef]
- Ilich, J.Z.; Kelly, O.J.; Inglis, J.E.; Panton, L.B.; Duque, G.; Ormsbee, M.J. Interrelationship among muscle, fat, and bone: Connecting the dots on cellular, hormonal, and whole body levels. Ageing Res. Rev. 2014, 15, 51–60. [Google Scholar] [CrossRef]
- Tereshina, E.V.; Ivanenko, S.I. Age-related obesity is a heritage of the evolutionary past. Biochemistry 2014, 79, 581–592. [Google Scholar] [CrossRef]
- Benton, M.J.; Whyte, M.D.; Dyal, B.W. Sarcopenic obesity: Strategies for management. Am. J. Nurs. 2011, 111, 38–44. [Google Scholar] [CrossRef]
- Zamboni, M.; Rubele, S.; Rossi, A.P. Sarcopenia and obesity. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 13–19. [Google Scholar] [CrossRef]
- Fan, J.; Kou, X.; Jia, S.; Yang, X.; Yang, Y.; Chen, N. Autophagy as a Potential Target for Sarcopenia. J. Cell. Physiol. 2016, 231, 1450–1459. [Google Scholar] [CrossRef]
- Ticinesi, A.; Lauretani, F.; Milani, C.; Nouvenne, A.; Tana, C.; Del Rio, D.; Maggio, M.; Ventura, M.; Meschi, T. Aging gut microbiota at the cross-road between nutrition, physical frailty, and sarcopenia: Is there a gut–muscle axis? Nutrients 2017, 9, 1303. [Google Scholar] [CrossRef]
- Zamboni, M.; Rossi, A.P.; Fantin, F.; Zamboni, G.; Chirumbolo, S.; Zoico, E.; Mazzali, G. Adipose tissue, diet and aging. Mech. Ageing Dev. 2014, 136–137, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Blomberg, B.B. Adipose tissue inflammation induces B cell inflammation and decreases B cell function in aging. Front. Immunol. 2017, 8, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Choi, K.M. Sarcopenic obesity, insulin resistance, and their implications in cardiovascular and metabolic consequences. Int. J. Mol. Sci. 2020, 21, 494. [Google Scholar] [CrossRef] [PubMed]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Abete, I.; Konieczna, J.; Zulet, M.A.; Galmés-Panades, A.M.; Ibero-Baraibar, I.; Babio, N.; Estruch, R.; Vidal, J.; Toledo, E.; Razquin, C.; et al. Association of lifestyle factors and inflammation with sarcopenic obesity: Data from the PREDIMED-Plus trial. J. Cachexia Sarcopenia Muscle 2019, 10, 974–984. [Google Scholar] [CrossRef]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef]
- Waters, D.L.; Baumgartner, R.N. Sarcopenia and Obesity. Clin. Geriatr. Med. 2011, 27, 401–421. [Google Scholar] [CrossRef]
- Schrauwen-Hinderling, V.B.; Kooi, M.E.; Hesselink, M.K.C.; Jeneson, J.A.L.; Backes, W.H.; Van Echteld, C.J.A.; Van Engelshoven, J.M.A.; Mensink, M.; Schrauwen, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Dobson, J.P.; Greene, N.P.; Wiggs, M.P.; Shimkus, K.L.; Wudeck, E.V.; Davis, A.R.; Laureano, M.L.; Fluckey, J.D. Abnormal protein turnover and anabolic resistance to exercise in sarcopenic obesity. FASEB J. 2013, 27, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, D.R.; Dionne, I.J.; Brochu, M. Sarcopenic/obesity and physical capacity in older men and women: Data from the nutrition as a determinant of successful aging (nuage)the quebec longitudinal study. Obesity 2009, 17, 2082–2088. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.N.; Wayne, S.J.; Waters, D.L.; Janssen, I.; Gallagher, D.; Morley, J.E. Sarcopenic obesity predicts instrumental activities of daily living disability in the elderly. Obes. Res. 2004, 12, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Roubenoff, R. Catabolism of aging: Is it an inflammatory process? Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 295–299. [Google Scholar] [CrossRef]
- Alway, S.E.; Degens, H.; Krishnamurthy, G.; Smith, C.A. Potential role for Id myogenic repressors in apoptosis and attenuation of hypertrophy in muscles of aged rats. Am. J. Physiol. Cell Physiol. 2002, 283, 66–76. [Google Scholar] [CrossRef]
- Shoji, H.; Takao, K.; Hattori, S.; Miyakawa, T. Age-related changes in behavior in C57BL/6J mice from young adulthood to middle age. Mol. Brain 2016, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Salvestrini, V.; Sell, C.; Lorenzini, A. Obesity may accelerate the aging process. Front. Endocrinol. 2019, 10, 226. [Google Scholar] [CrossRef]
- Pérez, L.M.; Pareja-Galeano, H.; Sanchis-Gomar, F.; Emanuele, E.; Lucia, A.; Gálvez, B.G. ‘Adipaging’: Ageing and obesity share biological hallmarks related to a dysfunctional adipose tissue. J. Physiol. 2016, 594, 3187–3207. [Google Scholar] [CrossRef]
- Choi, K.M. The impact of organokines on insulin resistance, inflammation, and atherosclerosis. Endocrinol. Metab. 2016, 31, 1–6. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.; Duan, Y.; Hu, C.A.A.; Tang, Y.; Yin, Y. Myokines and adipokines: Involvement in the crosstalk between skeletal muscle and adipose tissue. Cytokine Growth Factor Rev. 2017, 33, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Joulia, D.; Bernardi, H.; Garandel, V.; Rabenoelina, F.; Vernus, B.; Cabello, G. Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp. Cell Res. 2003, 286, 263–275. [Google Scholar] [CrossRef]
- Deng, Z.; Luo, P.; Lai, W.; Song, T.; Peng, J.; Wei, H.K. Myostatin inhibits eEF2K-eEF2 by regulating AMPK to suppress protein synthesis. Biochem. Biophys. Res. Commun. 2017, 494, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Markus, S.; Kathryn, R.; Leslie, E.; Christoph, H.; Thomas, R.; Wolfgang, K.; Thomas, B.; James, F.; Se Jin, L. Myostatin Mutation Associated with Gross Muscle Hypertrophy in a Child. N. Engl. J. Med. 2004, 350, 2682–2688. [Google Scholar] [CrossRef]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Hopkinson, N.S.; Kemp, P.R. Myostatin induces autophagy in skeletal muscle in vitro. Biochem. Biophys. Res. Commun. 2011, 415, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Lei, X.; Tan, S.Y.; Stanson, K.P.; Wei, Z.; Wong, G.W. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J. Biol. Chem. 2013, 288, 36073–36082. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jun, H.S. Role of myokines in regulating skeletal muscle mass and function. Front. Physiol. 2019, 10, 42. [Google Scholar] [CrossRef]
- Li, Y.; Li, F.; Lin, B.; Kong, X.; Tang, Y.; Yin, Y. Myokine IL-15 regulates the crosstalk of co-cultured porcine skeletal muscle satellite cells and preadipocytes. Mol. Biol. Rep. 2014, 41, 7543–7553. [Google Scholar] [CrossRef] [PubMed]
- Carbó, N.; López-Soriano, J.; Costelli, P.; Alvarez, B.; Busquets, S.; Baccino, F.M.; Quinn, L.B.S.; López-Soriano, F.J.; Argilés, J.M. Interleukin-15 mediates reciprocal regulation of adipose and muscle mass: A potential role in body weight control. Biochim. Biophys. Acta Gen. Subj. 2001, 1526, 17–24. [Google Scholar] [CrossRef]
- Quinn, L.S.; Anderson, B.G.; Strait-Bodey, L.; Stroud, A.M.; Argués, J.M. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E191–E202. [Google Scholar] [CrossRef] [PubMed]
- Barra, N.G.; Palanivel, R.; Denou, E.; Chew, M.V.; Gillgrass, A.; Walker, T.D.; Kong, J.; Richards, C.D.; Jordana, M.; Collins, S.M.; et al. Interleukin-15 modulates adipose tissue by altering mitochondrial mass and activity. PLoS ONE 2014, 9, e114799. [Google Scholar] [CrossRef] [PubMed]
- Le Quinn, B.S.; Anderson, B.G.; Conner, J.D.; Pistilli, E.E.; Wolden-Hanson, T. Overexpression of interleukin-15 in mice promotes resistance to diet-induced obesity, increased insulin sensitivity, and markers of oxidative skeletal muscle metabolism. Int. J. Interferon Cytokine Mediat. Res. 2011, 3, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Nelke, C.; Dziewas, R.; Minnerup, J.; Meuth, S.G.; Ruck, T. Skeletal muscle as potential central link between sarcopenia and immune senescence. EBioMedicine 2019, 49, 381–388. [Google Scholar] [CrossRef]
- Crane, J.D.; Macneil, L.G.; Lally, J.S.; Ford, R.J.; Bujak, A.L.; Brar, I.K.; Kemp, B.E.; Raha, S.; Steinberg, G.R.; Tarnopolsky, M.A. Exercise-stimulated interleukin-15 is controlled by AMPK and regulates skin metabolism and aging. Aging Cell 2015, 14, 625–634. [Google Scholar] [CrossRef]
- Omura, T.; Sano, M.; Omura, K.; Hasegawa, T.; Doi, M.; Sawada, T.; Nagano, A. Different expressions of BDNF, NT3, and NT4 in muscle and nerve after various types of peripheral nerve injuries. J. Peripher. Nerv. Syst. 2005, 10, 293–300. [Google Scholar] [CrossRef]
- Matthews, V.B.; Åström, M.B.; Chan, M.H.S.; Bruce, C.R.; Krabbe, K.S.; Prelovsek, O.; Åkerström, T.; Yfanti, C.; Broholm, C.; Mortensen, O.H.; et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 2009, 52, 1409–1418. [Google Scholar] [CrossRef]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.; Sharp, P.A.; et al. Sarcopenic obeisty and inflammation in the InCHIANTI study. PLoS ONE 2017, 32, 736–740. [Google Scholar] [CrossRef]
- Jimena Giudice, J.M.T. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef]
- So, B.; Kim, H.-J.; Kim, J.; Song, W. Exercise-induced myokines in health and metabolic diseases. Integr. Med. Res. 2014, 3, 172–179. [Google Scholar] [CrossRef]
- Nieto-Vazquez, I.; Fernandez-Veledo, S.; De Alvaro, C.; Lorenzo, M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 2008, 52, 3211–3221. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.A.; Brown, L.A.; Haynie, W.S.; Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Greene, N.P.; Washington, T.A. Cardiac hypertrophy in sarcopenic obese C57BL/6J mice is independent of Akt/mTOR cellular signaling. Exp. Gerontol. 2018, 111, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-α in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Kinney, B.; Yoo, H.S.; Lee, B.; Schaack, J.; Shao, J. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes 2012, 61, 1463–1470. [Google Scholar] [CrossRef][Green Version]
- Lee, B.; Shao, J. Adiponectin and lipid metabolism in skeletal muscle. Acta Pharm. Sin. B 2012, 2, 335–340. [Google Scholar] [CrossRef]
- Liu, Y.; Palanivel, R.; Rai, E.; Park, M.; Gabor, T.V.; Scheid, M.P.; Xu, A.; Sweeney, G. Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in Mice. Diabetes 2015, 64, 36–48. [Google Scholar] [CrossRef]
- Myeong, J.Y.; Gha, Y.L.; Chung, J.J.; Young, H.A.; Seung, H.H.; Jae, B.K. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor{alpha}. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef]
- Huh, J.Y.; Dincer, F.; Mesfum, E.; Mantzoros, C.S. Irisin stimulates muscle growth-related genes and regulates adipocyte differentiation and metabolism in humans. Int. J. Obes. 2014, 38, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Wente, W.; Efanov, A.M.; Brenner, M.; Kharitonenkov, A.; Köster, A.; Sandusky, G.E.; Sewing, S.; Treinies, I.; Zitzer, H.; Gromada, J. Fibroblast growth factor-21 improves pancreatic β-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes 2006, 55, 2470–2478. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. FGF21 activates AMPK signaling: Impact on metabolic regulation and the aging process. J. Mol. Med. 2017, 92, 123–131. [Google Scholar] [CrossRef]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef]
- Potes, Y.; de Luxán-Delgado, B.; Rodriguez-González, S.; Guimarães, M.R.M.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Boga, J.A.; Vega-Naredo, I.; Coto-Montes, A. Overweight in elderly people induces impaired autophagy in skeletal muscle. Free Radic. Biol. Med. 2017, 110, 31–41. [Google Scholar] [CrossRef]
- Riahi, Y.; Wikstrom, J.D.; Bachar-Wikstrom, E.; Polin, N.; Zucker, H.; Lee, M.S.; Quan, W.; Haataja, L.; Liu, M.; Arvan, P.; et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 2016, 59, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Whaley-Connell, A.T.; Sowers, J.R.; Ren, J. Autophagy as an Emerging Target in Cardiorenal Metabolic Disease: From Pathophysiology to Management. Pharmacol. Ther. 2018, 191, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. DMM Dis. Models Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Brian, J.; Morris, D.; Craig, W.; Timothy, A.; Donlon, B.J.W. FOXO3—A Major Gene for Human Longevity. Gerontology 2015, 61, 515–525. [Google Scholar]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- He, C.; Zhu, H.; Li, H.; Zou, M.H.; Xie, Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes 2013, 62, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- McMullen, C.A.; Ferry, A.L.; Gamboa, J.L.; Andrade, F.H.; Dupont-Versteegden, E.E. Age-related changes of cell death pathways in rat extraocular muscle. Exp. Gerontol. 2009, 44, 420–425. [Google Scholar] [CrossRef]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Current understanding of sarcopenia: Possible candidates modulating muscle mass. Pflug. Arch. Eur. J. Physiol. 2014, 467, 213–229. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Qiang, W.; Weiqiang, K.; Qing, Z.; Pengju, Z.; Yi, L. Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKα. Exp. Mol. Med. 2007, 39, 535–543. [Google Scholar] [CrossRef]
- Turdi, S.; Fan, X.; Li, J.; Zhao, J.; Huff, A.F.; Du, M.; Ren, J. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell 2010, 9, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, Y. Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Liu, H.W.; Chen, Y.T.; Chen, Y.A.; Chen, Y.J.; Chang, S.J. Resveratrol protects muscle cells against palmitate-induced cellular senescence and insulin resistance through ameliorating autophagic flux. J. Food Drug Anal. 2018, 26, 1066–1074. [Google Scholar] [CrossRef]
- Barlow, A.D.; Thomas, D.C. Autophagy in diabetes: β-cell dysfunction, insulin resistance, and complications. DNA Cell Biol. 2015, 34, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef]
- Brown, L.A.; Perry, R.A.; Haynie, W.; Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Greene, N.P.; Washington, T.A. Inflammatory signaling is altered in sarcopenic obese mice during skeletal muscle regeneration. FASEB J. 2017, 31, 1022. [Google Scholar]
- Zhang, N.; Cao, M.M.; Liu, H.; Xie, G.Y.; Li, Y.B. Autophagy regulates insulin resistance following endoplasmic reticulum stress in diabetes. J. Physiol. Biochem. 2015, 71, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Chen, X.; Chen, M.; Li, Y.; Li, Q.; Jiang, X.; Yang, Y.; Ling, W. Fish oil supplementation inhibits endoplasmic reticulum stress and improves insulin resistance: Involvement of AMP-activated protein kinase. Food Funct. 2017, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Li, L.; Liu, Y.; Pu, J.; Zhang, S.; Yu, J.; Zhao, J.; Liu, P. Oleate blocks palmitate-induced abnormal lipid distribution, endoplasmic reticulum expansion and stress, and insulin resistance in skeletal muscle. Endocrinology 2011, 152, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.N.; Choi, K.M. Sarcopenia: Definition, Epidemiology, and Pathophysiology. J. Bone Metab. 2013, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, M.; Mazzali, G.; Fantin, F.; Rossi, A.; Di Francesco, V. Sarcopenic obesity: A new category of obesity in the elderly. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Xu, Y. Association of sarcopenic obesity with the risk of all-cause mortality: A meta-analysis of prospective cohort studies. Geriatr. Gerontol. Int. 2016, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.; Liu, C.; Feng, W.; Xiao, X.; Tang, S.; Mo, Z. Role of AMPK in atherosclerosis via autophagy regulation. Sci. China Life Sci. 2018, 61, 1212–1221. [Google Scholar] [CrossRef]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef]
- Xue, Y.; Wei, Z.; Ding, H.; Wang, Q.; Zhou, Z.; Zheng, S.; Zhang, Y.; Hou, D.; Liu, Y.; Zen, K.; et al. MicroRNA-19b/221/222 induces endothelial cell dysfunction via suppression of PGC-1α in the progression of atherosclerosis. Atherosclerosis 2015, 241, 671–681. [Google Scholar] [CrossRef]
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1α in Vascular Regulation: Implications for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Kang, H.T.; Lee, D.C.; Lee, H.R.; Lee, Y.J. Body composition and its association with cardiometabolic risk factors in the elderly: A focus on sarcopenic obesity. Arch. Gerontol. Geriatr. 2013, 56, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Park, J.H.; Song, P.S.; Kim, D.K.; Kim, K.H.; Seol, S.H.; Kim, H.K.; Jang, H.J.; Lee, J.G.; Park, H.Y.; et al. Sarcopenic obesity as an independent risk factor of hypertension. J. Am. Soc. Hypertens. 2013, 7, 420–425. [Google Scholar] [CrossRef]
- Maliszewska, K.; Adamska-Patruno, E.; Krȩtowski, A. The interplay between muscle mass decline, obesity, and type 2 diabetes. Polish Arch. Intern. Med. 2019, 129, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Khadra, D.; Itani, L.; Tannir, H.; Kreidieh, D.; El Masri, D.; El Ghoch, M. Association between sarcopenic obesity and higher risk of type 2 diabetes in adults: A systematic review and meta-analysis. World J. Diabetes 2019, 10, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Cumming, R.; Naganathan, V.; Blyth, F.; Le Couteur, D.G.; Handelsman, D.J.; Seibel, M.; Waite, L.M.; Hirani, V. Associations of sarcopenic obesity with the metabolic syndrome and insulin resistance over five years in older men: The Concord Health and Ageing in Men Project. Exp. Gerontol. 2018, 108, 99–105. [Google Scholar] [CrossRef]
- Kim, T.N.; Choi, K.M. The implications of sarcopenia and sarcopenic obesity on cardiometabolic disease. J. Cell. Biochem. 2015, 116, 1171–1178. [Google Scholar] [CrossRef]
- Stephen, W.C.; Janssen, I. Sarcopenic-obesity and cardiovascular disease risk in the elderly. J. Nutr. Health Aging 2009, 13, 460–466. [Google Scholar] [CrossRef]
- Kim, J.H.; Cho, J.J.; Park, Y.S. Relationship between sarcopenic obesity and cardiovascular disease risk as estimated by the Framingham Risk Score. J. Korean Med. Sci. 2015, 30, 264–271. [Google Scholar] [CrossRef]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Matthew Morris, E.; Meers, G.M.E.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. Pgc-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.A.; Eagon, J.C.; LaRiviere, L.L.; Korenblat, K.M.; Klein, S.; Finck, B.N. Hepatic lipin 1β expression is diminished in insulin-resistant obese subjects and is reactivated by marked weight loss. Diabetes 2007, 56, 2395–2399. [Google Scholar] [CrossRef]
- Su, Z.; Nie, Y.; Huang, X.; Zhu, Y.; Feng, B.; Tang, L.; Zheng, G. Mitophagy in hepatic insulin resistance: Therapeutic potential and concerns. Front. Pharmacol. 2019, 10, 1193. [Google Scholar] [CrossRef]
- Koo, B.K.; Kim, D.; Joo, S.K.; Kim, J.H.; Chang, M.S.; Kim, B.G.; Lee, K.L.; Kim, W. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J. Hepatol. 2017, 66, 123–131. [Google Scholar] [CrossRef]
- Tovo, C.V.; Fernandes, S.A.; Buss, C.; De Mattos, A.A. Sarcopenia and non-alcoholic fatty liver disease: Is there a relationship? A systematic review. World J. Hepatol. 2017, 9, 326. [Google Scholar] [CrossRef]
- Pan, X.; Han, Y.; Zou, T.; Zhu, G.; Xu, K.; Zheng, J.; Zheng, M.; Cheng, X. Sarcopenia contributes to the progression of nonalcoholic fatty liver disease- related fibrosis: A meta-analysis. Dig. Dis. 2018, 36, 427–436. [Google Scholar] [CrossRef]
- Shida, T.; Oshida, N.; Oh, S.; Okada, K.; Shoda, J. Progressive reduction in skeletal muscle mass to visceral fat area ratio is associated with a worsening of the hepatic conditions of non-alcoholic fatty liver disease. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 495–503. [Google Scholar] [CrossRef]
- Merli, M.; Lattanzi, B.; Aprile, F. Sarcopenic obesity in fatty liver. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 185–190. [Google Scholar] [CrossRef]
- Hill, K.D.; Farrier, K.; Russell, M.; Burton, E. Dysmobility syndrome: Current perspectives. Clin. Interv. Aging 2017, 12, 145. [Google Scholar] [CrossRef]
- Binkley, N.; Krueger, D.; Buehring, B. What’s in a name revisited: Should osteoporosis and sarcopenia be considered components of “dysmobility syndrome?”. Osteoporos. Int. 2013, 24, 2955–2959. [Google Scholar] [CrossRef] [PubMed]
- Looker, A.C. Dysmobility syndrome and mortality risk in US men and women age 50 years and older. Osteoporos. Int. 2015, 26, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Liu, L.K.; Hwang, A.C.; Peng, L.N.; Lin, M.H.; Chen, L.K. Dysmobility Syndrome and Risk of Mortality for Community-Dwelling Middle-Aged and Older Adults: The Nexus of Aging and Body Composition. Sci. Rep. 2017, 7, 8785. [Google Scholar] [CrossRef] [PubMed]
- Hong, N.; Kim, C.O.; Youm, Y.; Choi, J.Y.; Kim, H.C.; Rhee, Y. Dysmobility syndrome is associated with prevalent morphometric vertebral fracture in older adults: The Korean Urban-Rural Elderly (KURE) study. Arch. Osteoporos. 2018, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Seibel, M.; Cumming, R.; Naganathan, V.; Blyth, F.; Le Couteur, D.G.; Handelsman, D.J.; Waite, L.M.; Hirani, V. Sarcopenic Obesity and Its Temporal Associations with Changes in Bone Mineral Density, Incident Falls, and Fractures in Older Men: The Concord Health and Ageing in Men Project. J. Bone Miner. Res. 2017, 32, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.; Chandrasekara, S.D.; Laslett, L.L.; Cicuttini, F.; Ebeling, P.R.; Jones, G. Associations of Sarcopenic Obesity and Dynapenic Obesity with Bone Mineral Density and Incident Fractures Over 5–10 Years in Community-Dwelling Older Adults. Calcif. Tissue Int. 2016, 99, 30–42. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Yang, B.; Lotz, M.; Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheumatol. 2011, 63, 1928–1937. [Google Scholar] [CrossRef]
- Zhou, S.; Lu, W.; Chen, L.; Ge, Q.; Chen, D.; Xu, Z.; Shi, D.; Dai, J.; Li, J.; Ju, H.; et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci. Rep. 2017, 7, 43245. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Zhang, Y.; Vasheghani, F.; Li, Y.H.; Blati, M.; Simeone, K.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Pelletier, J.P.; et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 2015, 74, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Godziuk, K.; Prado, C.M.; Woodhouse, L.J.; Forhan, M. Prevalence of sarcopenic obesity in adults with end-stage knee osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1735–1745. [Google Scholar] [CrossRef]
- Godziuk, K.; Prado, C.M.; Woodhouse, L.J.; Forhan, M. The impact of sarcopenic obesity on knee and hip osteoarthritis: A scoping review. BMC Musculoskelet. Disord. 2018, 19, 271. [Google Scholar] [CrossRef]
- Lee, S.; Kim, T.N.; Kim, S.H. Sarcopenic obesity is more closely associated with knee osteoarthritis than is nonsarcopenic obesity: A cross-sectional study. Arthritis Rheumatol. 2012, 64, 3947–3954. [Google Scholar] [CrossRef] [PubMed]
- Klingbeil, L.R.; Kim, P.; Piraino, G.; O’Connor, M.; Hake, P.W.; Wolfe, V.; Zingarelli, B. Age-dependent changes in AMPK metabolic pathways in the lung in a mouse model of hemorrhagic shock. Am. J. Respir. Cell Mol. Biol. 2017, 56, 585–596. [Google Scholar] [CrossRef]
- Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kong, M.H.; Kim, H.J. Implication of sarcopenia and sarcopenic obesity on lung function in healthy elderly: Using Korean national health and nutrition examination survey. J. Korean Med. Sci. 2015, 30, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Hamer, M.; Batty, G.D.; Kivimaki, M. Sarcopenic obesity and risk of new onset depressive symptoms in older adults: English Longitudinal Study of Ageing. Int. J. Obes. 2015, 39, 1717–1720. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.M.; Patterson, I.; McLoughlin, D.M. Peroxisome proliferator-activated receptor gamma co-activator-1 alpha in depression and the response to electroconvulsive therapy. Psychol. Med. 2019, 49, 1859–1868. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Lau, K.; Srivatsav, V.; Rizwan, A.; Nashed, A.; Liu, R.; Shen, R.; Akhtar, M. Bridging the gap between gut microbial dysbiosis and cardiovascular diseases. Nutrients 2017, 9, 859. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xia, J.; Jiang, C. Role of gut microbiota in the development of non-alcoholic fatty liver disease. Liver Res. 2019, 3, 25–30. [Google Scholar] [CrossRef]
- Schott, E.M.; Farnsworth, C.W.; Grier, A.; Lillis, J.A.; Soniwala, S.; Dadourian, G.H.; Bell, R.D.; Doolittle, M.L.; Villani, D.A.; Awad, H.; et al. Targeting the gut microbiome to treat the osteoarthritis of obesity. JCI Insight 2018, 3, e95997. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.H.; Paul, H.A.; Reimer, R.A.; Seerattan, R.A.; Hart, D.A.; Herzog, W. Relationship between inflammation, the gut microbiota, and metabolic osteoarthritis development: Studies in a rat model. Osteoarthr. Cartil. 2015, 23, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Mande, S.S. Diet, microbiota and gut-lung connection. Front. Microbiol. 2018, 9, 2147. [Google Scholar] [CrossRef]
- Nagpal, R.; Mainali, R.; Ahmadi, S.; Wang, S.; Singh, R.; Kavanagh, K.; Kitzman, D.W.; Kushugulova, A.; Marotta, F.; Yadav, H. Gut microbiome and aging: Physiological and mechanistic insights. Nutr. Healthy Aging 2018, 4, 267–285. [Google Scholar] [CrossRef]
- De Sire, R.; Rizzatti, G.; Ingravalle, F.; Pizzoferrato, M.; Petito, V.; Lopetuso, L.; Graziani, C.; De Sire, A.; Mentella, M.C.; Mele, M.C.; et al. Skeletal muscle-gut axis: Emerging mechanisms of sarcopenia for intestinal and extra intestinal diseases. Minerva Gastroenterol. Dietol. 2018, 64, 351–362. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Meehan, C.J.; Koenig, J.E.; Dhanani, A.S.; Rose, R.A.; Howlett, S.E.; Beiko, R.G. Microbial shifts in the aging mouse gut. Microbiome 2014, 2, 50. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Prakash, S. Longevity extension in Drosophila through gut-brain communication. Sci. Rep. 2018, 8, 8362. [Google Scholar] [CrossRef] [PubMed]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.L.; Zhou, Y.; Yi, L.; Gao, Y.X.; Ran, L.; Chen, S.H.; Zhang, T.; Zhou, X.; Zou, D.; et al. Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol. Nutr. Food Res. 2015, 59, 1443–1457. [Google Scholar] [CrossRef]
- Neff, F.; Flores-Dominguez, D.; Ryan, D.P.; Horsch, M.; Schröder, S.; Adler, T.; Afonso, L.C.; Aguilar-Pimentel, J.A.; Becker, L.; Garrett, L.; et al. Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Investig. 2013, 123, 3272–3291. [Google Scholar] [CrossRef]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Picca, A.; Fanelli, F.; Calvani, R.; Mulè, G.; Pesce, V.; Sisto, A.; Pantanelli, C.; Bernabei, R.; Landi, F.; Marzetti, E. Gut Dysbiosis and Muscle Aging: Searching for Novel Targets against Sarcopenia. Mediat. Inflamm. 2018, 2018, 7026198. [Google Scholar] [CrossRef]
- Lustgarten, M.S. The Role of the Gut Microbiome on Skeletal Muscle Mass and Physical Function: 2019 Update. Front. Physiol. 2019, 10, 1435. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, J.Y.; Choi, H.M.; Yang, H.-I.; Kim, K.S. Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link. Int. J. Mol. Sci. 2020, 21, 6887. https://doi.org/10.3390/ijms21186887
Ryu JY, Choi HM, Yang H-I, Kim KS. Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link. International Journal of Molecular Sciences. 2020; 21(18):6887. https://doi.org/10.3390/ijms21186887
Chicago/Turabian StyleRyu, Ji Yeon, Hyung Muk Choi, Hyung-In Yang, and Kyoung Soo Kim. 2020. "Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link" International Journal of Molecular Sciences 21, no. 18: 6887. https://doi.org/10.3390/ijms21186887
APA StyleRyu, J. Y., Choi, H. M., Yang, H.-I., & Kim, K. S. (2020). Dysregulated Autophagy Mediates Sarcopenic Obesity and Its Complications via AMPK and PGC1α Signaling Pathways: Potential Involvement of Gut Dysbiosis as a Pathological Link. International Journal of Molecular Sciences, 21(18), 6887. https://doi.org/10.3390/ijms21186887