
Maintaining Effective Beta Cell Function in the Face of Metabolic Syndrome-Associated Glucolipotoxicity—Nutraceutical Options


1
Catalytic Longevity Foundation, San Diego, CA 92109, USA
2
Department of Preventive Cardiology, Saint Luke’s Mid America Heart Institute, Kansas City, MO 64111, USA
*
Authors to whom correspondence should be addressed.
Healthcare 2022, 10(1), 3; https://doi.org/10.3390/healthcare10010003
Submission received: 6 December 2021
/
Revised: 16 December 2021
/
Accepted: 17 December 2021
/
Published: 21 December 2021
{kind=link}
Abstract
:In people with metabolic syndrome, episodic exposure of pancreatic beta cells to elevated levels of both glucose and free fatty acids (FFAs)—or glucolipotoxicity—can induce a loss of glucose-stimulated insulin secretion (GSIS). This in turn can lead to a chronic state of glucolipotoxicity and a sustained loss of GSIS, ushering in type 2 diabetes. Loss of GSIS reflects a decline in beta cell glucokinase (GK) expression associated with decreased nuclear levels of the pancreatic and duodenal homeobox 1 (PDX1) factor that drives its transcription, along with that of Glut2 and insulin. Glucolipotoxicity-induced production of reactive oxygen species (ROS), stemming from both mitochondria and the NOX2 isoform of NADPH oxidase, drives an increase in c-Jun N-terminal kinase (JNK) activity that promotes nuclear export of PDX1, and impairs autocrine insulin signaling; the latter effect decreases PDX1 expression at the transcriptional level and up-regulates beta cell apoptosis. Conversely, the incretin hormone glucagon-like peptide-1 (GLP-1) promotes nuclear import of PDX1 via cAMP signaling. Nutraceuticals that quell an increase in beta cell ROS production, that amplify or mimic autocrine insulin signaling, or that boost GLP-1 production, should help to maintain GSIS and suppress beta cell apoptosis in the face of glucolipotoxicity, postponing or preventing onset of type 2 diabetes. Nutraceuticals with potential in this regard include the following: phycocyanobilin—an inhibitor of NOX2; agents promoting mitophagy and mitochondrial biogenesis, such as ferulic acid, lipoic acid, melatonin, berberine, and astaxanthin; myo-inositol and high-dose biotin, which promote phosphatidylinositol 3-kinase (PI3K)/Akt activation; and prebiotics/probiotics capable of boosting GLP-1 secretion. Complex supplements or functional foods providing a selection of these agents might be useful for diabetes prevention.
Keywords:
type 2 diabetes; glucolipotoxicity; beta cells; GSIS; PDX1; JNK; GLP-1; nutraceuticals; pre-biotics; pro-biotics1. Failure of Beta Cell Glucose-Stimulated Insulin Secretion Initiates Onset of Type 2 Diabetes
As metabolic syndrome progresses and the insulin insensitivity of peripheral tissues worsens, glycemic control is maintained for some time owing to a compensatory increase in beta cell insulin secretion. However, the episodic exposure of beta cells to elevated levels of both glucose and free fatty acids (FFAs)—a phenomenon known as “glucolipotoxicity”—may eventually induce a loss of beta cell capacity to respond to an acute increase of plasma glucose with an appropriate compensatory increase in insulin secretion [1,2,3,4]. Baseline secretion of insulin may remain normal or high, but beta cells lose their ability to “sense” an increase in plasma glucose, such that postprandial glucose becomes inordinately high, and postprandial FFAs remain high—an effect that further worsens muscle insulin sensitivity. The resulting exacerbation of beta cell glucolipotoxicity may then progress to the point that fasting levels of glucose remain unduly elevated, at which point the patient is diagnosed with type 2 diabetes. The beta cells then remain locked in a dysfunctional state because the glucose and FFA levels are now chronically high.
The failure of glucose-stimulated insulin secretion (GSIS) that characterizes type 2 diabetes has been traced to a reduced expression of beta cell glucokinase activity [5,6]. Healthy beta cells detect an elevation of plasma glucose by increasing the rate at which they take up glucose and metabolize it via glycolysis. The evolved pyruvate is then oxidized in mitochondria, leading to an increase in the rate of mitochondrial ATP generation. The consequent increase in cytoplasmic ATP/ADP ratio inhibits ATP-sensitive potassium channels in the beta cell plasma membrane, such that plasma membrane potential declines. This in turn opens voltage-sensitive calcium channels, causing an influx of calcium that triggers increased exocytosis of beta cell insulin granules [7,8]. The resulting increase in plasma insulin promotes storage of the elevated plasma glucose, such that postprandial glucose eventually returns to its healthy baseline level.
GSIS is made possible by the fact that, unlike most tissues that rely on hexokinase to initiate glycolysis, glucose phosphorylation in beta cells is mediated largely by glucokinase (GK), which is sometimes referred to as the “glucose sensor” of the beta cell [7]. A similar phenomenon is seen in hepatocytes; in the context of elevated plasma glucose, glucose flux through hepatocyte glucokinase serves as a signal to suppress hepatic gluconeogenesis [9]. As contrasted to hexokinase, GK has a higher Michaelis–Menten constant (Km) for glucose, and is not subject to feedback inhibition by glucose-6-phosphate. For this reason, the rate of flux of glucose through GK is roughly proportionate to plasma glucose level, and hence can serve as a signal in beta cells and hepatocytes when plasma glucose is elevated [10]. The rate of ATP generation in healthy beta cells—which determines the rate of insulin secretion—thus varies directly with plasma glucose, which is a homeostatically appropriate response.
2. A Key Role for Loss of PDX1 Activity and Glucokinase Expression Driven by ROS
This mechanism fails in beta cells experiencing excessive glucolipotoxicity owing to a maladaptive reduction in beta cell GK expression [5,6] (expression of the chief beta cell glucose transporter Glut2 also falls, but this does not appear to be rate limiting for beta cell glycolytic activity). The mechanisms responsible for this decline in GK are now understood at least in part, and this understanding may make it feasible to define measures that could help to sustain beta cell GK expression.
The transcription factor PDX1 is a defining feature of beta cells; this transcription factor promotes expression of the key proteins Glut2, GK, and insulin [11]. The nuclear level of PDX1 in beta cells undergoing failure of GSIS notably declines, and this decline can largely account for the concomitant reduction in GK expression. Nuclear PDX1 declines both because of a reduction of its expression at the transcriptional level, and because PDX1 is exported from the nucleus [12,13,14].
The nuclear export of PDX1 in failing beta cells results from its phosphorylation by c-Jun N-terminal kinase (JNK); this activates a nuclear export signal in PDX1 [12]. The elevation of JNK activity that mediates this is driven by increased levels of oxidants, notably hydrogen peroxide [12]. The increase of reactive oxygen species (ROS) in beta cells exposed to glucolipotoxicity appears to reflect increased ROS production by both NADPH oxidase 2 (NOX2) and by mitochondria; the mechanisms driving this increase are still only partially understood, although the increase in mitochondrial oxidant production is at least partially a function of increased Krebs cycle activity, stemming from increased metabolism of glucose and of FFAs [15,16,17,18,19,20]. How ROS activate JNK in failing beta cells also requires further clarification; oxidant-induced ER stress, as well as activation of apoptosis signal-related kinase-1 (ASK-1) may play a role [21,22,23,24]. In any case, measures that suppress the activity of NOX2, and that optimize the structural and functional integrity of mitochondria, such that they can oxidize large amounts of substrate while keeping ROS production at a moderate level, have potential for lessening the JNK-driven nuclear export of PDX1.
3. Failure of Autocrine Insulin Signaling Promotes Apoptosis and Reduces PDX1 Expression
The reduction of PDX1 expression induced by glucolipotoxicity may stem largely from a reduction of autocrine insulin signaling within beta cells. Beta cells express insulin receptors, and the insulin-mediated activation of the insulin receptor substrate-2 (IRS-2)/PI3K/Akt pathway both opposes beta cell apoptosis and also supports homeostatically appropriate increases in beta cells’ mass in the context of peripheral insulin sensitivity [25,26,27,28,29]. In beta cells exposed to glucolipotoxicity, insulin-mediated activation of Akt is suppressed. This effect likewise is induced by beta cell oxidant stress; likely, this reflects JNK-mediated phosphorylation of IRS-2, which is known to disrupt its interaction with the insulin receptor and promote its proteolytic degradation [30,31].
The relationship of this phenomenon to PDX1 expression is as follows: appropriate Akt activity phosphorylates the forkhead box O1 (FOXO1) transcription factor, inducing its export from the nucleus [32]. On the promoter of PDX1 gene, FOXO1 competes with the binding of the transcription factor Foxa2, which promotes transcription of this gene [33]. Hence, nuclear FOXO1 acts as a transcriptional repressor of PDX1. Appropriate Akt activity, by inducing the expulsion of FOXO1 from the nucleus, disinhibits transcription of the PDX1 gene, and hence sustains PDX1 expression. The suppression of insulin signaling that characterizes glucolipotoxicity not only exposes beta cells to increased risk of apoptosis—ultimately leading to a maladaptive decrease in beta cell mass—but also compromises PDX1 expression.
In summary, the failure of GSIS induced by glucolipotoxicity, as well as the increased propensity for beta cell apoptosis, appears to reflect the interaction of increased ROS levels and the associated reduction in the efficiency of autocrine insulin signaling. On the positive side, the incretin hormone glucagon-like peptide-1 (GLP-1) acts to support effective GSIS [34]. GLP-1, via stimulation of cAMP production and consequent PKA activation, promotes the nuclear import of PDX1, counteracting the adverse effect of JNK in this regard [35]. Moreover, cAMP-stimulated EPAC (exchange protein activated by cAMP) somehow promotes PDX1 expression [36].
These considerations suggest that nutraceutical or dietary measures that lessen oxidant production by NOX2 or mitochondria, that suppress redox signaling, that reinforce signaling from the insulin receptor to Akt, or that boost intestinal production of GLP-1, could be expected to aid maintenance of GSIS and prevent apoptosis in beta cells challenged by glucolipotoxicity. Such measures may be doubly beneficial if they also counter the peripheral insulin resistance that gives rise to glucolipotoxicity.
4. Controlling Beta Cell Oxidant Stress—Focus on NOX2, Mitophagy, and Mitochondrial Biogenesis
With respect to NOX2-mediated oxidant production, the intracellular free bilirubin derived from heme oxygenase activity has been shown to function as a physiological inhibitor of NOX2 activity [37,38,39,40]. This likely explains why, in some prospective epidemiological studies and in Mendelian randomization analysis, higher serum levels of free bilirubin correlate with reduced risk for type 2 diabetes, and why oral administration of biliverdin (rapidly converted to bilirubin within cells) has been found to prevent or postpone the onset of hyperglycemia in db/db mice prone to this diabetes [41,42,43,44]. In this latter study, biliverdin treatment was associated with maintenance of PDX1 and insulin expression in beta cells, and a decrease in beta cell apoptosis, despite the fact that peripheral insulin sensitivity was only modestly influenced. Hence, the data suggested that biliverdin was directly protective to beta cells. Whereas biliverdin is expensive to synthesize and therefore impractical for widescale use as a nutraceutical, its metabolite phycocyanobilin (PCB), a light-absorbing chromophore present in high concentrations in cyanobacteria (such as the food spirulina) and certain blue-green algae, shares the ability of bilirubin to inhibit NOX2, likely because it is converted within cells to phycocyanorubin, a compound that is highly similar to bilirubin [45,46]. This may explain much of the versatile antioxidant and anti-inflammatory activity of spirulina or its chief protein phycocyanin (to which PCB is covalently attached) in a wide range of rodent studies [47,48]. Hence, regular ingestion of adequate amounts of spirulina, or of spirulina extracts enriched in PCB, may have potential for aiding the preservation of proper beta cell function and mass in patients with metabolic syndrome.
As noted, the increased mitochondrial oxidation of substrate that accompanies increased exposure to glucose and FFAs would be expected to increase generation of superoxide by the mitochondrial respiratory chain. However, the extent to which this gives rise to cellular oxidant stress should be modulated by the extent to which mitochondria are structurally and functionally intact. Oxidant-mediated damage to the mitochondrial respiratory chain, or improper formation of this chain owing to oxidant-mediated damage to mitochondrial DNA, could be expected to amplify the rate at which mitochondria release hydrogen peroxide in response to substrate overload. Conversely, increased expression of mitochondrial peroxidase activity would be expected to quell such release.
The process of mitophagy, coupled appropriately to mitochondrial biogenesis (MB), is the mechanism whereby structurally and functionally damaged mitochondria—as detected by a reduction in mitochondrial membrane potential—are replaced by new mitochondria that are more functionally competent and less likely to generate oxidants [49,50]. A previous discussion has proposed that drugs or nutraceuticals that activate sirtuin 1 (Sirt1), AMP-activated protein kinase (AMPK), Nrf2, and peroxisome proliferator-activated receptor alpha (PPARα) could be expected to collaborate in promotion of both mitophagy and MB [51]. Physiologically active nutraceuticals that can boost Sirt1 activity or expression include ferulic acid melatonin [52,53,54,55,56,57,58,59]. The herbal compound berberine, a nutraceutical long employed in diabetes treatment in China, shares the ability of metformin to activate AMPK [60,61,62,63,64]. Nutraceuticals that can amplify the transcriptional activity or expression of Nrf2 include lipoic acid, the sulforaphane generated from broccoli sprout extract, the neurohormone melatonin, and the xanthophyll carotenoid astaxanthin [58,65,66,67,68]. Moreover, astaxanthin can also serve as an agonist for PPARα and can act as a highly effective scavenging antioxidant in the mitochondrial inner membrane [68,69].
Activation of Nrf2 not only aids MB, but also increases mitochondrial expression of peroxidases and superoxide dismutase, which suppress mitochondrial release of hydrogen peroxide and, like astaxanthin, can protect the respiratory chain from oxidative damage [70]. Hence, nutraceuticals that boost Nrf2 activity can do double duty in protecting beta cells from the mitochondrial oxidant stress imposed by glucolipotoxicity. Additionally, by boosting the cytoplasmic expression of various antioxidant enzymes as well as glutathione, they can lessen the ability of hydrogen peroxide to provoke JNK activation [71]. With respect to glutathione, its cellular levels can be further boosted with supplemental N-acetylcysteine (NAC), particularly in the elderly [72,73,74]. In summary, nutraceutical programs featuring some or all of PCB, ferulic acid, melatonin, berberine, lipoic acid, NAC, and astaxanthin may have potential for lessening the activation of JNK induced by glucolipotoxicity. In this regard, each of these agents, excepting PCB, has been shown to protect beta cells from glucolipotoxicity in rodent or cells culture models [75,76,77,78,79,80,81,82,83,84,85,86,87].
Sirt1, in addition to promoting mitophagy and MB, can lessen glucotoxicity-induced mitochondrial oxidant production by preventing JNK-mediated phosphorylation and activation of p66Shc; activated p66Shc can migrate to mitochondria, where it oxidizes cytochrome c to generate hydrogen peroxide [88,89,90]. However, the interaction of JNK with p66Shc requires acetylation of the latter, which Sirt1 can reverse [88].
Support of Sirt1 activity in beta cells stressed by glucolipotoxicity may be all the more important in light of evidence that hyperglycemia can decrease Sirt1 protein expression in these cells [88]. JNK1 activated by glucolipotoxicity can confer a phosphorylation on Sirt1 (at Ser47) that marks it for ubiquitination and subsequent proteasomal degradation [91]. Moreover, recent studies show that USP22 deubiquitinase activity can raise protein levels of Sirt1 by reversing this ubiquitination [92,93,94,95]. The proximal promoter of the USP22 gene contains an Sp1-binding site, and Sp1 binding suppresses the gene’s transcription [96,97]. Activated p38 MAP kinase can confer a phosphorylation on Sp1 that expedites its binding to its response elements and has been found to inhibit Sp1 expression at the transcriptional level [97,98]. Both oxidative and ER stress can drive the activation of p38, in part via apoptosis signaling-regulated kinase (ASK1) [71,99,100]. Hence, oxidative and ER stress have the potential to decrease the protein expression of Sirt1 by inhibiting expression of USP22 deubiquitinase [90,97]. Indeed, in cell lines derived from human pancreatic beta cells or an insulinoma, hyperglycemia-induced NADPH oxidase activity, via p38, has been found to suppress transcription of the USP22 gene, and thereby down-regulate protein expression of Sirt1 [88].
With respect to AMPK activity in beta cells, this can also be stimulated by the hormone fibroblast hormone 21 (FGF21) [101]. Recent studies show that FGF21 levels are markedly elevated in people who habitually consume plant-based (vegan) diets of modest protein content—likely reflecting hepatic activation of GCN2, which detects a relative deficiency of essential amino acids [102,103,104]. An epidemiological study in Loma Linda, which has a high population of vegetarians, found that, after multivariate adjustment including BMI, long-term vegans, as opposed to omnivores, were 62% less likely to develop type 2 diabetes, without adjustment for BMI (which tends to be low in vegans), and 77% protection was observed [105]. The low proportion of total dietary fat provided as saturated fat in most vegan diets may also provide protection in this regard by improving peripheral insulin sensitivity and hence lessening glucose/lipid overexposure [106].
5. Amplifying or Mimicking the Insulin Signal
With respect to insulin/Akt signaling in beta cells, cGMP and protein kinase G (PKG) have been found to promote Akt activation in beta cells, mimicking the impact of insulin. The mechanistic basis of this effect remains unclear, but it is dependent on intact activity of PI3K [107]. cGMP production in beta cells can be directly stimulated by supraphysiological but clinically feasible concentrations of the B vitamin biotin; this can directly activate the soluble guanylate cyclase [108,109]. This phenomenon likely explains, at least in part, the utility of high-dose biotin for aiding glycemic control in rodent models of diabetes [110,111,112,113,114,115,116]. Increased expression of glucokinase and insulin in the beta-cells of diabetes-prone mice supplemented with high-dose biotin indicates that a protective effect of biotin on beta cell function in diabetic or pre-diabetic rodents [116,117]. High-dose biotin also exerts a trophic effect on beta cell proliferation in newborn mice [118]. Limited clinical data also point to a potential role for high-dose biotin in diabetes management, but the possibility that biotin might reduce risk for diabetes in patients with metabolic syndrome has not been assessed. While high-dose biotin is well tolerated clinically, it has the potential to interfere with certain laboratory tests employing biotinylated reagents, and hence should be discontinued temporarily prior to such tests.
Supplementation with myo-inositol can boost insulin/Akt signaling in circumstances in which the availability of this physiologically essential fact is limiting for the production of phosphatidyl-inositides, which are mediators of such signaling [119] (PI3K-mediated addition of a phosphate to the 3 position of the plasma membrane phosphatidyl-inositol-1,4,5-triphosphate is essential for insulin-stimulated Akt activation). Numerous clinical studies reveal that myo-inositol supplementation (typically 1–2 g twice daily) can aid insulin sensitivity in metabolic syndrome associated with polycystic ovarian syndrome (PCOS) and in gestational diabetes [120,121,122,123,124,125]. Intriguingly, such supplementation has been found to be highly effective for prevention of gestational diabetes in women known to be prone to this syndrome [126] (RR = 0.44, 95% CI (0.32, 0.62), p < 0.0001, as determined by meta-analysis of four randomized controlled studies). Although limited clinical evidence suggests that supplemental myo-inositol can aid metabolic control in patients with type 2 diabetes, the possibility that it might decrease risk for new onset diabetes in patients with metabolic syndrome, both by aiding maintenance of beta cell function and by lessening peripheral insulin resistance, has not been tested [127]. The strong utility of myo-inositol for the prevention of gestational diabetes encourages this hypothesis.
6. Boosting Glucagon-like Peptide-1 Production for Support of GSIS
With respect to intestinal GLP-1 production, this can be aided by insuring a good population of the colon with “friendly” bacteria that are capable of generating short-chain fatty acids (as by use of effective probiotics), and by the ingestion of prebiotics or of diets rich in soluble fiber and/or resistant starch that deliver fermentable carbohydrate to the distal intestine and colon [128,129,130,131,132]. Curiously, foods high in insoluble fibers (particularly wheat bran) that neither are fermentable nor influence glycemic index are linked to decreased diabetes risk; this may reflect the fact that such foods tend to be rich in ferulate conjugates that are partially cleaved during digestion, rendering the ferulic acid bioavailable [133,134,135,136,137]. Hence, such foods tend to be a source of delayed release ferulic acid, yielding a less episodic increase in plasma ferulic acid than the free compound.
7. Summing Up
A safe, practical strategy for preventing the conversion of metabolic syndrome to type 2 diabetes could be of tremendous benefit, given the horrendous medical and financial implications of the worldwide rise in diabetes prevalence. This essay has attempted to define—in a vastly simplified form—the molecular biology underlying the glucolipotoxicity-induced failure of beta cell GSIS and the apoptotic loss of beta cells that ushers in overt diabetes, and to postulate nutraceutical strategies with the potential for rectifying the mechanisms driving this beta cell failure. These strategies are intended to do the following: control beta cell ROS production via inhibition of NOX2 and maintenance of healthful mitochondrial function by promoting mitophagy and MB, boost or mimic autocrine insulin signaling, and enhance GLP-1 secretion. Nutraceuticals identified with potential for promoting one or more of these aims include PCB, ferulic acid, lipoic acid, melatonin, berberine, astaxanthin, myo-inositol, high-dose biotin, and pre/probiotics. Figure 1 outlines in schematic form the potential contribution of these nutraceuticals in this regard. Complex supplements and/or functional foods providing a selection of these nutraceuticals may in the future find a practical role in diabetes prevention.
It should be noted that some of the nutraceuticals discussed here may aid diabetes prevention not only through direct effects on beta cells, but also by improving peripheral insulin sensitivity or by aiding postprandial glycemic control, thereby mitigating episodes of glucolipotoxicity. These include berberine, myo-inositol, high-dose biotin, and PCB (from whole spirulina or phycocyanin) [58,66,111,112,113,114,115,125,138,139,140]. Evidently, appropriate weight loss, exercise training, and whole-food plant-based diets also have an important potential in this regard. Ample intakes of magnesium and zinc, and nutraceuticals such as chromium picolinate and cinnamon extract, may also be beneficial [141,142,143,144,145,146,147,148,149].
Author Contributions
M.F.M. conceived the review and wrote the initial draft; J.J.D. edited the draft and added additional pertinent material. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
None needed.
Informed Consent Statement
None needed.
Data Availability Statement
No original data.
Conflicts of Interest
Mark F. McCarty is co-inventor and co-owner of a USA patent on nutraceutical uses of phycocyanobilin oligopeptides derived from spirulina. James J. DiNicolantonio is Director of Scientific Affairs for AIDP.
References
- Vela-Guajardo, J.E.; Garza-González, S.; García, N. Glucolipotoxicity-induced oxidative stress is related to mitochondrial dysfunction and apoptosis of pancreatic β-cell. Curr. Diabetes Rev. 2021, 17, e031120187541. [Google Scholar] [CrossRef] [PubMed]
- Prentki, M.; Joly, E.; El-Assaad, W.; Roduit, R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes 2002, 51 (Suppl. 3), S405–S413. [Google Scholar] [CrossRef] [Green Version]
- Van Raalte, D.H.; Diamant, M. Glucolipotoxicity and beta cells in type 2 diabetes mellitus: Target for durable therapy? Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S37–S46. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Del Guera, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, B.; Kurmi, K.; Munoz-Gomez, M.; Ambuludi, E.J.J.; Tonne, J.M.; Rakshit, K.; Hitosugi, T.; Kudva, Y.C.; Matveyenko, A.V.; Ikeda, Y. Impaired β-cell glucokinase as an underlying mechanism in diet-induced diabetes. Dis. Model. Mech. 2018, 11, dmm033316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matschinsky, F.; Liang, Y.; Kesavan, P.; Wang, L.; Froguel, P.; Velho, G.; Cohen, D.; Permutt, M.A.; Tanizawa, Y.; Jetton, T.L. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J. Clin. Investig. 1993, 92, 2092–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.; Satin, L.S. Beta-cell ion channels and their role in regulating insulin secretion. Compr. Physiol. 2021, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nissim, I.; Horyn, O.; Nissim, I.; Daikhin, Y.; Wehrli, S.L.; Yudkoff, M.; Matschinsky, F.M. Effects of a glucokinase activator on hepatic intermediary metabolism: Study with 13C-isotopomer-based metabolomics. Biochem. J. 2012, 444, 537–551. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Parker, J.C.; Najafi, H.; Matschinsky, F.M. Control of glucose metabolism in pancreatic beta-cells by glucokinase, hexokinase, and phosphofructokinase. Model study with cell lines derived from beta-cells. Diabetes 1988, 37, 1524–1530. [Google Scholar] [CrossRef]
- Kaneto, H.; Miyatsuka, T.; Kawamori, D.; Yamamoto, K.; Kato, K.; Shiraiwa, T.; Katakami, N.; Yamasaki, Y.; Matsuhisa, M.; Matsuoka, T.-A. PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocr. J. 2008, 55, 235–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamori, D.; Kajimoto, Y.; Kaneto, H.; Umayahara, Y.; Fujitani, Y.; Miyatsuka, T.; Watada, H.; Leibiger, I.B.; Yamasaki, Y.; Hori, M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH2-terminal kinase. Diabetes 2003, 52, 2896–2904. [Google Scholar] [CrossRef] [Green Version]
- Olson, L.K.; Sharma, A.; Peshavaria, M.; Wright, C.V.; Towle, H.C.; Rodertson, R.P.; Stein, R. Reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to a supraphysiologic glucose concentration is associated with loss of STF-1 transcription factor expression. Proc. Natl. Acad. Sci. USA 1995, 92, 9127–9131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimer, M.K.; Ahrén, B. Altered beta-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes 2002, 51 (Suppl. 1), S138–S143. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53 pathways. PLoS ONE 2010, 5, e15726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Las, G.; Oliveira, M.F.; Shirihai, O.S. Emerging roles of β-cell mitochondria in type-2-diabetes. Mol. Asp. Med. 2020, 71, 100843. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Rashid, M.A.; Jang, M.; Kim, Y.; Won, H.; Lee, J.; Woo, J.T.; Kim, Y.S.; Murphy, M.P.; Ali, L.; et al. Mitochondria-targeted antioxidants protect pancreatic β-cells against oxidative stress and improve insulin secretion in glucotoxicity and glucolipotoxicity. Cell. Physiol. Biochem. 2011, 28, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Boas, E.A.; Nalbach, L.; Ampofo, E.; Lucena, C.F.; Naudet, L.; Ortis, F.; Carpinelli, A.R.; Morgan, B.; Roma, L.P. Transient NADPH oxidase 2-dependent H2O2 production drives early palmitate-induced lipotoxicity in pancreatic islets. Free Radic. Biol. Med. 2021, 162, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, A.; Kowluru, R.A. Phagocyte-like NADPH oxidase [Nox2] in cellular dysfunction in models of glucolipotoxicity and diabetes. Biochem. Pharmacol. 2014, 88, 275–283. [Google Scholar] [CrossRef]
- Sidarala, V.; Veluthakal, R.; Syeda, K.; Vlaar, C.; Newsholme, P.; Kowluru, A. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: Evidence for a requisite role of Ras-related C3 botulinum toxin substrate 1 (Rac1). Biochem. Pharmacol. 2015, 95, 301–310. [Google Scholar] [CrossRef]
- Singh, S.; Bhowmick, D.C.; Pany, S.; Joe, M.; Zaghlula, N.; Jeremic, A.M. Apoptosis signal regulating kinase-1 and NADPH oxidase mediate human amylin evoked redox stress and apoptosis in pancreatic beta-cells. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell. Sci. 2008, 121 Pt 14, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachar, E.; Ariav, Y.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 2009, 4, e4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Ma, J.; Lu, Y.; Zhou, C.; Zhao, T.; Ai, X.; Wei, X.; Lin, J.; Wang, W.; Yan, W.; et al. The protective role of the MKP-5-JNK/P38 pathway in glucolipotoxicity-induced islet β-cell dysfunction and apoptosis. Exp. Cell Res. 2019, 382, 111467. [Google Scholar] [CrossRef]
- Muller, D.; Huang, G.C.; Amiel, S.; Jones, P.M.; Persaud, S.J. Identification of insulin signaling elements in human beta-cells: Autocrine regulation of insulin gene expression. Diabetes 2006, 55, 2835–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevitch, D.; Boura-Halfon, S.; Isaac, R.; Shahaf, G.; Alberstein, M.; Ronen, D.; Lewis, E.C.; Zick, Y. Elimination of negative feedback control mechanisms along the insulin signaling pathway improves beta-cell function under stress. Diabetes 2010, 59, 2188–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamoto, I.; Terauchi, Y.; Kubota, N.; Ohsugi, M.; Ueki, K.; Kadowaki, T. Crucial role of insulin receptor substrate-2 in compensatory beta-cell hyperplasia in response to high fat diet-induced insulin resistance. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 147–156. [Google Scholar] [CrossRef]
- Rhodes, C.J. Type 2 diabetes-a matter of beta-cell life and death? Science 2005, 307, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Lingohr, M.K.; Dickson, L.M.; Wrede, C.E.; Briaud, I.; McCuaig, J.F.; Myers, M.G., Jr.; Rhodes, C.J. Decreasing IRS-2 expression in pancreatic beta-cells (INS-1) promotes apoptosis, which can be compensated for by introduction of IRS-4 expression. Mol. Cell. Endocrinol. 2003, 209, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, M.; Gotoh, Y.; Katagiri, H.; Sakoda, H.; Ogihara, T.; Anai, M.; Onishi, Y.; Ono, H.; Abe, M.; Shojima, N.; et al. Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol. Endocrinol. 2003, 17, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sharfi, H.; Eldar-Finkelman, H. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E307–E315. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gan, L.; Pan, H.; Guo, S.; Bencherif, M.; Olson, S.T.; Mesecar, A.; Adam, S.; Unterman, T.G. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 2002, 277, 45276–45284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Nakae, J.; Kitamura, Y.; Biggs, W.H.; Wright, C.V.; White, M.F.; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J. Clin. Investig. 2002, 110, 1839–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, M.E.; Egan, J.M. Glucagon-like peptide-1. Recent Prog. Horm. Res. 2001, 56, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, J.; Doyle, M.E.; Egan, J.M. Glucagon-like peptide-1 causes pancreatic duodenal homeobox-1 protein translocation from the cytoplasm to the nucleus of pancreatic beta-cells by a cyclic adenosine monophosphate/protein kinase A-dependent mechanism. Endocrinology 2001, 142, 1820–1827. [Google Scholar] [CrossRef]
- Kai, A.K.; Lam, A.K.; Chen, Y.; Tai, A.C.; Zhang, X.; Lai, A.K.; Yeung, P.K.; Tam, S.; Wang, J.; Lam, K.S.; et al. Exchange protein activated by cAMP 1 (Epac1)-deficient mice develop β-cell dysfunction and metabolic syndrome. FASEB J. 2013, 27, 4122–4135. [Google Scholar] [CrossRef]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taillé, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases NOS2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J. 2005, 19, 1890–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, H.; Ishikawa, K.; Itabe, H.; Maruyama, Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem. 2006, 291, 21–28. [Google Scholar] [CrossRef]
- Jiang, F.; Roberts, S.J.; Datla, S.R.; Dusting, G.J. NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension 2006, 48, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Datla, S.R.; Dusting, G.J.; Mori, T.A.; Taylor, C.J.; Croft, K.D.; Jiang, F. Induction of heme oxygenase-1 in vivo suppresses NADPH oxidase derived oxidative stress. Hypertension 2007, 50, 636–642. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Lee, M.J.; Kang, Y.M.; Hwang, J.Y.; Jang, J.E.; Leem, J.; Park, J.-Y.; Kim, H.-K.; Lee, W.J. Higher serum bilirubin level as a protective factor for the development of diabetes in healthy Korean men: A 4year retrospective longitudinal study. Metabolism 2014, 63, 87–93. [Google Scholar] [CrossRef]
- Yang, M.; Ni, C.; Chang, B.; Jiang, Z.; Zhu, Y.; Tang, Y.; Li, Z.; Li, C.; Li, B. Association between serum total bilirubin levels and the risk of type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2019, 152, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, A.; Deetman, P.E.; Corpeleijn, E.; Gansevoort, R.T.; Gans, R.O.; Hillege, H.L.; van der Harst, P.; Stolk, R.P.; Navis, G.; Alizadeh, B.Z.; et al. Bilirubin as a potential causal factor in type 2 diabetes risk: A Mendelian randomization study. Diabetes 2014, 64, 1459–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, N.; Inoguchi, T.; Sonoda, N.; Fujii, M.; Takei, R.; Hirata, E.; Yokomizo, H.; Zheng, J.; Maeda, Y.; Kobayashi, K.; et al. Biliverdin protects against the deterioration of glucose tolerance in db/db mice. Diabetologia 2011, 54, 2183–2191. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F. Clinical potential of Spirulina as a source of phycocyanobilin. J. Med. Food 2007, 10, 566–570. [Google Scholar] [CrossRef]
- Terry, M.J.; Maines, M.D.; Lagarias, J.C. Inactivation of phytochrome- and phycobiliprotein-chromophore precursors by rat liver biliverdin reductase. J. Biol. Chem. 1993, 268, 26099–26106. [Google Scholar] [CrossRef]
- Romay, C.; Gonzalez, R.; Ledon, N.; Remirez, D.; Rimbau, V. C-phycocyanin: A biliprotein with antioxidant, anti-inflammatory and neuroprotective effects. Curr. Protein Pept. Sci. 2003, 4, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Mysliwa-Kurdziel, B.; Solymosi, K. Phycobilins and phycobiliproteins used in food industry and medicine. Mini Rev. Med. Chem. 2017, 17, 1173–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, O.; Murakawa, T.; Nishida, K.; Otsu, K. Receptor-mediated mitophagy. J. Mol. Cell. Cardiol. 2016, 95, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Lujan, L.L.; Iloki-Assanga, S.; McCarty, M.F.; DiNicolantonio, J.J. Nutraceuticals/drugs promoting mitophagy and mitochondrial biogenesis may combat the mitochondrial dysfunction driving progression of dry age-related macular degeneration. Med. Hypotheses 2021. in submission. [Google Scholar]
- El-Mesallamy, H.O.; Gawish, R.A.; Sallam, A.M.; Fahmy, H.A.; Nada, A.S. Ferulic acid protects against radiation-induced testicular damage in male rats: Impact on SIRT1 and PARP1. Environ. Sci. Pollut. Res. 2018, 25, 6218–6227. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, F.H.; Mesbah-Ardakani, M.; Nasr-Esfahani, M.-H. Ferulic acid exerts concentration-dependent anti-apoptotic and neuronal differentiation-inducing effects in PC12 and mouse neural stem cells. Eur. J. Pharmacol. 2018, 841, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Zhang, L.; Yang, X. Ferulic acid, a natural polyphenol, protects against osteoporosis by activating SIRT1 and NF-κB in neonatal rats with glucocorticoid-induced osteoporosis. Biomed. Pharm. 2019, 120, 109205. [Google Scholar] [CrossRef]
- Xu, T.; Song, Q.; Zhou, L.; Yang, W.; Wu, X.; Qian, Q.; Chai, H.; Han, Q.; Pan, H.; Dou, X.; et al. Ferulic acid alleviates lipotoxicity-induced hepatocellular death through the SIRT1-regulated autophagy pathway and independently of AMPK and Akt in AML-12 hepatocytes. Nutr. Metab. 2021, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Huang, Z.; Chen, D.; Yu, B.; Chen, H.; Yu, J.; Luo, Y.; Zheng, P.; He, J. Effects of dietary ferulic acid supplementation on growth performance and skeletal muscle fiber type conversion in weaned piglets. J. Sci. Food Agric. 2021, 101, 5116–5123. [Google Scholar] [CrossRef]
- Du, K.; Fang, X.; Li, Z. Ferulic acid suppresses interleukin-1β-induced degeneration of chondrocytes isolated from patients with osteoarthritis through the SIRT1/AMPK/PGC-1α signaling pathway. Immun. Inflamm. Dis. 2021, 9, 710–720. [Google Scholar] [CrossRef]
- Fang, J.; Yan, Y.; Teng, X.; Wen, X.; Li, N.; Peng, S.; Liu, W.; Donadeu, F.X.; Zhao, S.; Hua, J. Melatonin prevents senescence of canine adipose-derived mesenchymal stem cells through ac-tivating NRF2 and inhibiting ER stress. Aging (Albany N. Y.) 2018, 10, 2954–2972. [Google Scholar]
- Wang, Z.; Ma, C.; Meng, C.J.; Zhu, G.-Q.; Sun, X.-B.; Huo, L.; Zhang, J.; Liu, H.-X.; He, W.-C.; Shen, X.-M.; et al. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J. Pineal Res. 2012, 53, 129–137. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Kim, W.S.; Kim, K.H.; Yoon, M.J.; Cho, H.J.; Shen, Y.; Ye, J.-M.; Lee, C.H.; Oh, W.K.; Kim, C.T.; et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 2006, 55, 2256–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Li, J.Y.; Gosby, A.; To, S.W.; Cheng, Z.; Miyoshi, H.; Taketo, M.M.; Cooney, G.J.; Kraegen, E.W.; James, D.E.; et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: A mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 2008, 57, 1414–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Liu, X.; Wu, N.; Han, Y.; Wang, J.; Yu, Y.; Chen, Q. Efficacy and safety of berberine alone for several metabolic disorders: A systematic review and meta-analysis of randomized clinical trials. Front. Pharmacol. 2021, 12, 653887. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Xu, X.; Yin, M.; Zhang, Y.; Huang, L.; Chen, R.; Ni, J. Effects of berberine on blood glucose in patients with type 2 diabetes mellitus: A systematic literature review and a meta-analysis. Endocr. J. 2019, 66, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Fratantonio, D.; Speciale, A.; Molonia, M.S.; Bashallari, R.; Palumbo, M.; Saija, A.; Cimino, F.; Monastra, G.; Virgili, F. Alpha-lipoic acid, but not di-hydrolipoic acid, activates Nrf2 response in primary human umbilical-vein endothelial cells and protects against TNF-α induced endothelium dysfunction. Arch. Biochem. Biophys. 2018, 655, 18–25. [Google Scholar] [CrossRef]
- Kensler, T.W.; Egner, P.A.; Agyeman, A.S.; Visvanathan, K.; Groopman, J.D.; Chen, J.-G.; Chen, T.-Y.; Fahey, J.W.; Talalay, P. Keap1–Nrf2 signaling: A target for cancer prevention by sulforaphane. Top Curr. Chem. 2012, 329, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Kohandel, Z.; Farkhondeh, T.; Aschner, M.; Samarghandian, S. Nrf2 a molecular therapeutic target for Astaxanthin. Biomed. Pharmacother. 2021, 137, 111374. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wu, C.; Kim, J.; Kim, B.; Lee, S.-J. Astaxanthin reduces hepatic lipid accumulations in high-fat-fed C57BL/6J mice via activation of peroxisome proliferator-activated receptor (PPAR) alpha and inhibition of PPAR gamma and Akt. J. Nutr. Biochem. 2016, 28, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharm. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Matsukawa, J.; Matsuzawa, A.; Takeda, K.; Ichijo, H. The ASK1-MAP kinase cascades in mammalian stress response. J. Biochem. 2004, 136, 261–265. [Google Scholar] [CrossRef]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharm. 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, S.; Dean, O.; Copolov, D.L.; Malhi, G.S.; Berk, M. N-acetylcysteine for antioxidant therapy: Pharmacology and clinical utility. Expert Opin. Biol. Ther. 2008, 8, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V. GlyNAC supplementation improves glutathione deficiency, oxidative stress, mitochondrial dysfunction, inflammation, aging hallmarks, metabolic defects, muscle strength, cognitive decline, and body composition: Implications for healthy aging. J. Nutr. 2021, 151, 3606–3616. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Boss, M.; Thomas, A.P.; Matveyenko, A.V. Activation of melatonin signaling promotes β-cell survival and function. Mol. Endocrinol. 2015, 29, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shim, H.M.; Na, A.Y.; Bae, K.-C.; Bae, J.-H.; Im, S.-S.; Cho, H.-C.; Song, D.-K. Melatonin prevents pancreatic β -cell loss due to glucotoxicity: The relationship between oxidative stress and endoplasmic reticulum stress. J. Pineal Res. 2014, 56, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Kozuka, C.; Okamoto, S.; Yonamine, M.; Tanaka, H.; Shimabukuro, M. Brown rice-specific γ-oryzanol as a promising prophylactic avenue to protect against diabetes mellitus and obesity in humans. J. Diabetes Investig. 2019, 10, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Gao, J.; Li, H. Ferulic acid confers protection on islet β cells and placental tissues of rats with gestational diabetes mellitus. Cell. Mol. Biol. (Noisy-Le-Grand) 2020, 66, 37–41. [Google Scholar] [CrossRef]
- Nobakht-Haghighi, N.; Rahimifard, M.; Baeeri, M.; Rezvanfar, M.A.; Nodeh, S.M.; Haghi-Aminjan, H.; Hamurtekin, E.; Abdollahi, M. Regulation of aging and oxidative stress pathways in aged pancreatic islets using alpha-lipoic acid. Mol. Cell. Biochem. 2018, 449, 267–276. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, W.; Liu, Y.; Guo, T.; Chen, P.; Ma, K.; Zhou, C. α-lipoic acid inhibits high glucose-induced apoptosis in HIT-T15 cells. Dev. Growth Differ. 2012, 54, 557–565. [Google Scholar] [CrossRef]
- Shen, W.; Liu, K.; Tian, C.; Yang, L.; Li, X.; Ren, J.; Packer, L.; Head, E.; Sharman, E.; Liu, J. Protective effects of R-alpha-lipoic acid and acetyl-L-carnitine in MIN6 and isolated rat islet cells chronically exposed to oleic acid. J. Cell. Biochem. 2008, 104, 1232–1243. [Google Scholar] [CrossRef]
- Song, K.H.; Lee, W.J.; Koh, J.M.; Kim, H.S.; Youn, J.-Y.; Park, H.-S.; Koh, E.H.; Kim, M.-S.; Youn, J.H.; Lee, K.-U.; et al. α-lipoic acid prevents diabetes mellitus in diabetes-prone obese rats. Biochem. Biophys. Res. Commun. 2004, 326, 197–202. [Google Scholar] [CrossRef]
- Uchiyama, K.; Naito, Y.; Hasegawa, G.; Nakamura, N.; Takahashi, J.; Yoshikawa, T. Astaxanthin protects beta-cells against glucose toxicity in diabetic db/db mice. Redox Rep. 2002, 7, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, S.; Tang, J.; Zhang, K.; Guang, L.; Huang, Y.; Xu, Y.; Ying, Y.; Zhang, L.; Li, D. Protective effect of berberine on beta cells in streptozotocin- and high-carbohydrate/high-fat diet-induced diabetic rats. Eur. J. Pharmacol. 2009, 606, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Alnahdi, A.; John, A.; Raza, H. Mitigation of glucolipotoxicity-induced apoptosis, mitochondrial dysfunction, and metabolic stress by N-acetyl cysteine in pancreatic β-cells. Biomolecules 2020, 10, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zou, S.; Cui, Z.; Guo, P.; Meng, Q.; Shi, X.; Gao, Y.; Yang, G.; Han, Z. Zerumbone protects INS-1 rat pancreatic beta cells from high glucose-induced apoptosis through generation of reactive oxygen species. Biochem. Biophys. Res. Commun. 2015, 460, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.A.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Karunakaran, U.; Moon, J.S.; Won, K.C. High glucose-induced PRDX3 acetylation contributes to glucotoxicity in pancreatic β-cells: Prevention by Teneligliptin. Free Radic. Biol. Med. 2020, 160, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schultz, R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharmacol. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhang, J.; Kheterpal, I.; Kennedy, N.; Davis, R.J.; Ye, J. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 2011, 286, 22227–22234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Yang, H.; Kong, Q.; Li, J.; Lee, S.-M.; Gao, B.; Dong, H.; Wei, J.; Song, J.; Zhang, D.D.; et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 2012, 46, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Ao, N.; Liu, Y.; Feng, H.; Bian, X.; Li, Z.; Gu, B.; Zhao, X.; Liu, Y. Ubiquitin-specific peptidase USP22 negatively regulates the STAT signaling pathway by deubiquitinating SIRT1. Cell. Physiol. Biochem. 2014, 33, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Yang, Y.M.; Han, C.Y.; Koo, J.H.; Oh, H.; Kim, S.S.; You, B.H.; Choi, Y.H.; Park, T.S.; Lee, C.H.; et al. Gα12 ablation exacerbates liver steatosis and obesity by suppressing USP22/SIRT1-regulated mitochondrial respiration. J. Clin. Investig. 2018, 128, 5587–5602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Sun, L.; Wu, W.; Wu, J.; Sun, Z.; Ren, J. USP22 protects against myocardial ischemia-reperfusion injury via the SIRT1-p53/SLC7A11-dependent inhibition of ferroptosis-induced cardiomyocyte death. Front. Physiol. 2020, 11, 551318. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Che, X.; Li, X.; Yu, H.; Gong, Z.; Li, W. Cloning and characterization of the human USP22 gene promoter. PLoS ONE 2012, 7, e52716. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Gong, Z.; Zhou, X.; Liu, J.; Jiang, H.; Wu, P.; Li, W. p38 mitogen-activated protein kinase inhibits USP22 transcription in HeLa cells. Biomed. Rep. 2015, 3, 461–467. [Google Scholar] [CrossRef] [Green Version]
- D’Addario, M.; Arora, P.D.; McCulloch, C.A. Role of p38 in stress activation of Sp1. Gene 2006, 379, 51–61. [Google Scholar] [CrossRef]
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Nishitoh, H.; Tobiume, K.; Takeda, K.; Ichijo, H. Physiological roles of ASK1-Mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: Advanced findings from ASK1 knockout mice. Antioxid. Redox Signal. 2002, 4, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; So, W.Y.; Li, X.Y.; Leung, P.S. Fibroblast growth factor 21 protects against lipotoxicity-induced pancreatic β-cell dysfunction via regulation of AMPK signaling and lipid metabolism. Clin. Sci. (Lond.) 2019, 133, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Laeger, T.; Albarado, D.C.; Burke, S.J.; Trosclair, L.; Hedgepeth, J.W.; Berthoud, H.-R.; Gettys, T.W.; Collier, J.J.; Münzberg, H.; Morrison, C.D. Metabolic responses to dietary protein restriction require an increase in FGF21 that is delayed by the absence of GCN2. Cell Rep. 2016, 16, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Castaño-Martinez, T.; Schumacher, F.; Schumacher, S.; Kochlik, B.; Weber, D.; Grune, T.; Biemann, R.; McCann, A.; Abraham, K.; Weikert, C.; et al. Methionine restriction prevents onset of type 2 diabetes in NZO mice. FASEB J. 2019, 33, 7092–7102. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. GCN2 and FGF21 are likely mediators of the protection from cancer, autoimmunity, obesity, and diabetes afforded by vegan diets. Med. Hypotheses 2014, 83, 365–371. [Google Scholar] [CrossRef]
- Tonstad, S.; Stewart, K.; Oda, K.; Batech, M.; Herring, R.P.; Fraser, G.E. Vegetarian diets and incidence of diabetes in the Adventist Health Study-2. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 292–299. [Google Scholar] [CrossRef] [Green Version]
- McCarty, M.F. Dietary saturate/unsaturate ratio as a determinant of adiposity. Med. Hypotheses 2010, 75, 14–16. [Google Scholar] [CrossRef]
- You, H.; Laychock, S.G. Atrial natriuretic peptide promotes pancreatic islet beta-cell growth and Akt/Foxo1a/cyclin D2 signaling. Endocrinology 2009, 150, 5455–5465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesely, D.L. Biotin enhances guanylate cyclase activity. Science 1982, 216, 1329–1330. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Kamiyama, M.; Kamiyama, S.; Horiuchi, K.; Ohinata, K.; Shirakawa, H.; Furukawa, Y.; Komai, M. Antihypertensive effect of biotin in stroke-prone spontaneously hypertensive rats. Br. J. Nutr. 2008, 99, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Reddi, A.; DeAngelis, B.; Frank, O.; Lasker, N.; Baker, H. Biotin supplementation improves glucose and insulin tolerances in genetically diabetic KK mice. Life Sci. 1988, 42, 1323–1330. [Google Scholar] [CrossRef]
- Zhang, H.; Osada, K.; Maebashi, M.; Ito, M.; Komai, M.; Furukawa, Y. A high biotin diet improves the impaired glucose tolerance of long-term spontaneously hyperglycemic rats with non-insulin-dependent diabetes mellitus. J. Nutr. Sci. Vitaminol. (Tokyo) 1996, 42, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Osada, K.; Sone, H.; Furukawa, Y. Biotin administration improves the impaired glucose tolerance of streptozotocin-induced diabetic Wistar rats. J. Nutr Sci Vitam. (Tokyo) 1997, 43, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y. Enhancement of glucose-induced insulin secretion and modification of glucose metabolism by biotin]. Nihon Rinsho. Jpn. J. Clin. Med. 1999, 57, 2261–2269. [Google Scholar]
- Sasaki, Y.; Sone, H.; Kamiyama, S.; Shimizu, M.; Shirakawa, H.; Kagawa, Y.; Komai, M.; Furukawa, Y. Administration of biotin prevents the development of insulin resistance in the skeletal muscles of Otsuka Long-Evans Tokushima fatty rats. Food Funct. 2012, 3, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Liu, Y.; Zhang, X.; Zhang, W.; Wang, Z. [Effects of biotin on blood glucose regulation in type 2 diabetes rat model]. Wei Sheng Yan Jiu(J. Hyg. Res.) 2015, 44, 185–189, 195. [Google Scholar]
- McCarty, M.F. cGMP may have trophic effects on beta cell function comparable to those of cAMP, implying a role for high-dose biotin in prevention/treatment of diabetes. Med. Hypotheses 2006, 66, 323–328. [Google Scholar] [CrossRef]
- Lazo de la Vega-Monroy, M.L.; Larrieta, E.; German, M.S.; Baez-Saldana, A.; Fernandez-Mejia, C. Effects of biotin supplementation in the diet on insulin secretion, islet gene expression, glucose homeostasis and beta-cell proportion. J. Nutr. Biochem. 2013, 24, 169–177. [Google Scholar] [CrossRef]
- Tixi-Verdugo, W.; Contreras-Ramos, J.; Sicilia-Argumedo, G.; German, M.S.; Fernandez-Mejia, C. Effects of biotin supplementation during the first week postweaning increases pancreatic islet area, beta-cell proportion, islets number, and beta-cell proliferation. J. Med. Food 2018, 21, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Lepore, E.; Lauretta, R.; Bianchini, M.; Mormando, M.; Di Lorenzo, C.; Unfer, V. Inositols depletion and resistance: Principal mechanisms and therapeutic strategies. Int. J. Mol. Sci. 2021, 22, 6796. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cheng, H.; Wang, X.; Zheng, L. Effectiveness and acceptability of myoinositol in prevention of gestational diabetes mellitus: A protocol for systematic review and meta-analysis. Medicine 2021, 100, e25673. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Wong, M.M.H.; Pang, S.S.H.; Lo, K.K.H. Dietary supplementation for gestational diabetes prevention and management: A meta-analysis of randomized controlled trials. Arch. Gynecol. Obstet. 2021, 303, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Miñambres, I.; Cuixart, G.; Gonçalves, A.; Corcoy, R. Effects of inositol on glucose homeostasis: Systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. 2019, 38, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, R.; Ostadmohammadi, V.; Lankarani, K.B.; Peymani, P.; Akbari, M.; Kolahdooz, F.; Asemi, Z. The effects of inositol supplementation on lipid profiles among patients with metabolic diseases: A systematic review and meta-analysis of randomized controlled trials. Lipids Health Dis. 2018, 17, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Yang, K. Effectiveness of myoinositol for polycystic ovary syndrome: A systematic review and meta-analysis. Endocrine 2018, 59, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Unfer, V.; Facchinetti, F.; Orrù, B.; Giordani, B.; Nestler, J. Myo-inositol effects in women with PCOS: A meta-analysis of randomized controlled trials. Endocr. Connect. 2017, 6, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Guo, S.; Miao, Z.; Li, Z.; Zhang, H. Myo-inositol lowers the risk of developing gestational diabetic mellitus in pregnancies: A systematic review and meta-analysis of randomized controlled trials with trial sequential analysis. J. Diabetes Complicat. 2018, 32, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Pintaudi, B.; Di Vieste, G.; Bonomo, M. The effectiveness of Myo-Inositol and D-chiro inositol treatment in type 2 diabetes. Int. J. Endocrinol. 2016, 2016, 9132052. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.A.; Keogh, J.B.; Clifton, P.M. Probiotics, prebiotics, synbiotics and insulin sensitivity. Nutr. Res. Rev. 2018, 31, 35–51. [Google Scholar] [CrossRef]
- Guo, J.; Tan, L.; Kong, L. Impact of dietary intake of resistant starch on obesity and associated metabolic profiles in human: A systematic review of the literature. Crit. Rev. Food Sci. Nutr. 2020, 61, 889–905. [Google Scholar] [CrossRef]
- Keenan, M.J.; Zhou, J.; Hegsted, M.; Pelkman, C.; Durham, H.A.; Coulon, D.B.; Martin, R.J. Role of resistant starch in improving gut health, adiposity, and insulin resistance. Adv. Nutr. 2015, 6, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Ríos, J.L.; Andújar, I.; Schinella, G.R.; Francini, F. Modulation of diabetes by natural products and medicinal plants via incretins. Planta Med. 2019, 85, 825–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Besten, G.; Gerding, A.; van Dijk, T.H.; Ciapaite, J.; Bleeker, A.; van Eunen, K.; Havinga, R.; Groen, A.K.; Reijngoud, D.J.; Bakker, B.M. Protection against the metabolic syndrome by guar gum-derived short-chain fatty acids depends on peroxisome proliferator-activated receptor γ and glucagon-like peptide-1. PLoS ONE 2015, 10, e0136364. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of dietary fiber consumption on insulin resistance and the prevention of type 2 diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondini, L.; Peyrat-Maillard, M.N.; Marsset-Baglieri, A.; Fromentin, G.; Durand, P.; Tomé, D.; Prost, M.; Berset, C. Bound ferulic acid from bran is more bioavailable than the free compound in rat. J. Agric. Food Chem. 2004, 52, 4338–4343. [Google Scholar] [CrossRef] [PubMed]
- Mateo, A.N.; Aura, A.M.; Selinheimo, E.; Mattila, I.; Poutanen, K.; Van Den Berg, R.; Havenaar, R.; Bast, A.; Haenen, G.R. Bioprocessing of wheat bran in whole wheat bread increases the bioavailability of phenolic acids in men and exerts antiinflammatory effects ex vivo. J. Nutr. 2011, 141, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Neacsu, M.; McMonagle, J.; Fletcher, R.J.; Hulshof, T.; Duncan, S.; Scobbie, L.; Duncan, G.J.; Cantlay, L.; Horgan, G.; de Roos, B.; et al. Availability and dose response of phytophenols from a wheat bran rich cereal product in healthy human volunteers. Mol. Nutr. Food Res. 2017, 61, 1600202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, A.L.; Michaelson, L.V.; Shewry, P.R.; Lovegrove, A.; Spencer, J.P.E. Increased bioavailability of phenolic acids and enhanced vascular function following intake of feruloyl esterase-processed high fibre bread: A randomized, controlled, single blind, crossover human intervention trial. Clin. Nutr. 2021, 40, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Hamedifard, Z.; Milajerdi, A.; Željko, R.; Taghizadeh, M.; Kolahdooz, F.; Asemi, Z. The effects of Spirulina on glycemic control and serum lipoproteins in patients with metabolic syndrome and related disorders: A systematic review and meta-analysis of randomized controlled trials. Phytother. Res. 2019, 33, 2609–2621. [Google Scholar] [CrossRef] [PubMed]
- Szulinska, M.; Gibas-Dorna, M.; Miller-Kasprzak, E.; Suliburska, J.; Miczke, A.; Walczak-Gałezewska, M.; Stelmach-Mardas, M.; Walkowiak, J.; Bogdanski, P. Spirulina maxima improves insulin sensitivity, lipid profile, and total antioxidant status in obese patients with well-treated hypertension: A randomized double-blind placebo-controlled study. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2473–2481. [Google Scholar]
- Ou, Y.; Lin, L.; Yang, X.; Pan, Q.; Cheng, X. Antidiabetic potential of phycocyanin: Effects on KKAy mice. Pharm. Biol. 2013, 51, 539–544. [Google Scholar] [CrossRef]
- Veronese, N.; Watutantrige-Fernando, S.; Luchini, C.; Solmi, M.; Sartore, G.; Sergi, G.; Manzato, E.; Barbagallo, M.; Maggi, S.; Stubbs, B. Effect of magnesium supplementation on glucose metabolism in people with or at risk of diabetes: A systematic review and meta-analysis of double-blind randomized controlled trials. Eur. J. Clin. Nutr. 2016, 70, 1354–1359. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Xun, P.; Tang, Q.; Cai, W.; He, K. Circulating magnesium levels and incidence of coronary heart diseases, hypertension, and type 2 diabetes mellitus: A meta-analysis of prospective cohort studies. Nutr. J. 2017, 16, 60. [Google Scholar] [CrossRef]
- Fang, X.; Wang, K.; Han, D.; He, X.; Wei, J.; Zhao, L.; Imam, M.U.; Ping, Z.; Li, Y.; Xu, Y.; et al. Dietary magnesium intake and the risk of cardiovascular disease, type 2 diabetes, and all-cause mortality: A dose–response meta-analysis of prospective cohort studies. BMC Med. 2016, 14, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wu, W.; Zheng, W.; Fang, X.; Chen, L.; Rink, L.; Min, J.; Wang, F. Zinc supplementation improves glycemic control for diabetes prevention and management: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2019, 110, 76–90. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.A.; Cheng, N.; Bryden, N.A.; Polansky, M.M.; Cheng, N.; Chi, J.; Feng, J. Elevated intakes of supplemental chromium improve glucose and insulin variables in individuals with type 2 diabetes. Diabetes 1997, 46, 1786–1791. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Chromium supplementation in human health, metabolic syndrome, and diabetes. Met. Ions Life Sci. 2019, 19. [Google Scholar] [CrossRef]
- Anderson, R.A. Chromium and polyphenols from cinnamon improve insulin sensitivity. Proc. Nutr. Soc. 2008, 67, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, C.; Lin, H.; Fang, X.; Wang, Y.; Jiang, Z.; Qu, Y.; Xiang, M.; Shen, Z.; Xin, L.; Lu, Y.; et al. Beneficial effects of cinnamon and its extracts in the management of cardiovascular diseases and diabetes. Food Funct. 2021, 12, 12194. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Li, K.; Wang, T.; Ji, J.; Wang, Y.; Chen, K.-X.; Jia, Q.; Li, Y.-M.; Wang, H.-Y. Procyanidin C1, a component of cinnamon extracts, is a potential insulin sensitizer that targets adipocytes. J. Agric. Food Chem. 2019, 67, 8839–8846. [Google Scholar] [CrossRef] [PubMed]
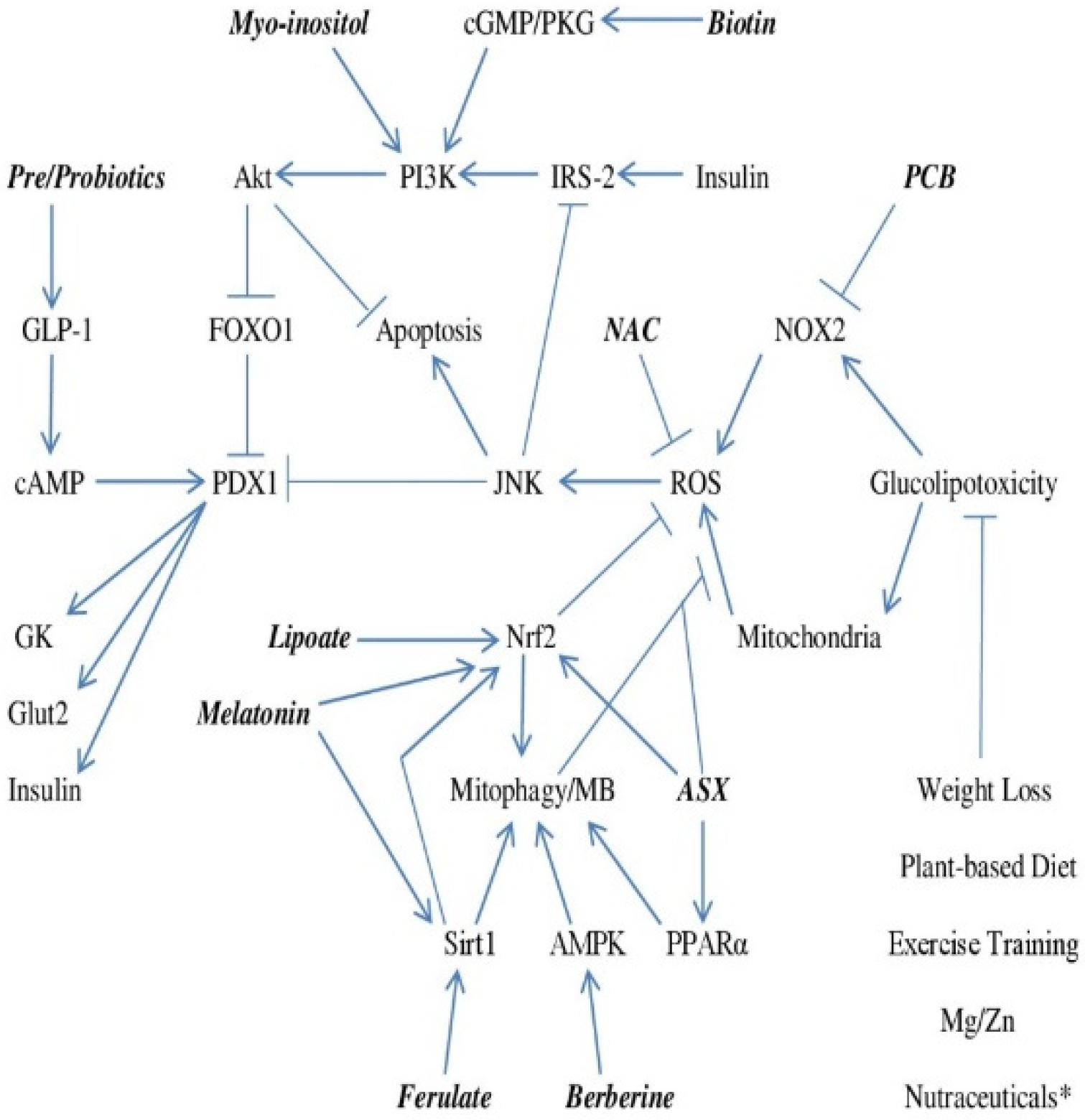
Figure 1.
Regulation of GSIS and Apoptosis in Beta Cells Exposed to Glucolipotoxicity–and How Nutraceuticals and Lifestyle May Support Effective Beta Cell Function and Survival. * Nutraceuticals including berberine, myo-inositol, biotin, PCB, cinnamon extract and chromium may diminish beta cell exposure to glucose and free fatty acids by aiding peripheral insulin sensitivity or decreasing hepatic glucose output.
Figure 1.
Regulation of GSIS and Apoptosis in Beta Cells Exposed to Glucolipotoxicity–and How Nutraceuticals and Lifestyle May Support Effective Beta Cell Function and Survival. * Nutraceuticals including berberine, myo-inositol, biotin, PCB, cinnamon extract and chromium may diminish beta cell exposure to glucose and free fatty acids by aiding peripheral insulin sensitivity or decreasing hepatic glucose output.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
McCarty, M.F.; DiNicolantonio, J.J. Maintaining Effective Beta Cell Function in the Face of Metabolic Syndrome-Associated Glucolipotoxicity—Nutraceutical Options. Healthcare 2022, 10, 3. https://doi.org/10.3390/healthcare10010003
AMA Style
McCarty MF, DiNicolantonio JJ. Maintaining Effective Beta Cell Function in the Face of Metabolic Syndrome-Associated Glucolipotoxicity—Nutraceutical Options. Healthcare. 2022; 10(1):3. https://doi.org/10.3390/healthcare10010003
Chicago/Turabian StyleMcCarty, Mark F., and James J. DiNicolantonio. 2022. "Maintaining Effective Beta Cell Function in the Face of Metabolic Syndrome-Associated Glucolipotoxicity—Nutraceutical Options" Healthcare 10, no. 1: 3. https://doi.org/10.3390/healthcare10010003
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.