Detection of Inflammation-Related Melanoma Small Extracellular Vesicle (sEV) mRNA Content Using Primary Melanocyte sEVs as a Reference


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
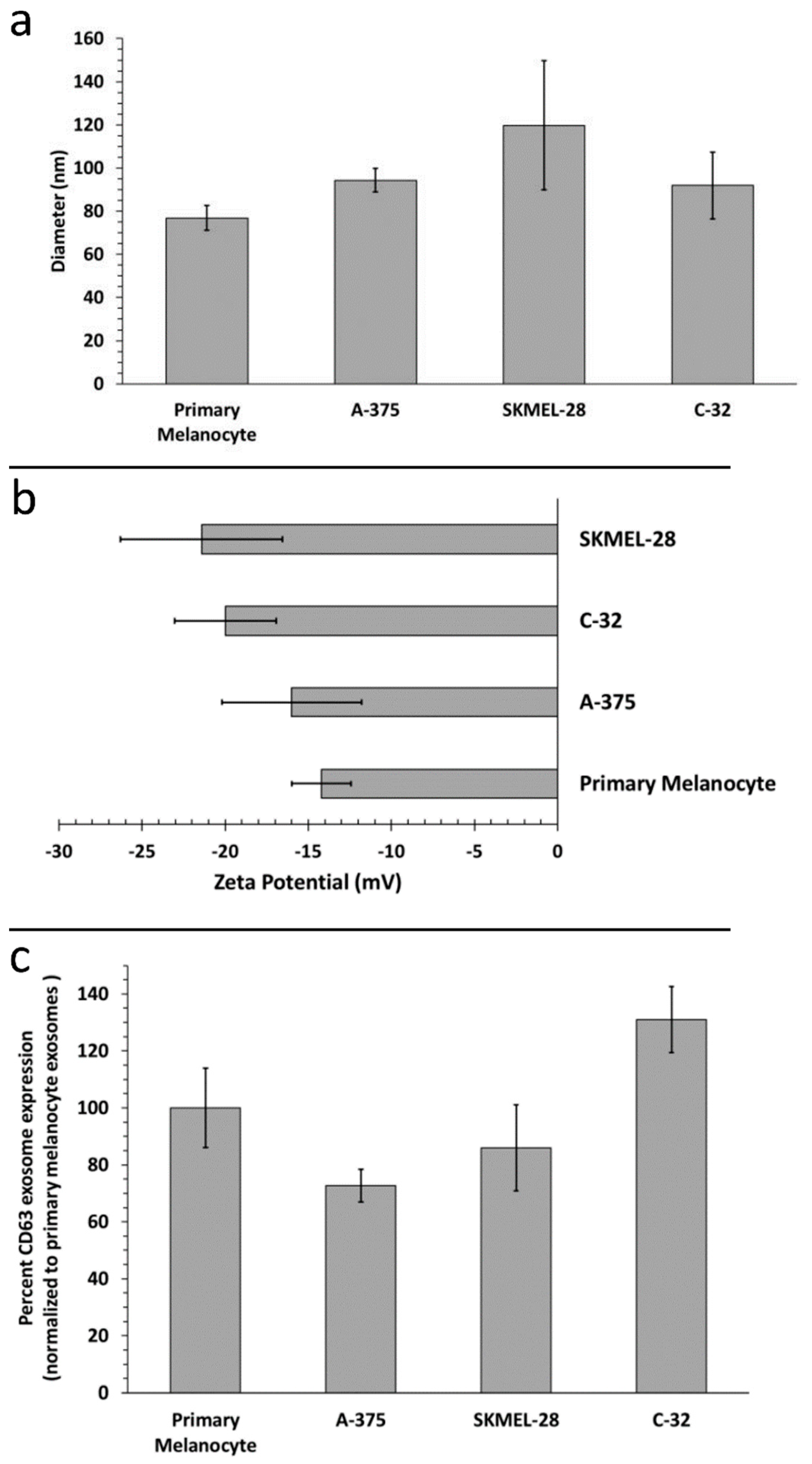
2.1. Biophysical Characterization of Primary Melanocyte sEVs and Melanoma sEVs
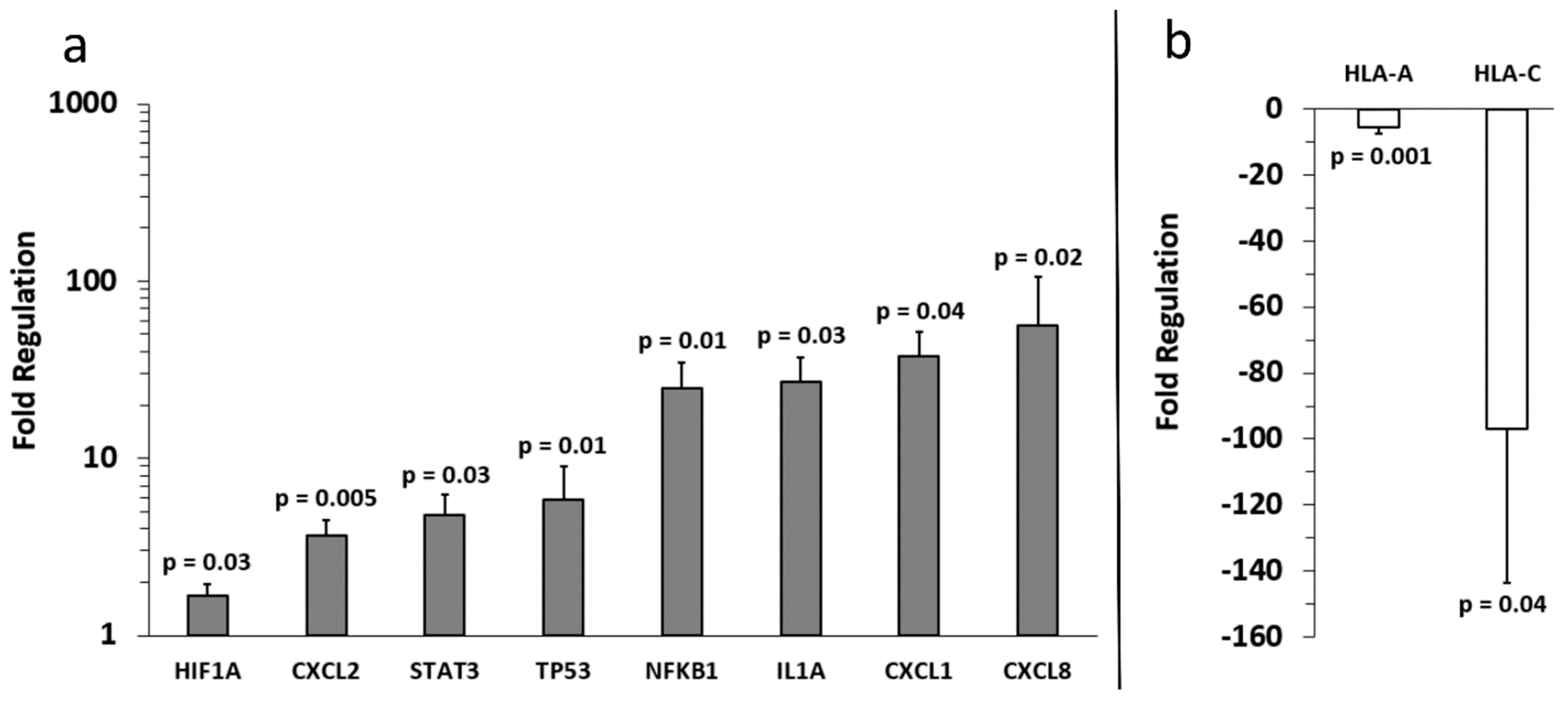
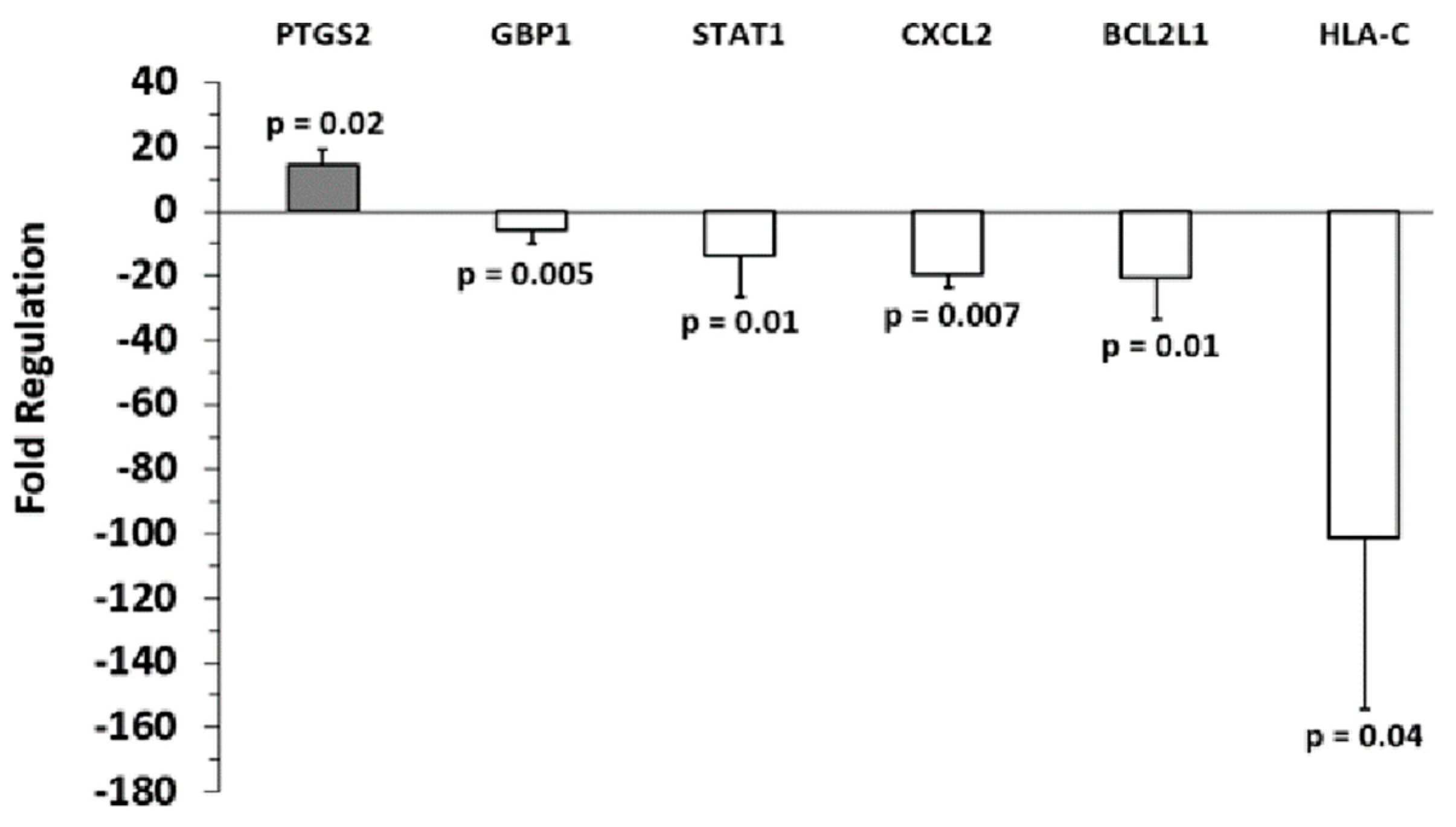
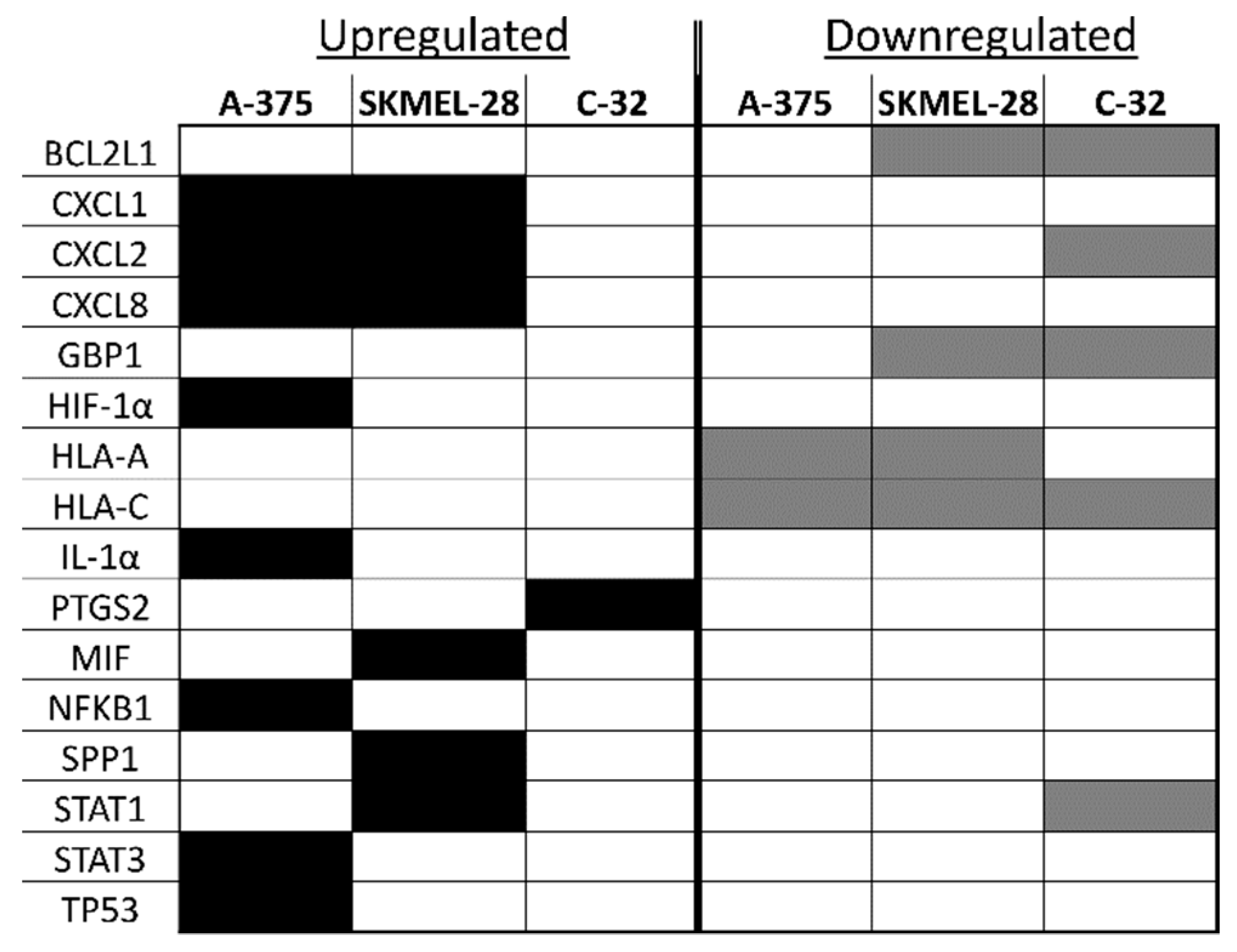
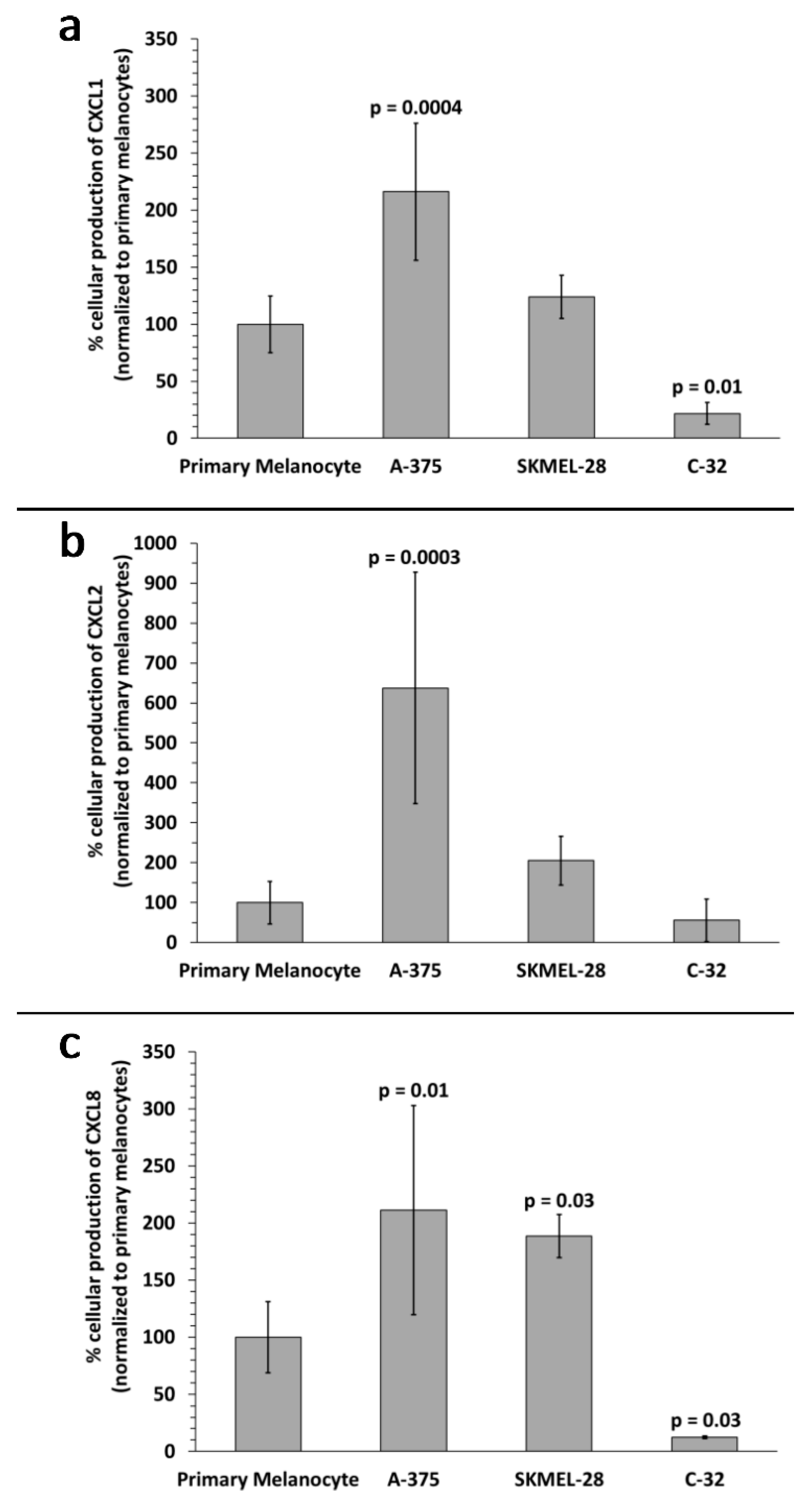
2.2. Identification of Inflammation-Related mRNA Content of Primary Melanocyte sEVs and Melanoma sEVs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Small EV Isolation by Standard Differential Centrifugation
4.3. Determination of sEV Size and Zeta Potential Properties
4.4. Qualitative Determination of Melanoma sEV CD63 Marker Expression
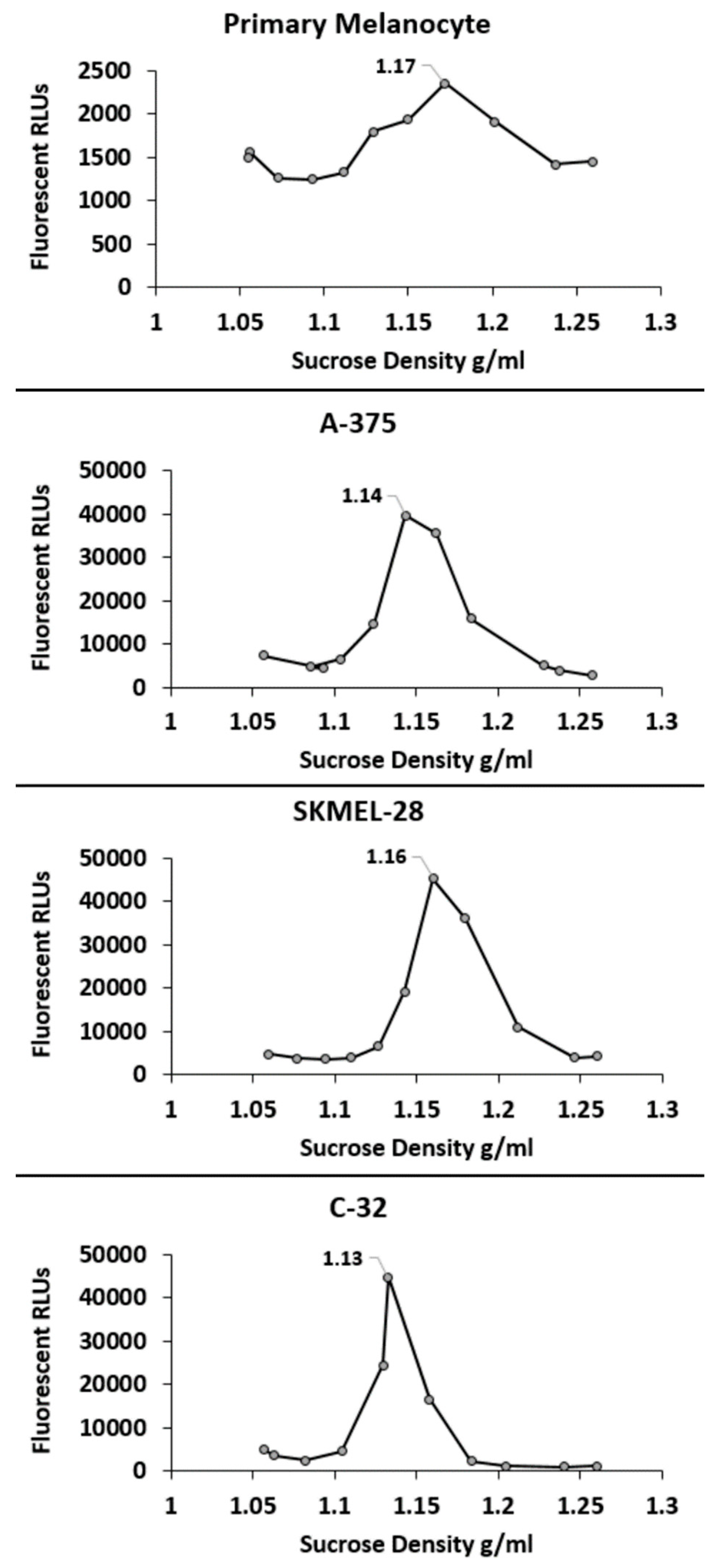
4.5. Determination of sEV Density
4.6. Detection of sEV Inflammation-Related mRNA Content by Quantitative Reverse Transcription PCR (RT-qPCR)
4.7. Enzyme-Linked Immunosorbent Assays (ELISAs)
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rinderknecht, M.; Detmar, M. Tumor lymphangiogenesis and melanoma metastasis. J. Cell. Physiol. 2008, 216, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L. Pre-analytical influences on the population heterogeneity of human extracellular vesicles sourced for nanomedicine uses. Nanomedicine 2018, 13, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (misev2018): A position statement of the international society for extracellular vesicles and update of the misev2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L. Post isolation modification of exosomes for nanomedicine applications. Nanomedicine 2016, 11, 1745–1756. [Google Scholar] [CrossRef]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through met. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Bardi, G.T.; Smith, M.A.; Hood, J.L. Melanoma exosomes promote mixed m1 and m2 macrophage polarization. Cytokine 2018, 105, 63–72. [Google Scholar] [CrossRef]
- Ekstrom, E.J.; Bergenfelz, C.; von Bulow, V.; Serifler, F.; Carlemalm, E.; Jonsson, G.; Andersson, T.; Leandersson, K. Wnt5a induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. Microrna signatures of tumor-derived exosomes as diagnositic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L.; Wickline, S.A. A systematic approach to exosome-based translational nanomedicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.L. The association of exosomes with lymph nodes. Semin. Cell Dev. Biol. 2017, 67, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Kobayashi, M.; Hanamura, N.; Akagi, T.; Kosaka, N.; Ochiya, T.; Ichiki, T. Electrokinetic evaluation of individual exosomes by on-chip microcapillary electrophoresis with laser dark-field microscopy. Jpn. J. Appl. Phys. 2013, 52, 06GK10. [Google Scholar] [CrossRef]
- Weiner, B.B. Measuring the Size & Surface Charge of Exosomes, Microvesicles and Liposomes. Available online: http://www.brookhaveninstruments.com/literature/pdf/Measuring%20the%20Size%20and%20Surface%20Charge%20of%20Exosomes.pdf (accessed on 18 November 2013).
- Hood, J.L.; Scott, M.J.; Wickline, S.A. Maximizing exosome colloidal stability following electroporation. Anal. Biochem. 2014, 448, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Kato, K.; Kobayashi, M.; Kosaka, N.; Ochiya, T.; Ichiki, T. On-chip immunoelectrophoresis of extracellular vesicles released from human breast cancer cells. PLoS ONE 2015, 10, e0123603. [Google Scholar] [CrossRef]
- Hood, J.L.; Pan, H.; Lanza, G.M.; Wickline, S.A. Paracrine induction of endothelium by tumor exosomes. Lab. Investig. 2009, 89, 1317–1328. [Google Scholar] [CrossRef]
- Petersen, K.E.; Manangon, E.; Hood, J.L.; Wickline, S.A.; Fernandez, D.P.; Johnson, W.P.; Gale, B.K. A review of exosome separation techniques and characterization of b16-f10 mouse melanoma exosomes with af4-uv-mals-dls-tem. Anal. Bioanal. Chem. 2014, 406, 7855–7866. [Google Scholar] [CrossRef]
- Petersen, K.E.; Shiri, F.; White, T.; Bardi, G.T.; Sant, H.; Gale, B.K.; Hood, J.L. Exosome isolation: Cyclical electrical field flow fractionation in low-ionic-strength fluids. Anal. Chem. 2018, 90, 12783–12790. [Google Scholar] [CrossRef]
- Dhawan, P.; Richmond, A. Role of cxcl1 in tumorigenesis of melanoma. J. Leukoc. Biol. 2002, 72, 9–18. [Google Scholar]
- Sarvaiya, P.J.; Guo, D.; Ulasov, I.; Gabikian, P.; Lesniak, M.S. Chemokines in tumor progression and metastasis. Oncotarget 2013, 4, 2171–2185. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.; Thomas, H.G. Melanoma growth stimulatory activity: Isolation from human melanoma tumors and characterization of tissue distribution. J. Cell. Biochem. 1988, 36, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Han, X.; Sun, Y.; Shang, C.; Wei, M.; Ba, X.; Zeng, X. Chemokine (c-x-c motif) ligand 1 and cxcl2 produced by tumor promote the generation of monocytic myeloid-derived suppressor cells. Cancer Sci. 2018, 109, 3826–3839. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.S.; Cornelius, L.A. The role of chemokines in melanoma tumor growth and metastasis. J. Investig. Dermatol. 2002, 118, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Palena, C.; Hamilton, D.H.; Fernando, R.I. Influence of il-8 on the epithelial–mesenchymal transition and the tumor microenvironment. Future Oncol. 2012, 8, 713–722. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The il-8/il-8r axis: A double agent in tumor immune resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Mikula-Pietrasik, J.; Sosinska, P.; Maksin, K.; Kucinska, M.G.; Piotrowska, H.; Murias, M.; Wozniak, A.; Szpurek, D.; Ksiazek, K. Colorectal cancer-promoting activity of the senescent peritoneal mesothelium. Oncotarget 2015, 6, 29178–29195. [Google Scholar] [CrossRef]
- Mikula-Pietrasik, J.; Sosinska, P.; Naumowicz, E.; Maksin, K.; Piotrowska, H.; Wozniak, A.; Szpurek, D.; Ksiazek, K. Senescent peritoneal mesothelium induces a pro-angiogenic phenotype in ovarian cancer cells in vitro and in a mouse xenograft model in vivo. Clin. Exp. Metastasis 2016, 33, 15–27. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mrnas and micrornas is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–672. [Google Scholar] [CrossRef]
- Botti, G.; Fratangelo, F.; Cerrone, M.; Liguori, G.; Cantile, M.; Anniciello, A.M.; Scala, S.; D’Alterio, C.; Trimarco, C.; Ianaro, A.; et al. Cox-2 expression positively correlates with pd-l1 expression in human melanoma cells. J. Transl. Med. 2017, 15, 46. [Google Scholar] [CrossRef]
- Zhuang, L.; Lee, C.S.; Scolyer, R.A.; McCarthy, S.W.; Zhang, X.D.; Thompson, J.F.; Hersey, P. Mcl-1, bcl-xl and stat3 expression are associated with progression of melanoma whereas bcl-2, ap-2 and mitf levels decrease during progression of melanoma. Mod. Pathol. 2007, 20, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, C.; Lesinski, G.B. The jak-stat signal transduction pathway in melanoma. In Breakthroughs in Melanoma Research; Tanaka, D.Y., Ed.; InTech: Houston, TX, USA, 2011. [Google Scholar]
- Maeurer, M.J.; Gollin, S.M.; Storkus, W.J.; Swaney, W.; Karbach, J.; Martin, D.; Castelli, C.; Salter, R.; Knuth, A.; Lotze, M.T. Tumor escape from immune recognition: Loss of hla-a2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin. Cancer Res. 1996, 2, 641–652. [Google Scholar] [PubMed]
- Wang, Z.; Seliger, B.; Mike, N.; Momburg, F.; Knuth, A.; Ferrone, S. Molecular analysis of the hla-a2 antigen loss by melanoma cells sk-mel-29.1.22 and sk-mel-29.1.29. Cancer Res. 1998, 58, 2149–2157. [Google Scholar] [PubMed]
- Muenst, S.; Laubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the dependency of cellular protein levels on mrna abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227. [Google Scholar] [CrossRef]
- Janas, T.; Janas, M.M.; Sapoń, K.; Janas, T. Mechanisms of rna loading into exosomes. FEBS Lett. 2015, 589, 1391–1398. [Google Scholar] [CrossRef]
- Orlikowsky, T.W.; Neunhoeffer, F.; Goelz, R.; Eichner, M.; Henkel, C.; Zwirner, M.; Poets, C.F. Evaluation of il-8-concentrations in plasma and lysed edta-blood in healthy neonates and those with suspected early onset bacterial infection. Pediatr. Res. 2004, 56, 804–809. [Google Scholar] [CrossRef]
- Tsui, N.B.; Ng, E.K.; Lo, Y.M. Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clin. Chem. 2002, 48, 1647–1653. [Google Scholar]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754. [Google Scholar] [CrossRef]
- Ramachandran, S.; Palanisamy, V. Horizontal transfer of rnas: Exosomes as mediators of intercellular communication. Wiley Interdiscip. Rev. RNA 2012, 3, 286–293. [Google Scholar] [CrossRef]
- Thery, C.; Clayton, A.; Amigorena, S.; Raposo, G. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell. Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Mantel, P.Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.; Bonebreak, C.M.; Saracoglu, E.; et al. Current methods for the isolation of extracellular vesicles. Biol. Chem. 2013, 394, 1253–1262. [Google Scholar] [CrossRef]
- Boriachek, K.; Islam, M.N.; Moller, A.; Salomon, C.; Nguyen, N.T.; Hossain, M.S.A.; Yamauchi, Y.; Shiddiky, M.J.A. Biological functions and current advances in isolation and detection strategies for exosome nanovesicles. Small 2018, 14, 1702153. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar]
- Hu, L.; Wickline, S.A.; Hood, J.L. Magnetic resonance imaging of melanoma exosomes in lymph nodes. Magn. Reson. Med. 2015, 74, 266–271. [Google Scholar] [CrossRef]
- The GeneGlobe Data Analysis Center. Available online: http://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/rt2-profiler-pcr-arrays-data-analysis-center (accessed on 17 December 2017).







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bardi, G.T.; Al-Rayan, N.; Richie, J.L.; Yaddanapudi, K.; Hood, J.L. Detection of Inflammation-Related Melanoma Small Extracellular Vesicle (sEV) mRNA Content Using Primary Melanocyte sEVs as a Reference. Int. J. Mol. Sci. 2019, 20, 1235. https://doi.org/10.3390/ijms20051235
Bardi GT, Al-Rayan N, Richie JL, Yaddanapudi K, Hood JL. Detection of Inflammation-Related Melanoma Small Extracellular Vesicle (sEV) mRNA Content Using Primary Melanocyte sEVs as a Reference. International Journal of Molecular Sciences. 2019; 20(5):1235. https://doi.org/10.3390/ijms20051235
Chicago/Turabian StyleBardi, Gina T., Numan Al-Rayan, Jamaal L. Richie, Kavitha Yaddanapudi, and Joshua L. Hood. 2019. "Detection of Inflammation-Related Melanoma Small Extracellular Vesicle (sEV) mRNA Content Using Primary Melanocyte sEVs as a Reference" International Journal of Molecular Sciences 20, no. 5: 1235. https://doi.org/10.3390/ijms20051235
APA StyleBardi, G. T., Al-Rayan, N., Richie, J. L., Yaddanapudi, K., & Hood, J. L. (2019). Detection of Inflammation-Related Melanoma Small Extracellular Vesicle (sEV) mRNA Content Using Primary Melanocyte sEVs as a Reference. International Journal of Molecular Sciences, 20(5), 1235. https://doi.org/10.3390/ijms20051235