Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT)

Abstract
:1. Telomeres and Telomere Maintenance Mechanisms
2. Telomerase and ALT in Cancer
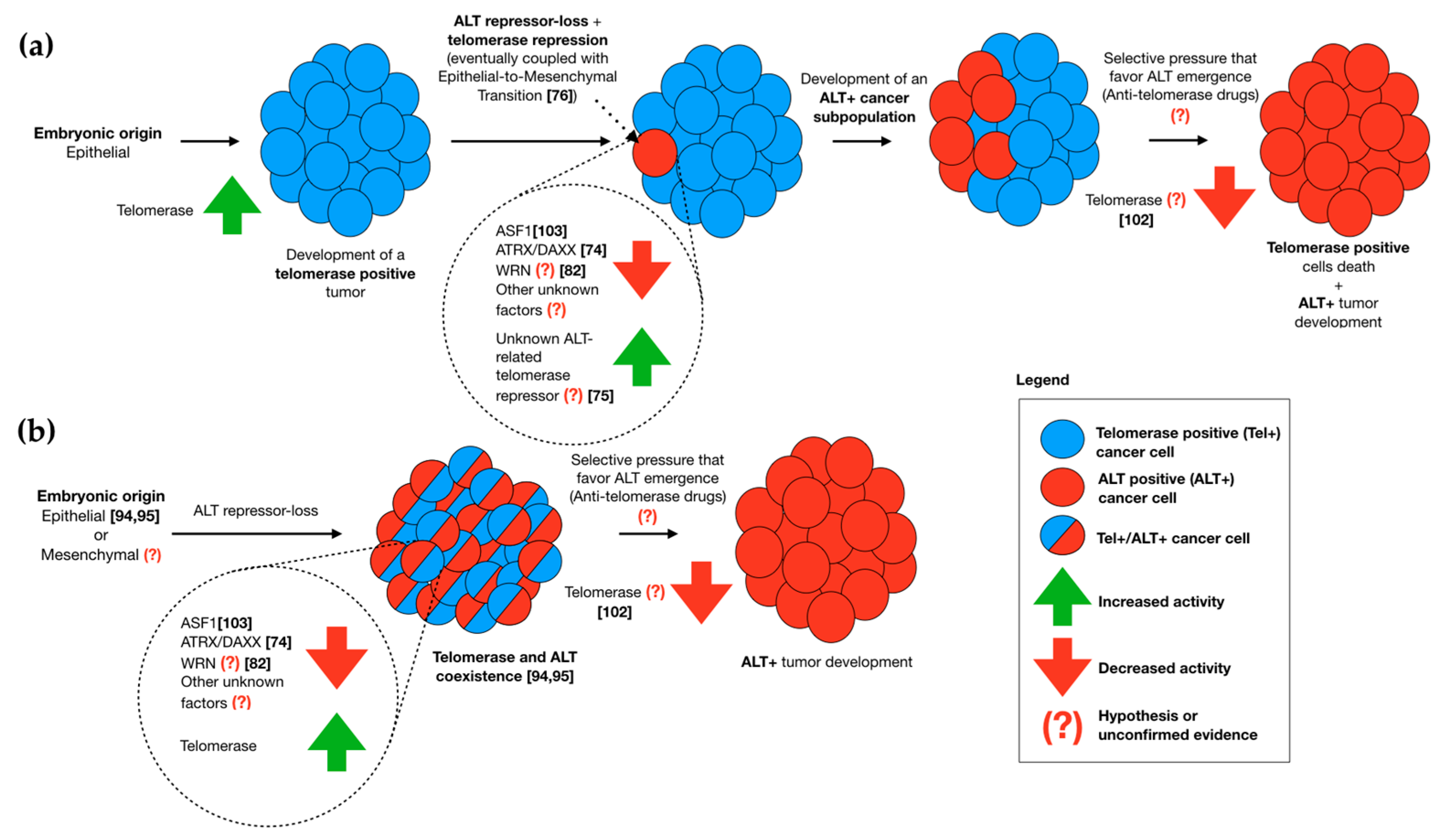
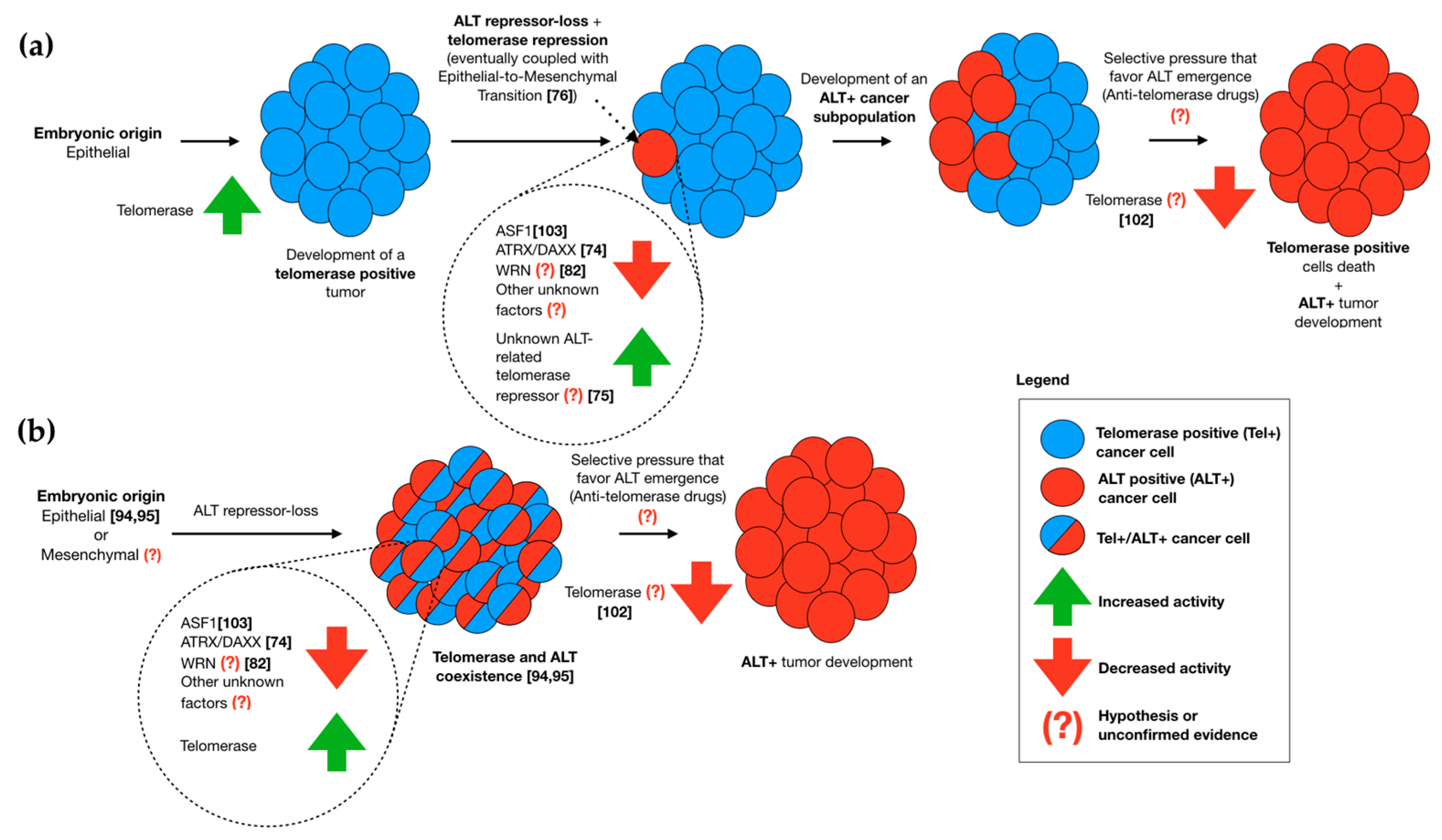
2.1. The Role of Embryonic Origin in ALT Activation
2.2. Telomerase and ALT Coexistence In Vitro
2.3. Telomerase and ALT Coexistence in Cancer
2.4. Telomerase/ALT Switching
3. Neither Telomerase nor ALT When Tumors Do Not Maintain Their Telomeres
4. Conclusions
Conflicts of Interest
Abbreviations
| ALT | Alternative Lengthening of Telomeres |
| APB | ALT-Associated PML Body |
| ATRX | α-Thalassemia/Mental Retardation Syndrome X-Linked Protein |
| CNS | Central Nervous System |
| DAXX | Death Domain-Associated Protein |
| DDR | DNA Damage Response |
| ECTR | Extra Chromosomal Telomere Repeat |
| EMT | Epithelial-to-Mesenchymal Transition |
| HR | Homologous Recombination |
| MSI-H | Microsatellite Instability-High |
| PanNET | Pancreatic Neuroendocrine Tumor |
| PML | Promyelocytic Leukemia Protein |
| PNS | Peripheral Nervous System |
| SCC | Squamous Cell Carcinoma |
| T-SCE | Telomere-Sister Chromatid Exchange |
| TMM | Telomere Maintenance Mechanism |
| WRN | Werner Protein |
References
- Moyzis, R.K.; Buckingham, J.M.; Cram, L.S.; Dani, M.; Deaven, L.L.; Jones, M.D.; Meyne, J.; Ratliff, R.L.; Wu, J.R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 1988, 85, 6622–6626. [Google Scholar] [CrossRef] [PubMed]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- d'Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Kim, N.W.; Effros, R.B.; Chiu, C.P. Mechanism of telomerase induction during T cell activation. Exp. Cell Res. 1996, 228, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Kurz, D.J.; Hong, Y.; Trivier, E.; Huang, H.L.; Decary, S.; Zang, G.H.; Luscher, T.F.; Erusalimsky, J.D. Fibroblast growth factor-2, but not vascular endothelial growth factor, upregulates telomerase activity in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Hapangama, D.K.; Kamal, A.; Saretzki, G. Implications of telomeres and telomerase in endometrial pathology. Hum. Reprod. Update 2017, 23, 166–187. [Google Scholar] [CrossRef] [PubMed]
- Opresko, P.L.; Shay, J.W. Telomere-associated aging disorders. Ageing Res. Rev. 2017, 33, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Akincilar, S.C.; Unal, B.; Tergaonkar, V. Reactivation of telomerase in cancer. Cell. Mol. Life Sci. 2016, 73, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Stewart, S.A.; Hahn, W.C.; O’Connor, B.F.; Banner, E.N.; Lundberg, A.S.; Modha, P.; Mizuno, H.; Brooks, M.W.; Fleming, M.; Zimonjic, D.B.; et al. Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 12606–12611. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Draskovic, I.; Londono Vallejo, A. Telomere recombination and alternative telomere lengthening mechanisms. Front. Biosci. 2013, 18, 1–20. [Google Scholar]
- Londono-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wu, S.; Liu, H.; Stratt, R.; Barak, O.G.; Shiekhattar, R.; Picketts, D.J.; Yang, X. A novel transcription regulatory complex containing death domain-associated protein and the ATR-X syndrome protein. J. Biol. Chem. 2004, 279, 20369–20377. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Greenberg, R.A. Alternative telomere maintenance and cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sobinoff, A.P.; Pickett, H.A. Alternative lengthening of telomeres: DNA repair pathways converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.P.; Sabatier, L.; Marder, B.A.; Morgan, W.F. Telomere dynamics in an immortal human cell line. EMBO J. 1994, 13, 4953–4962. [Google Scholar] [PubMed]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef] [PubMed]
- Nabetani, A.; Ishikawa, F. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol. Cell. Biol. 2009, 29, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar] [PubMed]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Brummendorf, T.H.; Balabanov, S. Telomere length dynamics in normal hematopoiesis and in disease states characterized by increased stem cell turnover. Leukemia 2006, 20, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Sarin, K.Y.; Cheung, P.; Gilison, D.; Lee, E.; Tennen, R.I.; Wang, E.; Artandi, M.K.; Oro, A.E.; Artandi, S.E. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 2005, 436, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, M.; Ohshima, K.; Nakamura, H.; Suzumiya, J.; Nakayama, Y.; Kanda, M.; Haraoka, S.; Kikuchi, M. Decreased expression of telomerase-associated RNAs in the proliferation of stem cells in comparison with continuous expression in malignant tumors. Int. J. Oncol. 1999, 15, 1075–1080. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Tatsumoto, N.; Kodama, T.; Hiyama, K.; Shay, J.; Yokoyama, T. Telomerase activity in human intestine. Int. J. Oncol. 1996, 9, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Dan, Y.Y.; Riehle, K.J.; Lazaro, C.; Teoh, N.; Haque, J.; Campbell, J.S.; Fausto, N. Isolation of multipotent progenitor cells from human fetal liver capable of differentiating into liver and mesenchymal lineages. Proc. Natl. Acad. Sci. USA 2006, 103, 9912–9917. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Reddel, R.R. Assaying and investigating alternative lengthening of telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Giordano, T.J.; Hammer, G.D. Evaluation of telomere length maintenance mechanisms in adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 2008, 93, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The role of ATRX in the alternative lengthening of telomeres (ALT) phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2015, 6, 7493–7503. [Google Scholar] [CrossRef] [PubMed]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.D.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive proliferation of human cancer cells with ever-shorter telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [PubMed]
- Sanders, R.P.; Drissi, R.; Billups, C.A.; Daw, N.C.; Valentine, M.B.; Dome, J.S. Telomerase expression predicts unfavorable outcome in osteosarcoma. J. Clin. Oncol. 2004, 22, 3790–3797. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Hoffman, A.R.; Otero, J.; Huang, H.Y.; Zhao, Z.; Mazumdar, M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Ladanyi, M. Divergent patterns of telomere maintenance mechanisms among human sarcomas: Sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in ewing’s sarcomas and osteosarcomas. Genes Chromosomes Cancer 2004, 41, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Huang, H.Y.; Otero, J.; Zhao, Z.; Ben-Porat, L.; Satagopan, J.M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Huvos, A.G.; et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res. 2003, 63, 1759–1763. [Google Scholar] [PubMed]
- Subhawong, A.P.; Heaphy, C.M.; Argani, P.; Konishi, Y.; Kouprina, N.; Nassar, H.; Vang, R.; Meeker, A.K. The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod. Pathol. 2009, 22, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Abedalthagafi, M.; Phillips, J.J.; Kim, G.E.; Mueller, S.; Haas-Kogen, D.A.; Marshall, R.E.; Croul, S.E.; Santi, M.R.; Cheng, J.; Zhou, S.; et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: A multi-institutional study of 214 astrocytomas. Mod. Pathol. 2013, 26, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Heaphy, C.M.; de Wilde, R.F.; Orr, B.A.; Odia, Y.; Eberhart, C.G.; Meeker, A.K.; Rodriguez, F.J. Molecular and morphologic correlates of the alternative lengthening of telomeres phenotype in high-grade astrocytomas. Brain Pathol. 2013, 23, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hakin-Smith, V.; Jellinek, D.A.; Levy, D.; Carroll, T.; Teo, M.; Timperley, W.R.; McKay, M.J.; Reddel, R.R.; Royds, J.A. Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 2003, 361, 836–838. [Google Scholar] [CrossRef]
- McDonald, K.L.; McDonnell, J.; Muntoni, A.; Henson, J.D.; Hegi, M.E.; von Deimling, A.; Wheeler, H.R.; Cook, R.J.; Biggs, M.T.; Little, N.S.; et al. Presence of alternative lengthening of telomeres mechanism in patients with glioblastoma identifies a less aggressive tumor type with longer survival. J. Neuropathol. Exp. Neurol. 2010, 69, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Boardman, L.A.; Johnson, R.A.; Viker, K.B.; Hafner, K.A.; Jenkins, R.B.; Riegert-Johnson, D.L.; Smyrk, T.C.; Litzelman, K.; Seo, S.; Gangnon, R.E.; et al. Correlation of chromosomal instability, telomere length and telomere maintenance in microsatellite stable rectal cancer: A molecular subclass of rectal cancer. PLoS ONE 2013, 8, e80015. [Google Scholar] [CrossRef] [PubMed]
- Marinoni, I.; Kurrer, A.S.; Vassella, E.; Dettmer, M.; Rudolph, T.; Banz, V.; Hunger, F.; Pasquinelli, S.; Speel, E.J.; Perren, A. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 2014, 146, 453–460.e5. [Google Scholar] [CrossRef] [PubMed]
- Viceconte, N.; Dheur, M.S.; Majerova, E.; Pierreux, C.E.; Baurain, J.F.; van Baren, N.; Decottignies, A. Highly aggressive metastatic melanoma cells unable to maintain telomere length. Cell Rep. 2017, 19, 2529–2543. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Lee, J.C.; Tsai, J.H.; Yang, C.Y.; Liu, T.L.; Ke, Z.L.; Hsu, H.H.; Jeng, Y.M. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Mod. Pathol. 2015, 28, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Shay, J.W.; Wright, W.E.; Hiyama, E.; Shimose, S.; Kubo, T.; Sugita, T.; Yasunaga, Y.; Ochi, M. Telomere-maintenance mechanisms in soft-tissue malignant fibrous histiocytomas. J. Bone Joint Surg. Am. Vol. 2009, 91, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Tsai, J.H.; Jeng, Y.M.; Lee, J.C.; Hsu, H.H.; Yang, C.Y. Leiomyosarcoma with alternative lengthening of telomeres is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am. J. Surg. Pathol. 2015, 39, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Daidone, M.G.; Daprai, L.; Villa, R.; Cantu, S.; Pilotti, S.; Mariani, L.; Gronchi, A.; Henson, J.D.; Reddel, R.R.; et al. Telomere maintenance mechanisms in liposarcomas: Association with histologic subtypes and disease progression. Cancer Res. 2006, 66, 8918–8924. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Varkonyi, R.J.; Schwalm, J.; Cragle, R.; Klein-Szanto, A.; Patchefsky, A.; Cukierman, E.; von Mehren, M.; Broccoli, D. Multiple mechanisms of telomere maintenance exist in liposarcomas. Clin. Cancer Res. 2005, 11, 5347–5355. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Jeng, Y.M.; Liau, J.Y.; Tsai, J.H.; Hsu, H.H.; Yang, C.Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Venturini, L.; Motta, R.; Gronchi, A.; Daidone, M.; Zaffaroni, N. Prognostic relevance of alt-associated markers in liposarcoma: A comparative analysis. BMC Cancer 2010, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Tsai, J.H.; Yang, C.Y.; Lee, J.C.; Liang, C.W.; Hsu, H.H.; Jeng, Y.M. Alternative lengthening of telomeres phenotype in malignant vascular tumors is highly associated with loss of ATRX expression and is frequently observed in hepatic angiosarcomas. Hum. Pathol. 2015, 46, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Venturini, L.; Daidone, M.G.; Motta, R.; Cimino-Reale, G.; Hoare, S.F.; Gronchi, A.; Folini, M.; Keith, W.N.; Zaffaroni, N. Telomere maintenance mechanisms in malignant peripheral nerve sheath tumors: Expression and prognostic relevance. Neuro-Oncology 2012, 14, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Nakayama, F.; Li, D.; Kanemitsu, K.; Semba, S.; Ito, A.; Yokozaki, H. Alternative lengthening of telomeres frequently occurs in mismatch repair system-deficient gastric carcinoma. Cancer Sci. 2009, 100, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, D.; Sofiadis, A.; Hoog, A.; Vukojevic, V.; Backdahl, M.; Zedenius, J.; Larsson, C. Telomerase-dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E1571–E1579. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Voss, M.; Kaiser, S.; Kapp, U.; Waller, C.F.; Martens, U.M. Lack of telomerase activity in human mesenchymal stem cells. Leukemia 2003, 17, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.W.; Williams, E.D. Emt and met in carcinoma—Clinical observations, regulatory pathways and new models. Clin. Exp. Metastasis 2008, 25, 591–592. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; El-Naggar, S.; Darling, D.S.; Higashi, Y.; Dean, D.C. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 2008, 135, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic instability is induced by persistent proliferation of cells undergoing epithelial-to-mesenchymal transition. Cell Rep. 2016, 17, 2632–2647. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Li, L.; Zhang, D.; Wu, K.; Chen, Y.; Zeng, J.; Wang, X.; He, D. Twisted epithelial-to-mesenchymal transition promotes progression of surviving bladder cancer t24 cells with htert-dysfunction. PLoS ONE 2011, 6, e27748. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londono-Vallejo, A.; Decottignies, A. Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Gonzalo, S.; Jaco, I.; Schotta, G.; Klatt, P.; Jenuwein, T.; Blasco, M.A. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J. Cell Biol. 2007, 178, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cao, M.; O’Sullivan, R.; Peters, A.H.; Jenuwein, T.; Blasco, M.A. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat. Genet. 2004, 36, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, S.; Jaco, I.; Fraga, M.F.; Chen, T.; Li, E.; Esteller, M.; Blasco, M.A. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat. Cell Biol. 2006, 8, 416–424. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Almouzni, G. Assembly of telomeric chromatin to create alternative endings. Trends Cell Biol. 2014, 24, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Voon, H.P.J.; Collas, P.; Wong, L.H. Compromised telomeric heterochromatin promotes alternative lengthening of telomeres. Trends Cancer 2016, 2, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Perrem, K.; Bryan, T.M.; Englezou, A.; Hackl, T.; Moy, E.L.; Reddel, R.R. Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids. Oncogene 1999, 18, 3383–3390. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Zhang, J.; Lu, C.; Parker, M.; Bahrami, A.; Tickoo, S.K.; Heguy, A.; Pappo, A.S.; Federico, S.; Dalton, J.; et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012, 307, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef] [PubMed]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M.; Kameyama, M.; Kugoh, H.; Shimizu, M.; Oshimura, M. A repressor function for telomerase activity in telomerase-negative immortal cells. Mol. Carcinog. 1998, 21, 17–25. [Google Scholar] [CrossRef]
- Cerone, M.A.; Londono-Vallejo, J.A.; Bacchetti, S. Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum. Mol. Genet. 2001, 10, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Perrem, K.; Colgin, L.M.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Coexistence of alternative lengthening of telomeres and telomerase in htert-transfected gm847 cells. Mol. Cell. Biol. 2001, 21, 3862–3875. [Google Scholar] [CrossRef] [PubMed]
- Grobelny, J.V.; Kulp-McEliece, M.; Broccoli, D. Effects of reconstitution of telomerase activity on telomere maintenance by the alternative lengthening of telomeres (ALT) pathway. Hum. Mol. Genet. 2001, 10, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.C.; Zakian, V.A. Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 8083–8093. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.; Zhao, Y.; Balajee, A.S. Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells. Cancer Lett. 2018, 413, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laud, P.R.; Multani, A.S.; Bailey, S.M.; Wu, L.; Ma, J.; Kingsley, C.; Lebel, M.; Pathak, S.; DePinho, R.A.; Chang, S. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005, 19, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Siddiqa, A.; Cavazos, D.; Chavez, J.; Long, L.; Marciniak, R.A. Modulation of telomeres in alternative lengthening of telomeres type I like human cells by the expression of werner protein and telomerase. J. Oncol. 2012, 2012, 806382. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An alternative pathway for yeast telomere maintenance rescues EST1- senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Basenko, E.; Topcu, Z.; McEachern, M.J. Recombination can either help maintain very short telomeres or generate longer telomeres in yeast cells with weak telomerase activity. Eukaryotic Cell 2011, 10, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Morrish, T.A.; Greider, C.W. Short telomeres initiate telomere recombination in primary and tumor cells. PLoS Genet. 2009, 5, e1000357. [Google Scholar] [CrossRef] [PubMed]
- Brault, M.E.; Autexier, C. Telomeric recombination induced by dysfunctional telomeres. Mol. Biol. Cell 2011, 22, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Gupta, J.; Han, L.P.; Wang, P.; Gallie, B.L.; Bacchetti, S. Development of retinoblastoma in the absence of telomerase activity. J. Natl. Cancer Inst. 1996, 88, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Benhattar, J.; Coindre, J.M.; Guillou, L. Telomerase activity and hTERT mRNA expression can be heterogeneous and does not correlate with telomere length in soft tissue sarcomas. Int. J. Cancer 2002, 98, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, E.; Argani, P.; Hicks, J.L.; DeMarzo, A.M.; Meeker, A.K. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am. J. Pathol. 2004, 164, 1523–1529. [Google Scholar] [CrossRef]
- Villa, R.; Daidone, M.G.; Motta, R.; Venturini, L.; De Marco, C.; Vannelli, A.; Kusamura, S.; Baratti, D.; Deraco, M.; Costa, A.; et al. Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin. Cancer Res. 2008, 14, 4134–4140. [Google Scholar] [CrossRef] [PubMed]
- Venturini, L.; Daidone, M.G.; Motta, R.; Collini, P.; Spreafico, F.; Terenziani, M.; Piva, L.; Radice, P.; Perotti, D.; Zaffaroni, N. Telomere maintenance in wilms tumors: First evidence for the presence of alternative lengthening of telomeres mechanism. Genes Chromosomes Cancer 2011, 50, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: Implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Peng, M.; Song, Q. The co-expression of telomerase and alt pathway in human breast cancer tissues. Tumour Biol. 2014, 35, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Bojovic, B.; Booth, R.E.; Jin, Y.; Zhou, X.; Crowe, D.L. Alternative lengthening of telomeres in cancer stem cells in vivo. Oncogene 2015, 34, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Zhou, S.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef] [PubMed]
- Bechter, O.E.; Zou, Y.; Walker, W.; Wright, W.E.; Shay, J.W. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004, 64, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, S.M.; Yu, Y.; Xiao, B.K.; Huang, Z.W.; Tao, Z.Z. Telomerase inhibition alters telomere maintenance mechanisms in laryngeal squamous carcinoma cells. J. Laryngol. Otol. 2010, 124, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Reddel, R.R.; Wright, W.E. Cancer and telomeres—An alternative to telomerase. Science 2012, 336, 1388–1390. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Bower, K.; Napier, C.E.; Cole, S.L.; Dagg, R.A.; Lau, L.M.; Duncan, E.L.; Moy, E.L.; Reddel, R.R. Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of alternative lengthening of telomeres. PLoS ONE 2012, 7, e50062. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant h3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. DAXX is an h3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Shiels, C.; Freemont, P.S. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001, 20, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Kim, D.Y.; Lee, J.M.; Kim, S.T.; Han, T.H.; Ahn, J.H. Requirement of the coiled-coil domain of PML-RARalpha oncoprotein for localization, sumoylation, and inhibition of monocyte differentiation. Biochem. Biophys. Res. Commun. 2005, 330, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Yong, J.W.; Yeo, X.; Khan, M.M.; Lee, M.B.; Hande, M.P. Stable expression of promyelocytic leukaemia (PML) protein in telomerase positive MCF7 cells results in alternative lengthening of telomeres phenotype. Genome Integr. 2012, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Fasching, C.L.; Bower, K.; Reddel, R.R. Telomerase-independent telomere length maintenance in the absence of alternative lengthening of telomeres-associated promyelocytic leukemia bodies. Cancer Res. 2005, 65, 2722–2729. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, J.N.; Mendez-Bermudez, A.; Zaffaroni, N.; Dubrova, Y.E.; Royle, N.J. Evidence for alternative lengthening of telomeres in liposarcomas in the absence of ALT-associated PML bodies. Int. J. Cancer 2008, 122, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- Gagos, S.; Papaioannou, G.; Chiourea, M.; Merk-Loretti, S.; Jefford, C.E.; Mikou, P.; Irminger-Finger, I.; Liossi, A.; Blouin, J.L.; Dahoun, S. Unusually stable abnormal karyotype in a highly aggressive melanoma negative for telomerase activity. Mol. Cytogenet. 2008, 1, 20. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Hewitt, G.; Passos, J.F. Telomeres, oxidative stress and inflammatory factors: Partners in cellular senescence? Longev. Healthspan 2014, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Meierjohann, S. Oxidative stress in melanocyte senescence and melanoma transformation. Eur. J. Cell Biol. 2014, 93, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Ulaner, G.A.; Giudice, L.C. Developmental regulation of telomerase activity in human fetal tissues during gestation. Mol. Hum. Reprod. 1997, 3, 769–773. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Tissue Origin 1 | %ALT+ | %ALT−/Tel− | References |
|---|---|---|---|
| Adrenal gland/PNS | |||
| Adrenocortical carcinoma | 12 | - | [34,35] |
| Ganglioneuroblastoma | 14 | - | [35,36] |
| Neuroblastoma | 34 | 6 | [35,36,37,38] |
| Pheochromocytoma | 3 | 88 2 | [25,35,36] |
| Bone | |||
| Osteosarcoma | 64 | 18 | [35,39,40,41,42] |
| Synovial Sarcoma | 9 | - | [35,39] |
| Breast | 2 | - | [35,36,43] |
| CNS | |||
| Astrocytoma | 42 | - | [35,36,39,44,45] |
| Glioblastoma | 28 | 46 | [35,36,45,46,47,48] |
| Other | 13 | - | [35,36] |
| Colorectal | 6 | - | [19,36,49] |
| Hematopoietic | 0 | - | [19,36] |
| Kidney | 5 | 75 2 | [25,35,36] |
| Liver | 7 | - | [19,36] |
| Lung | 1 | - | [35,36] |
| Neuroendocrine | |||
| Carcinoid tumor | 6 | - | [35,36] |
| PanNET | 53 | - | [19,50] |
| Paraganglioma | 13 | - | [35,36] |
| Ovary | 1 | - | [35,36] |
| Pancreas | 0 | - | [35,36] |
| Prostate | 0 | - | [35,36] |
| Skin | |||
| Basal cell carcinoma | 0 | - | [35,36] |
| Melanoma | 7 | 11 | [35,36,51] |
| Skin basal and squamous cell carcinoma | 0 | - | [35,36] |
| Soft tissue | |||
| Malignant fibrous histiocytoma | 62 | - | [35,39,52,53] |
| Leiomyosarcoma | 58 | - | [35,36,39,54] |
| Liposarcoma | 25 | 50 | [35,36,39,55,56,57,58] |
| Other | 22 | - | [35,36,39,52,59,60] |
| Stomach | |||
| Gastric carcinoma | 19 | - | [35,36,61] |
| MSI-H Gastric carcinoma | 57 | - | [35,61] |
| Non-MSI-H Gastric carcinoma | 19 | - | [35,61] |
| Testis | 8 | - | [35,36] |
| Thyroid | |||
| Follicular-cell derived | 0 | 79 2 | [25,35,36] |
| Medullary thyroid carcinoma | 28 | - | [35,62] |
| Urinary bladder | 4 | - | [35,36] |
| Uterus | 2 | - | [35,36] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. https://doi.org/10.3390/ijms19020606
De Vitis M, Berardinelli F, Sgura A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). International Journal of Molecular Sciences. 2018; 19(2):606. https://doi.org/10.3390/ijms19020606
Chicago/Turabian StyleDe Vitis, Marco, Francesco Berardinelli, and Antonella Sgura. 2018. "Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT)" International Journal of Molecular Sciences 19, no. 2: 606. https://doi.org/10.3390/ijms19020606