
Pathophysiology-Based Management of Acute Heart Failure



Abstract
:1. Introduction
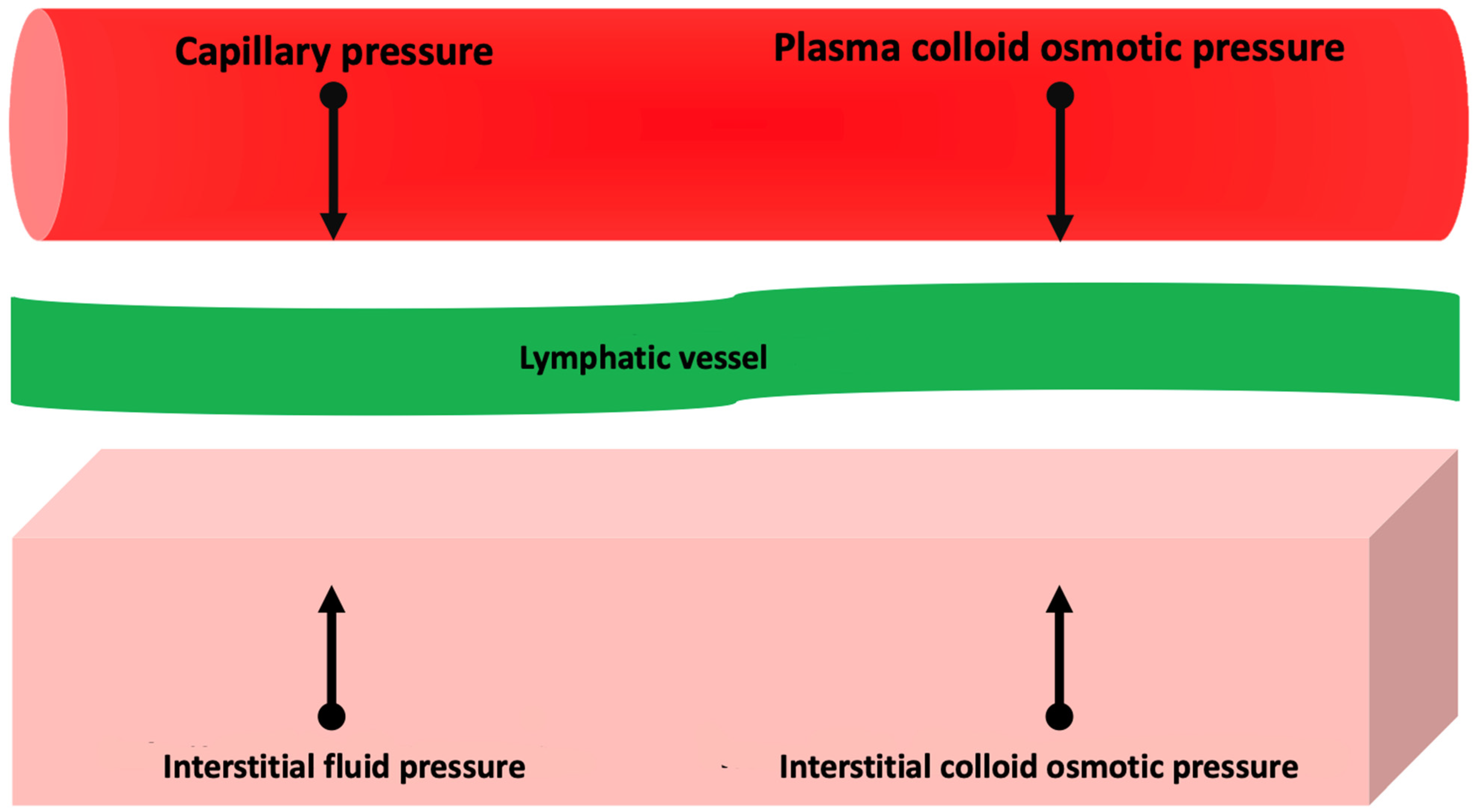
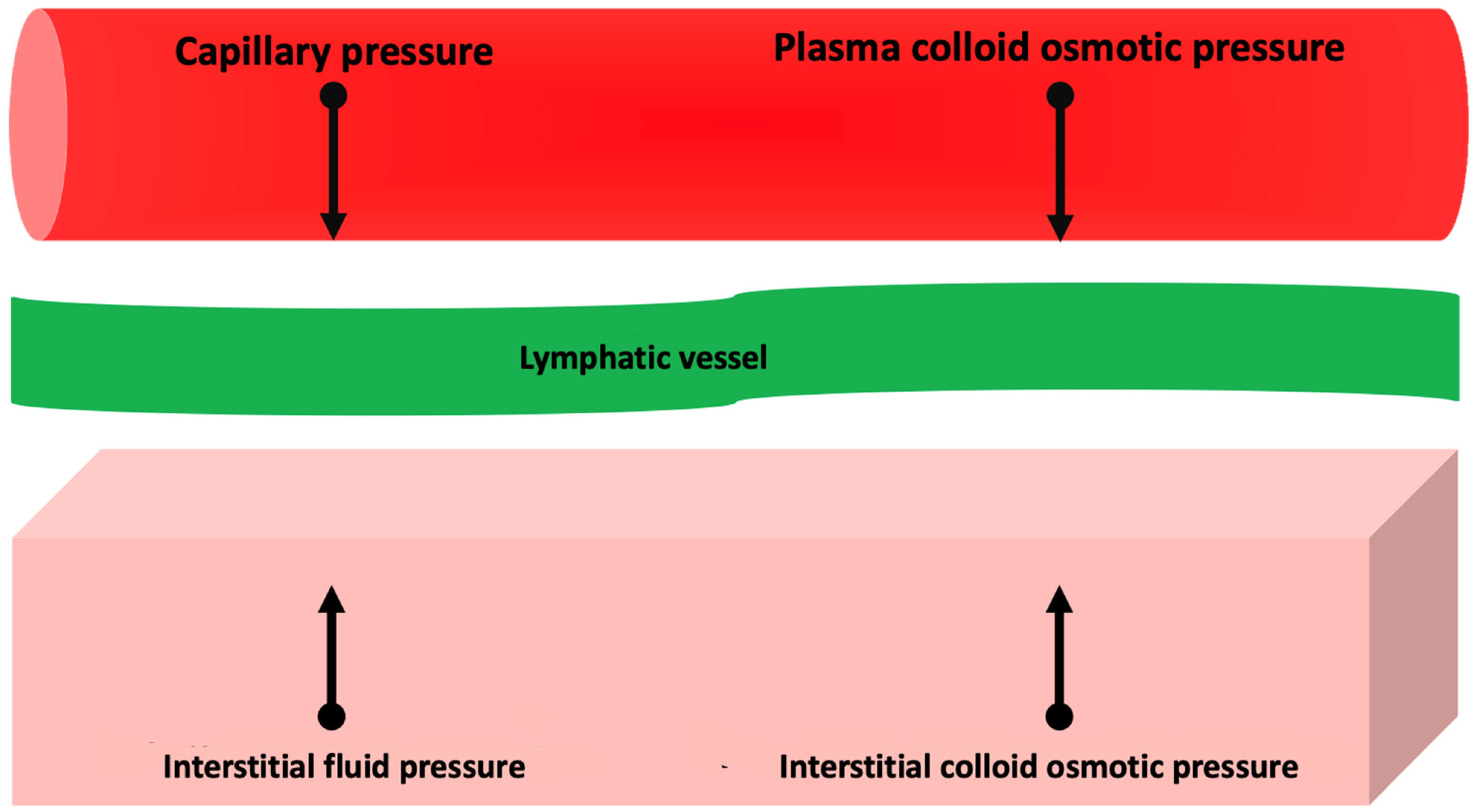
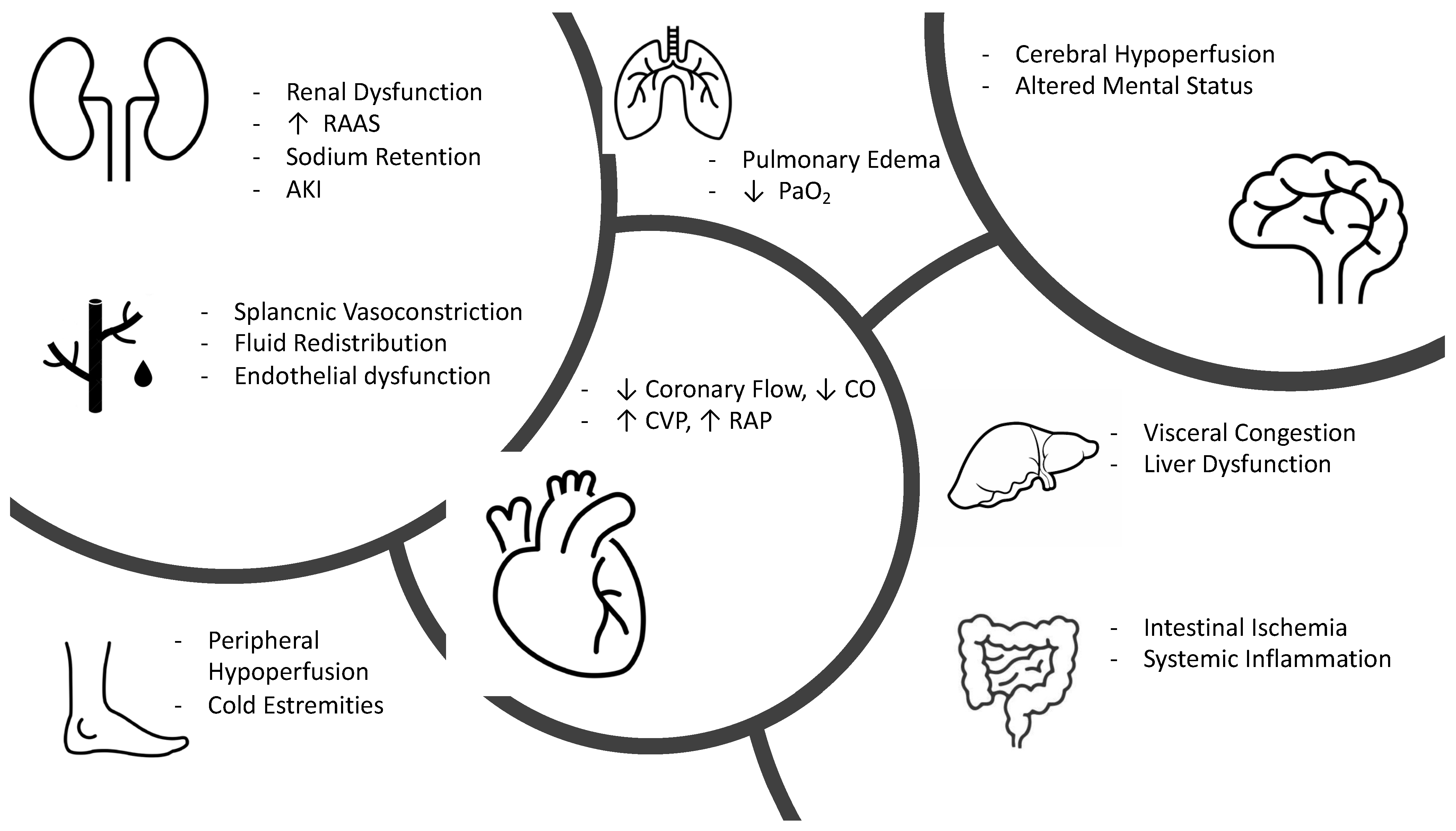
2. Pathophysiology of Congestion
2.1. Congestion Related to Cardiac Failure
2.2. Congestion Related to Vascular Failure
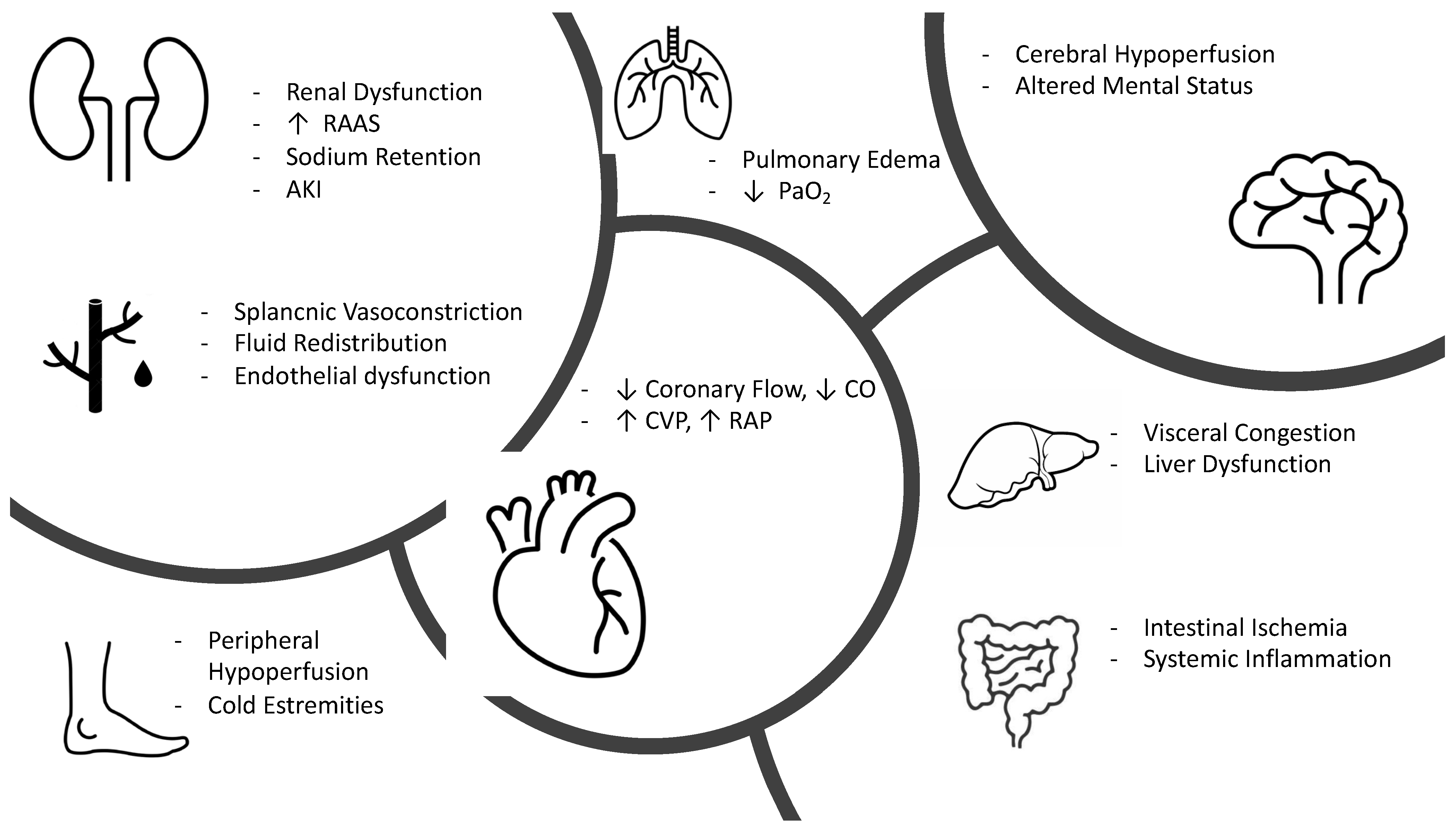
3. Clinical Pathophysiology of Hypoperfusion
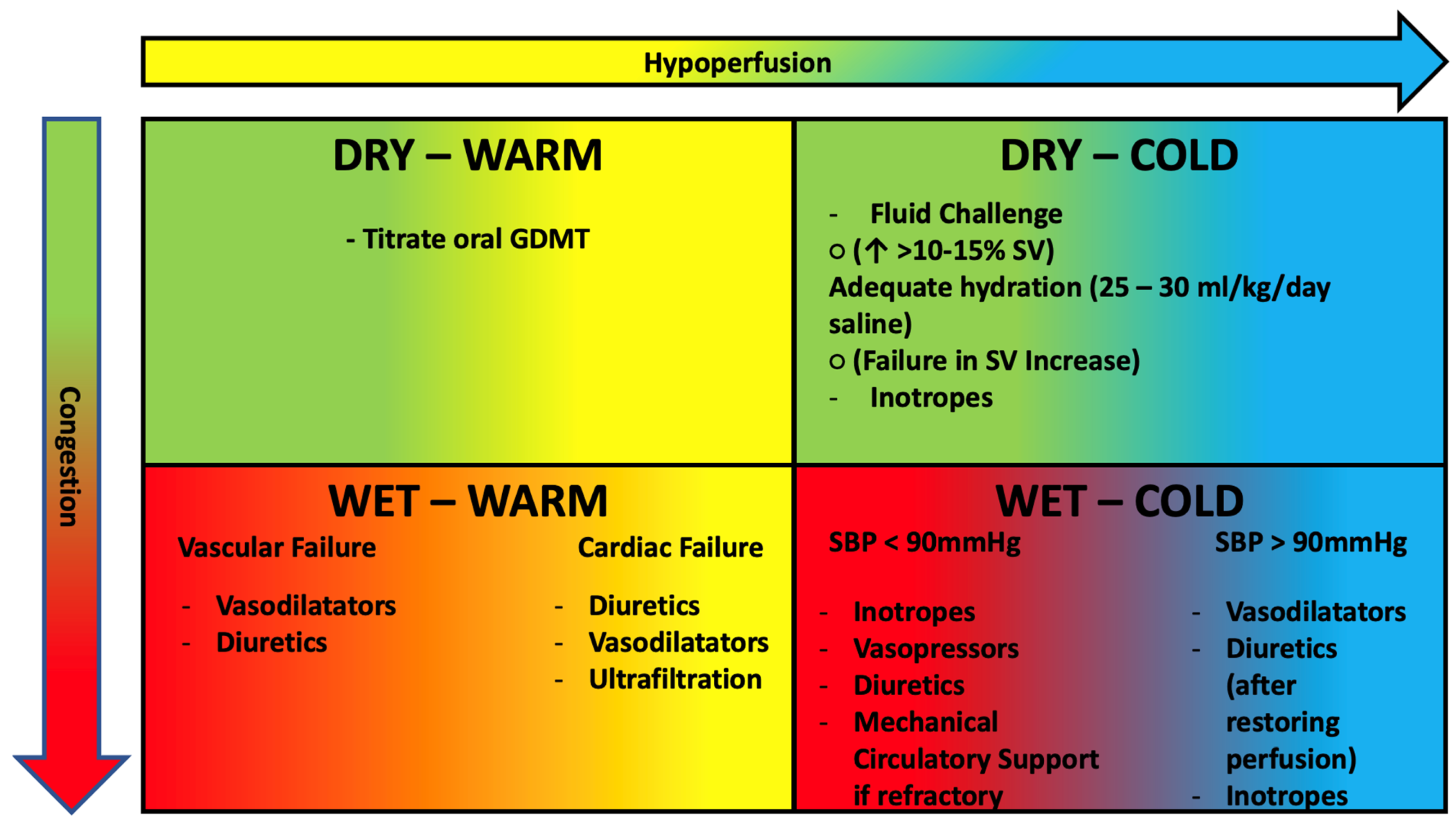
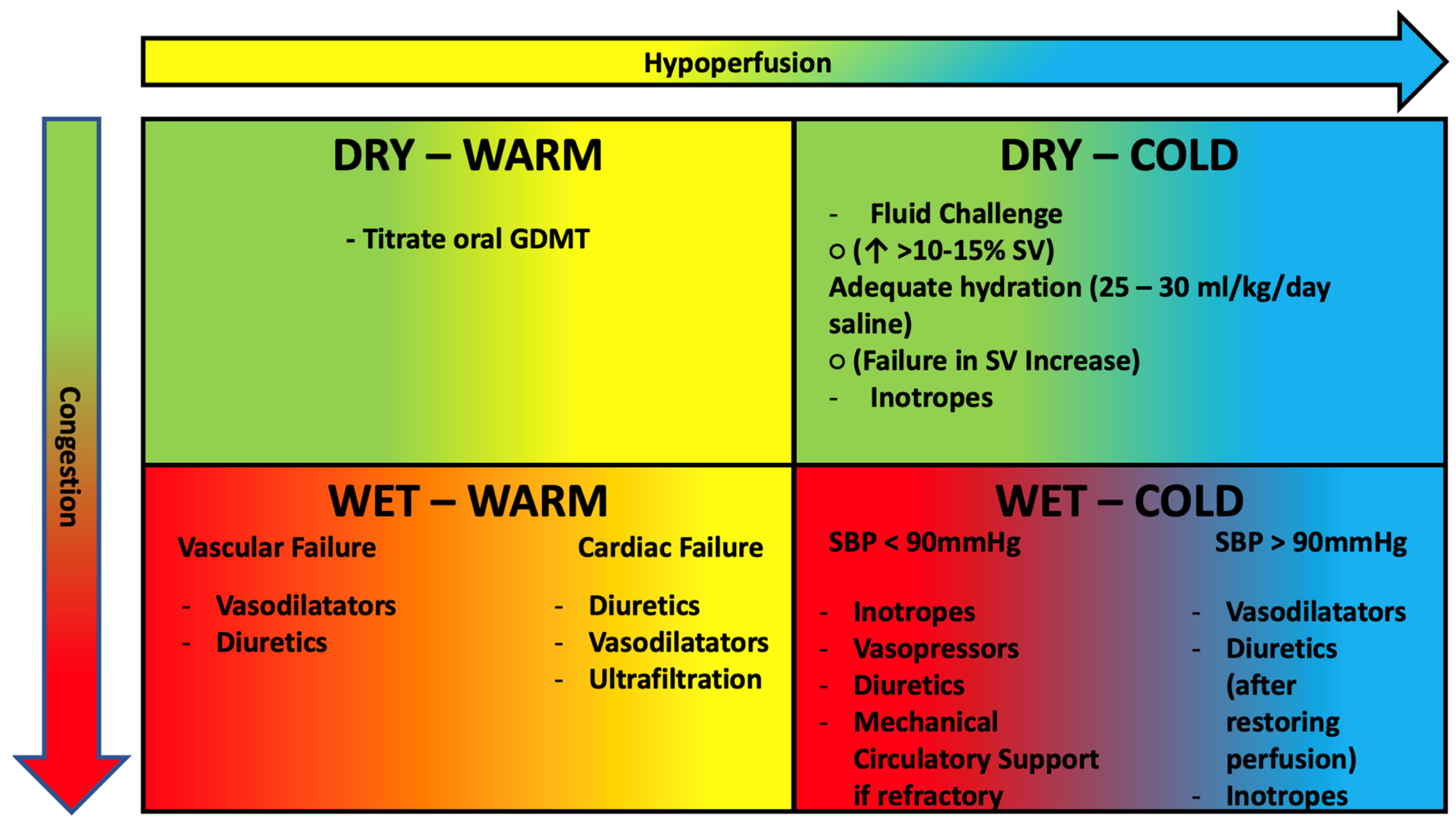
4. Pathophysiology-Based Management of AHF
- -
- SBP > 90 mmHg: the patient benefits from intravenous administration of diuretics and nitroderivatives. It is important to remember that in cases of hypoperfusion, the use of diuretics should be considered after perfusion is restored. If insufficient, the use of positive inotropic drugs such as levosimendan (particularly in patients treated with ß-blockers) or dobutamine should be considered [93].
- -
- -
- Oxygen therapy is not routinely indicated in patients with AHF but only in patients with documentation of hypoxemia (SPo2 < 90%, PaO2 < 60 mmHg); in such patients, the target to be achieved is a Pa02 between 60 and 90 mmHg [96] (generally corresponding to a SaO2 > 90% in chronic hypoxics and a SaO2 > 95 mmHg in other subjects), avoiding hyperoxia that could lead increase peripheral vascular resistance lowering cardiac output [97].
- -
- Disease-modifying drug therapy should be continued in cases of HF flare-ups, except in the patient with hemodynamic instability (symptomatic hypotension or bradycardia, cardiogenic shock), pre-renal acute renal failure, and severe hyperkalemia. In these cases, one should first try to reduce therapy without discontinuing it all together until the patient is stabilized.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [PubMed]
- Endorsed by the European Society of Intensive Care Medicine; Nieminen, M.S.; Böhm, M.; Cowie, M.; Drexler, H.; Filippatos, G.S.; Jondeau, G.; Hasin, Y.; Lopez-Sendon, J.; Mebazaa, A.; et al. Executive summary of the guidelines on the diagnosis and treatment of acute heart failure: The Task Force on Acute Heart Failure of the European Society of Cardiology. Eur. Heart J. 2005, 26, 384–416. [Google Scholar] [CrossRef] [Green Version]
- Nieminen, M.S.; Brutsaert, D.; Dickstein, K.; Drexler, H.; Follath, F.; Harjola, V.-P.; Hochadel, M.; Komajda, M.; Lassus, J.; Lopez-Sendon, J.L.; et al. EuroHeart Failure Survey II (EHFS II): A survey on hospitalized acute heart failure patients: Description of population. Eur. Heart J. 2006, 27, 2725–2736. [Google Scholar] [CrossRef] [Green Version]
- Chioncel, O.; Mebazaa, A.; Harjola, V.-P.; Coats, A.J.; Piepoli, M.F.; Crespo-Leiro, M.G.; Laroche, C.; Seferovic, P.M.; Anker, S.D.; Ferrari, R.; et al. Clinical phenotypes and outcome of patients hospitalized for acute heart failure: The ESC Heart Failure Long-Term Registry. Eur. J. Heart Fail. 2017, 19, 1242–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollenberg, S.M.; Warner Stevenson, L.; Ahmad, T.; Amin, V.J.; Bozkurt, B.; Butler, J.; Davis, L.L.; Drazner, M.H.; Kirkpatrick, J.N.; Peterson, P.N.; et al. 2019 ACC expert consensus decision pathway on risk assessment, management, and clinical trajectory of patients hospitalized with heart failure: A report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2019, 74, 1966–2011. [Google Scholar] [CrossRef]
- Sokolska, J.M.; Sokolski, M.; Zymliński, R.; Biegus, J.; Siwołowski, P.; Nawrocka-Millward, S.; Swoboda, K.; Gajewski, P.; Jankowska, E.A.; Banasiak, W.; et al. Distinct clinical phenotypes of congestion in acute heart failure: Characteristics, treatment response, and outcomes. ESC Heart Fail. 2020, 7, 3830–3840. [Google Scholar] [CrossRef] [PubMed]
- Boorsma, E.M.; Ter Maaten, J.M.; Damman, K.; Dinh, W.; Gustafsson, F.; Goldsmith, S.; Burkhoff, D.; Zannad, F.; Udelson, J.E.; Voors, A.A. Congestion in heart failure: A contemporary look at physiology, diagnosis and treatment. Nat. Rev. Cardiol. 2020, 17, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Viau, D.M.; Sala-Mercado, J.A.; Spranger, M.D.; O’Leary, D.S.; Levy, P.D. The pathophysiology of hypertensive acute heart failure. Heart 2015, 101, 1861–1867. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Evangelista, I.; Nuti, R. Congestion occurrence and evaluation in acute heart failure scenario: Time to reconsider different pathways of volume overload. Heart Fail. Rev. 2019, 25, 119–131. [Google Scholar] [CrossRef]
- Fudim, M.; Hernandez, A.F.; Felker, G.M. Role of Volume Redistribution in the Congestion of Heart Failure. J. Am. Heart Assoc. 2017, 6, e006817. [Google Scholar] [CrossRef]
- Schrier, R.W.; Abraham, W.T. Hormones and Hemodynamics in Heart Failure. N. Engl. J. Med. 1999, 341, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W.; Berl, T.; Anderson, R.J. Osmotic and nonosmotic control of vasopressin release. Am. J. Physiol. Physiol. 1979, 236, F321–F332. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Li, J. The role of the renin-angiotensin-aldosterone system in heart failure. J. Renin-Angiotensin-Aldosterone Syst. 2004, 5, S7–S10. [Google Scholar] [CrossRef]
- Weber, K.T. Aldosterone in congestive heart failure. N. Engl. J. Med. 2001, 345, 1689–1697. [Google Scholar] [CrossRef]
- Malpas, S.C. Sympathetic Nervous System Overactivity and Its Role in the Development of Cardiovascular Disease. Physiol. Rev. 2010, 90, 513–557. [Google Scholar] [CrossRef] [PubMed]
- Charloux, A.; Piquard, F.; Doutreleau, S.; Brandenberger, G.; Geny, B. Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur. J. Clin. Investig. 2003, 33, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, M.; Parissis, J.T.; Akiyama, E.; Mebazaa, A. Understanding acute heart failure: Pathophysiology and diagnosis. Eur. Heart J. Suppl. 2016, 18, G11–G18. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.L.; Cleland, J.G.F. Causes and treatment of oedema in patients with heart failure. Nat. Rev. Cardiol. 2013, 10, 156–170. [Google Scholar] [CrossRef]
- Nijst, P.; Verbrugge, F.H.; Grieten, L.; Dupont, M.; Steels, P.; Tang, W.W.; Mullens, W. The Pathophysiological Role of Interstitial Sodium in Heart Failure. J. Am. Coll. Cardiol. 2015, 65, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Mullens, W.; Abrahams, Z.; Skouri, H.N.; Francis, G.S.; Taylor, D.O.; Starling, R.C.; Paganini, E.; Tang, W.W. Elevated intra-abdominal pressure in acute decompensated heart failure: A potential contributor to worsening renal function? J. Am. Coll. Cardiol. 2008, 51, 300–306. [Google Scholar] [CrossRef]
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart Failure and Liver Disease: Cardiohepatic Interactions. JACC Heart Fail. 2019, 7, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Nijst, P. Cardiac Output and Renal Dysfunction: Definitely More Than Impaired Flow. J. Am. Coll. Cardiol. 2016, 67, 2209–2212. [Google Scholar] [CrossRef]
- Hanberg, J.S.; Sury, K.; Wilson, F.P.; Brisco, M.A.; Ahmad, T.; ter Maaten, J.M.; Broughton, J.S.; Assefa, M.; Tang, W.W.; Parikh, C.R.; et al. Reduced Cardiac Index Is Not the Dominant Driver of Renal Dysfunction in Heart Failure. J. Am. Coll. Cardiol. 2016, 67, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- èMullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 2009, 53, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbrugge, F.H.; Dupont, M.; Steels, P.; Grieten, L.; Malbrain, M.; Wilson Tang, W.H.; Mullens, W. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J. Am. Coll. Cardiol. 2013, 62, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Taylor, D.O.; Starling, R.C.; Tang, W.W. Prompt Reduction in Intra-Abdominal Pressure Following Large-Volume Mechanical Fluid Removal Improves Renal Insufficiency in Refractory Decompensated Heart Failure. J. Card. Fail. 2008, 14, 508–514. [Google Scholar] [CrossRef]
- Costanzo, M.R.; Guglin, M.E.; Saltzberg, M.T.; Jessup, M.L.; Bart, B.A.; Teerlink, J.R.; Jaski, B.E.; Fang, J.C.; Feller, E.D.; Haas, G.J.; et al. Ultrafiltration Versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Heart Failure. J. Am. Coll. Cardiol. 2007, 49, 675–683. [Google Scholar] [CrossRef]
- Bart, B.A.; Goldsmith, S.R.; Lee, K.L.; Givertz, M.M.; O’Connor, C.M.; Bull, D.A.; Redfield, M.M.; Deswal, A.; Rouleau, J.L.; LeWinter, M.M.; et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N. Engl. J. Med. 2012, 367, 2296–2304. [Google Scholar] [CrossRef] [Green Version]
- Felker, G.M.; Lee, K.L.; Bull, D.A.; Redfield, M.M.; Stevenson, L.W.; Goldsmith, S.R.; LeWinter, M.M.; Deswal, A.; Rouleau, J.L.; Ofili, E.O.; et al. Diuretic strategies in patients with acute decompensated heart failure. N. Engl. J. Med. 2011, 64, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Mullens, W.; Damman, K.; Harjola, V.-P.; Mebazaa, A.; Rocca, H.-P.B.-L.; Martens, P.; Testani, J.M.; Tang, W.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef]
- Mullens, W.; Verbrugge, F.H.; Nijst, P.; Tang, W.H.W. Renal sodium avidity in heart failure: From pathophysiology to treatment strategies. Eur. Heart J. 2017, 38, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Møller, S.; Bernardi, M. Interactions of the heart and the liver. Eur. Heart J. 2013, 34, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Arques, S.; Ambrosi, P. Human Serum Albumin in the Clinical Syndrome of Heart Failure. J. Card. Fail. 2011, 17, 451–458. [Google Scholar] [CrossRef]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Colombo, P.C.; Banchs, J.E.; Celaj, S.; Talreja, A.; Lachmann, J.; Malla, S.; DuBois, N.B.; Ashton, A.W.; Latif, F.; Jorde, U.P.; et al. Endothelial Cell Activation in Patients With Decompensated Heart Failure. Circulation 2005, 111, 58–62. [Google Scholar] [CrossRef] [Green Version]
- Colombo, P.C.; Onat, D.; Sabbah, H.N. Acute heart failure as “acute endothelitis”—Interaction of fluid overload and endothelial dysfunction. Eur. J. Heart Fail. 2008, 10, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, G.P.; Song, H.; Tressel, S.L.; Hwang, J.; Dikalov, S.; Smith, D.A.; Boyd, N.L.; Platt, M.O.; Lassègue, B.; Griendling, K.K.; et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ. Res. 2004, 95, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M.; Naruse, K.; Komatsu, S.; Kobayashi, S.; Nagino, M.; Nimura, Y.; Sokabe, M. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: Clinical and experimental studies. J. Hepatol. 2002, 37, 240–246. [Google Scholar] [CrossRef]
- Wang, B.-W.; Chang, H.; Lin, S.; Kuan, P.; Shyu, K.-G. Induction of matrix metalloproteinases-14 and -2 by cyclical mechanical stretch is mediated by tumor necrosis factor-alpha in cultured human umbilical vein endothelial cells. Cardiovasc. Res. 2003, 59, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Colombo, P.C.; Rastogi, S.; Onat, D.; Zacà, V.; Gupta, R.C.; Jorde, U.P.; Sabbah, H.N. Activation of endothelial cells in conduit veins of dogs with heart failure and veins of normal dogs after vascular stretch by acute volume loading. J. Card. Fail. 2009, 15, 457–463. [Google Scholar] [CrossRef]
- Colombo, P.C.; Onat, D.; Harxhi, A.; Demmer, R.T.; Hayashi, Y.; Jelic, S.; LeJemtel, T.H.; Bucciarelli, L.; Kebschull, M.; Papapanou, P.N.; et al. Peripheral venous congestion causes inflammation, neurohormonal, and endothelial cell activation. Eur. Heart J. 2013, 35, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganda, A.; Onat, D.; Demmer, R.T.; Wan, E.; Vittorio, T.J.; Sabbah, H.N.; Colombo, P.C. Venous congestion and endothelial cell activation in acute decompensated heart failure. Curr. Heart Fail. Rep. 2010, 7, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, L.W. Are hemodynamic goals viable in tailoring heart failure therapy? Hemodynamic goals are relevant. Circulation 2006, 113, 1020–1027. [Google Scholar] [CrossRef] [Green Version]
- Drexler, H. Endothelium as a Therapeutic Target in Heart Failure. Circulation 1998, 98, 2652–2655. [Google Scholar] [CrossRef] [Green Version]
- Gimbrone, M.A., Jr.; Nagel, T.; Topper, J.N. Biomechanical activation: An emerging paradigm in endothelial adhesion biology. J. Clin. Investig. 1997, 99, 1809–1813. [Google Scholar] [CrossRef] [PubMed]
- Luxán, G.; Dimmeler, S. The vasculature: A therapeutic target in heart failure? Cardiovasc. Res. 2022, 118, 53–64. [Google Scholar] [CrossRef]
- Whaley-Connell, A.T.; Chowdhury, N.A.; Hayden, M.R.; Stump, C.S.; Habibi, J.; Wiedmeyer, C.E.; Gallagher, P.E.; Tallant, E.A.; Cooper, S.A.; Link, C.D.; et al. Oxidative stress and glomerular filtration barrier injury: Role of the renin-angiotensin system in the Ren2 transgenic rat. Am. J. Physiol. Physiol. 2006, 291, F1308–F1314. [Google Scholar] [CrossRef]
- Scagliola, R.; Brunelli, C. Venous Congestion and Systemic Hypoperfusion in Cardiorenal Syndrome: Two Sides of the Same Coin. Rev. Cardiovasc. Med. 2022, 23, 111. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.I.; Wang, Y.; Concato, J.; Gill, T.; Krumholz, H.M. Patterns of Weight Change Preceding Hospitalization for Heart Failure. Circulation 2007, 116, 1549–1554. [Google Scholar] [CrossRef]
- Cotter, G.; Metra, M.; Milo-Cotter, O.; Dittrich, H.C.; Gheorghiade, M. Fluid overload in acute heart failure—Re-distribution and other mechanisms beyond fluid accumulation. Eur. J. Heart Fail. 2008, 10, 165–169. [Google Scholar] [CrossRef]
- Fallick, C.; Sobotka, P.A.; Dunlap, M.E. Sympathetically mediated changes in capacitance: Redistribution of the venous reservoir as a cause of decompensation. Circ. Heart Fail. 2011, 4, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Balmain, S.; Padmanabhan, N.; Ferrell, W.R.; Morton, J.J.; Mcmurray, J. Differences in arterial compliance, microvascular function and venous capacitance between patients with heart failure and either preserved or reduced left ventricular systolic function. Eur. J. Heart Fail. 2007, 9, 865–871. [Google Scholar] [CrossRef]
- Gelman, S.; Mushlin, P.S. Catecholamine-induced Changes in the Splanchnic Circulation Affecting Systemic Hemodynamics. Anesthesiology 2004, 100, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Bishu, K.; Redfield, M.M. Acute Heart Failure with Preserved Ejection Fraction: Unique Patient Characteristics and Targets for Therapy. Curr. Heart Fail. Rep. 2013, 10, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Mentz, R.J.; Kjeldsen, K.; Rossi, G.P.; Voors, A.A.; Cleland, J.G.; Anker, S.D.; Gheorghiade, M.; Fiuzat, M.; Rossignol, P.; Zannad, F.; et al. Decongestion in acute heart failure. Eur. J. Heart Fail. 2014, 16, 471–482. [Google Scholar] [CrossRef]
- Chioncel, O.; Mebazaa, A.; Maggioni, A.P.; Harjola, V.P.; Rosano, G.; Laroche, C.; Piepoli, M.F.; Crespo-Leiro, M.G.; Lainscak, M.; Ponikowski, P.; et al. Acute heart failure congestion and perfusion status—Impact of the clinical classification on in-hospital and long-term outcomes; insights from the ESC-EORP-HFA Heart Failure Long-Term Registry. Eur. J. Heart Fail. 2019, 21, 1338–1352. [Google Scholar] [CrossRef]
- Harjola, V.-P.; Mullens, W.; Banaszewski, M.; Bauersachs, J.; Brunner-La Rocca, H.P.; Chioncel, O.; Collins, S.P.; Doehner, W.; Filippatos, G.S.; Flammer, A.J.; et al. Organ dysfunction, injury and failure in acute heart failure: From pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2017, 19, 821–836. [Google Scholar] [CrossRef] [Green Version]
- Vahdatpour, C.; Collins, D.; Goldberg, S. Cardiogenic Shock. J. Am. Heart Assoc. 2019, 8, e011991. [Google Scholar] [CrossRef] [Green Version]
- Nagatomo, Y.; Tang, W.H.W. Intersections between Microbiome and Heart Failure: Revisiting the Gut Hypothesis. J. Card. Fail. 2015, 21, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Chioncel, O.; Parissis, J.; Mebazaa, A.; Thiele, H.; Desch, S.; Bauersachs, J.; Harjola, V.; Antohi, E.; Arrigo, M.; Ben Gal, T.; et al. Epidemiology, pathophysiology and contemporary management of cardiogenic shock—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 1315–1341. [Google Scholar] [CrossRef]
- Ghionzoli, N.; Sciaccaluga, C.; Mandoli, G.; Vergaro, G.; Gentile, F.; D’Ascenzi, F.; Mondillo, S.; Emdin, M.; Valente, S.; Cameli, M. Cardiogenic shock and acute kidney injury: The rule rather than the exception. Heart Fail. Rev. 2020, 26, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, B.A. Low-Flow Acute Kidney Injury: The Pathophysiology of Prerenal Azotemia, Abdominal Compartment Syndrome, and Obstructive Uropathy. Clin. J. Am. Soc. Nephrol. 2022, 17, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Ebert, E.C. Hypoxic liver injury. Mayo Clin. Proc. 2006, 81, 1232–1236. [Google Scholar] [CrossRef] [Green Version]
- Knauf, H.; Mutschler, E. Sequential Nephron Blockade Breaks Resistance to Diuretics in Edematous States. J. Cardiovasc. Pharmacol. 1997, 29, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gracia, J.; Demissei, B.G.; Ter Maaten, J.M.; Cleland, J.G.; O’Connor, C.M.; Metra, M.; Ponikowski, P.; Teerlink, J.R.; Cotter, G.; Davison, B.A.; et al. Prevalence, predictors and clinical outcome of residual congestion in acute decompensated heart failure. Int. J. Cardiol. 2018, 258, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Coiro, S.; Rossignol, P.; Ambrosio, G.; Carluccio, E.; Alunni, G.; Murrone, A.; Tritto, I.; Zannad, F.; Girerd, N. Prognostic value of residual pulmonary congestion at discharge assessed by lung ultrasound imaging in heart failure. Eur. J. Heart Fail. 2015, 17, 1172–1181. [Google Scholar] [CrossRef]
- Lala, A.; McNulty, S.E.; Mentz, R.J.; Dunlay, S.M.; Vader, J.M.; AbouEzzeddine, O.F.; DeVore, A.D.; Khazanie, P.; Redfield, M.M.; Goldsmith, S.R.; et al. Relief and Recurrence of Congestion During and After Hospitalization for Acute Heart Failure: Insights From Diuretic Optimization Strategy Evaluation in Acute Decompensated Heart Failure (DOSE-AHF) and Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARESS-HF). Circ. Heart Fail. 2015, 8, 741–748. [Google Scholar]
- O’Connor, C.M.; Stough, W.G.; Gallup, D.S.; Hasselblad, V.; Gheorghiade, M. Demographics, Clinical Characteristics, and Outcomes of Patients Hospitalized for Decompensated Heart Failure: Observations from the IMPACT-HF Registry. J. Card. Fail. 2005, 11, 200–205. [Google Scholar] [CrossRef]
- Rivas-Lasarte, M.; Maestro, A.; Fernández-Martínez, J.; López-López, L.; Solé-González, E.; Vives-Borrás, M.; Montero, S.; Mesado, N.; Pirla, M.J.; Mirabet, S.; et al. Prevalence and prognostic impact of subclinical pulmonary congestion at discharge in patients with acute heart failure. ESC Heart Fail. 2020, 21, 2621–2628. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Greene, S.J.; Fonarow, G.; Voors, A.A.; Butler, J.; Gheorghiade, M. Hemoconcentration-guided Diuresis in Heart Failure. Am. J. Med. 2014, 127, 1154–1159. [Google Scholar] [CrossRef]
- Januzzi, J.L.; van Kimmenade, R.; Lainchbury, J.; Bayes-Genis, A.; Ordonez-Llanos, J.; Santalo-Bel, M.; Pinto, Y.M.; Richards, M. NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart failure: An international pooled analysis of 1256 patients: The International Collaborative of NT-proBNP Study. Eur. Heart J. 2006, 27, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; McDonald, K.; de Boer, R.A.; Maisel, A.; Cleland, J.G.; Kozhuharov, N.; Coats, A.J.; Metra, M.; Mebazaa, A.; Ruschitzka, F.; et al. Heart Failure Association of the European Society of Cardiology practical guidance on the use of natriuretic peptide concentrations. Eur. J. Heart Fail. 2019, 21, 715–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, S.U.; Mueller, T.; Januzzi, J.L., Jr. Characteristics of the Novel Interleukin Family Biomarker ST2 in Patients with Acute Heart Failure. J. Am. Coll. Cardiol. 2008, 52, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Januzzi, J.L., Jr.; Peacock, W.F.; Maisel, A.S.; Chae, C.U.; Jesse, R.L.; Baggish, A.L.; O’Donoghue, M.; Sakhuja, R.; Chen, A.A.; van Kimmenade, R.R.; et al. Measurement of the interleukin family member ST2 in patients with acute dyspnea: Results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J. Am. Coll. Cardiol. 2007, 50, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Mueller, T.; Dieplinger, B.; Gegenhuber, A.; Poelz, W.; Pacher, R.; Haltmayer, M. Increased Plasma Concentrations of Soluble ST2 are Predictive for 1-Year Mortality in Patients with Acute Destabilized Heart Failure. Clin. Chem. 2008, 54, 752–756. [Google Scholar] [CrossRef] [Green Version]
- Aimo, A.; Vergaro, G.; Ripoli, A.; Bayes-Genis, A.; Figal, D.A.P.; de Boer, R.A.; Lassus, J.; Mebazaa, A.; Gayat, E.; Breidthardt, T.; et al. Meta-Analysis of Soluble Suppression of Tumorigenicity-2 and Prognosis in Acute Heart Failure. JACC Heart Fail. 2017, 5, 287–296. [Google Scholar] [CrossRef]
- Wollert, K.C.; Kempf, T.; Wallentin, L. Growth Differentiation Factor 15 as a Biomarker in Cardiovascular Disease. Clin. Chem. 2017, 63, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Demissei, B.G.; Cotter, G.; Prescott, M.F.; Felker, G.M.; Filippatos, G.; Greenberg, B.H.; Pang, P.S.; Ponikowski, P.; Severin, T.M.; Wang, Y.; et al. A multimarker multi-time point-based risk stratification strategy in acute heart failure: Results from the RELAX-AHF trial. Eur. J. Heart Fail. 2017, 19, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Bouabdallaoui, N.; Claggett, B.; Zile, M.; McMurray, J.J.; O’Meara, E.; Packer, M.; Prescott, M.F.; Swedberg, K.; Solomon, S.D.; Rouleau, J.L.; et al. Growth differentiation factor-15 is not modified by sacubitril/valsartan and is an independent marker of risk in patients with heart failure and reduced ejection fraction: The PARADIGM-HF trial. Eur. J. Heart Fail. 2018, 20, 1701–1709. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, A.; Florescu, R.; Kneizeh, K.; Cornelissen, C.; Brandenburg, V.; Liehn, E.; Schuh, A. Intact fibroblast growth factor 23 levels and outcome prediction in patients with acute heart failure. Sci. Rep. 2021, 11, 15507. [Google Scholar] [CrossRef]
- Pöss, J.; Mahfoud, F.; Seiler, S.; Heine, G.H.; Fliser, D.; Böhm, M.; Link, A. FGF-23 is associated with increased disease severity and early mortality in cardiogenic shock. Eur. Heart J. Acute Cardiovasc. Care 2013, 2, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Elkayam, U.; Janmohamed, M.; Habib, M.; Hatamizadeh, P. Vasodilators in the management of acute heart failure. Crit. Care Med. 2008, 36, S95–S105. [Google Scholar] [CrossRef] [PubMed]
- Alzahri, M.S.; Rohra, A.; Peacock, W.F. Nitrates as a Treatment of Acute Heart Failure. Card. Fail. Rev. 2016, 2, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Piper, S.; McDonagh, T. The Role of Intravenous Vasodilators in Acute Heart Failure Management. Eur. J. Heart Fail. 2014, 16, 827–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo-Leiro, M.G.; Anker, S.D.; Maggioni, A.P.; Coats, A.J.; Filippatos, G.; Ruschitzka, F.; Ferrari, R.; Piepoli, M.F.; Delgado Jimenez, J.F.; Metra, M.; et al. European Society of Cardiology Heart Failure Long-Term Registry (ESC-HF-LT): 1-year follow-up outcomes and differences across regions. Eur. J. Heart Fail. 2016, 18, 613–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonarow, G.C.; Stough, W.G.; Abraham, W.T.; Albert, N.M.; Gheorghiade, M.; Greenberg, B.H.; O’Connor, C.M.; Sun, J.L.; Yancy, C.W.; Young, J.B.; et al. Characteristics, treatments, and outcomes of patients with preserved systolic function hospitalized for heart failure: A report from the OPTIMIZE-HF Registry. J. Am. Coll. Cardiol. 2007, 50, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, E.J.; Morrow, D.A.; DeVore, A.D.; Duffy, C.I.; Ambrosy, A.P.; McCague, K.; Rocha, R.; Braunwald, E. Angiotensin–Neprilysin Inhibition in Acute Decompensated Heart Failure. N. Engl. J. Med. 2019, 380, 539–548. [Google Scholar] [CrossRef]
- Wachter, R.; Senni, M.; Belohlavek, J.; Straburzynska-Migaj, E.; Witte, K.K.; Kobalava, Z.; Fonseca, C.; Goncalvesova, E.; Cavusoglu, Y.; Fernandez, A.; et al. Initiation of sacubitril/valsartan in haemodynamically stabilised heart failure patients in hospital or early after discharge: Primary results of the randomised transition study. Eur. J. Heart Fail. 2019, 21, 998–1007. [Google Scholar] [CrossRef] [Green Version]
- Voors, A.A.; Angermann, C.E.; Teerlink, J.R.; Collins, S.P.; Kosiborod, M.; Biegus, J.; Ferreira, J.P.; Nassif, M.E.; Psotka, M.A.; Tromp, J.; et al. The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: A multinational randomized trial. Nat. Med. 2022, 28, 568–574. [Google Scholar] [CrossRef]
- Miller, R.J.; Howlett, J.G.; Fine, N.M. A Novel Approach to Medical Management of Heart Failure with Reduced Ejection Fraction. Can. J. Cardiol. 2021, 37, 632–643. [Google Scholar] [CrossRef]
- Prins, K.; Neill, J.; Tyler, J.; Eckman, P.; Duval, S. Effects of Beta-Blocker Withdrawal in Acute Decompensated Heart Failure: A Systematic Review and Meta-Analysis. JACC Heart Fail. 2015, 3, 647–653. [Google Scholar] [CrossRef]
- Takagi, K.; Kimmoun, A.; Sato, N.; Mebazaa, A. Management of Acute Heart Failure during an Early Phase. Int. J. Heart Fail. 2020, 2, 91–110. [Google Scholar] [CrossRef]
- Mebazaa, A.; Nieminen, M.S.; Filippatos, G.S.; Cleland, J.G.; Salon, J.E.; Thakkar, R.; Padley, R.J.; Huang, B.; Cohen-Solal, A. Levosimendan vs. dobutamine: Outcomes for acute heart failure patients on beta-blockers in survive. Eur. J. Heart Fail. 2009, 11, 304–311. [Google Scholar] [CrossRef]
- van Diepen, S.; Katz, J.N.; Albert, N.M.; Henry, T.D.; Jacobs, A.K.; Kapur, N.K.; Kilic, A.; Menon, V.; Ohman, E.M.; Sweitzer, N.K.; et al. Contemporary Management of Cardiogenic Shock: A Scientific Statement From the American Heart Association. Circulation 2017, 136, e232–e268. [Google Scholar] [CrossRef]
- Cecconi, M.; Parsons, A.K.; Rhodes, A. What is a fluid challenge? Curr. Opin. Crit. Care 2011, 17, 290–295. [Google Scholar] [CrossRef]
- Girardis, M.; Busani, S.; Damiani, E.; Donati, A.; Rinaldi, L.; Marudi, A.; Morelli, A.; Antonelli, M.; Singer, M. Effect of Conservative vs Conventional Oxygen Therapy on Mortality Among Patients in an Intensive Care Unit: The Oxygen-ICU Randomized Clinical Trial. JAMA 2016, 316, 1583–1589. [Google Scholar] [CrossRef]
- Reinhart, K.; Bloos, F.; König, F.; Bredle, D.; Hannemann, L. Reversible Decrease of Oxygen Consumption by Hyperoxia. Chest 1991, 99, 690–694. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Parameters | Peripheral Congestion | Pulmonary Congestion | Notes |
|---|---|---|---|
| JVP > 8 cm | Yes | No | Difficult to assess (particularly in obese patients) |
| Hepatomegaly | Yes | No | Also due to non HF causes |
| Bilateral legs edema | Yes | No | Also due to non HF causes |
| Rales with base-apex gradient | No | Yes | Also due to non HF causes |
| Bendopnea | Yes | Yes | Also due to non HF causes |
| Inferior vena cava collapse < 50% with sniff | Yes | No | Difficult to assess in positive pressure ventilated patients |
| Deceleration time < 130 msec | No | Yes | Unassessable in tachycardic patients and in patients with PR interval > 200 msec |
| Lateral E/e’ > 12 | No | Yes | Inaccurate in patients with advanced heart failure |
| B lines on lung ultrasound | No | Yes | Also due to non HF causes |
| NT-proBNP | Yes | No | Elevation also due to non HF causes (caveats), less accurate in obese patients |
| Measurement | Mild | Moderate | Severe |
|---|---|---|---|
| Orthopnea | Absent | Moderate | Severe |
| Hepatomegaly | Absent | Moderate Enlargement | Severe Enlargement |
| JVP | <8 cm | 11–15 cm | >16 cm |
| Edema | Absent | 1 | >+2 |
| 6MWT | >300 m | 200–300 m | <200 m |
| BNP | <100 | 100–299 | >300 |
| NT-proBNP | <400 | 400–1500 | >1500 |
| Chest X-Ray | Clear | Cardiomegaly |
|
| Vena Cava | None of two:
| One of two:
| Both:
|
| Lung | <15 B Lines | 15–30 B Lines | >30 B Lines |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falco, L.; Martucci, M.L.; Valente, F.; Verrengia, M.; Pacileo, G.; Masarone, D. Pathophysiology-Based Management of Acute Heart Failure. Clin. Pract. 2023, 13, 206-218. https://doi.org/10.3390/clinpract13010019
Falco L, Martucci ML, Valente F, Verrengia M, Pacileo G, Masarone D. Pathophysiology-Based Management of Acute Heart Failure. Clinics and Practice. 2023; 13(1):206-218. https://doi.org/10.3390/clinpract13010019
Chicago/Turabian StyleFalco, Luigi, Maria Luigia Martucci, Fabio Valente, Marina Verrengia, Giuseppe Pacileo, and Daniele Masarone. 2023. "Pathophysiology-Based Management of Acute Heart Failure" Clinics and Practice 13, no. 1: 206-218. https://doi.org/10.3390/clinpract13010019
APA StyleFalco, L., Martucci, M. L., Valente, F., Verrengia, M., Pacileo, G., & Masarone, D. (2023). Pathophysiology-Based Management of Acute Heart Failure. Clinics and Practice, 13(1), 206-218. https://doi.org/10.3390/clinpract13010019