1. Introduction
Endothelial cells form a barrier, effectively separating the luminal from the abluminal side of the vessels and responding to a variety of cues coming from continuous intercellular communication, both locally and systemically. They also function as an endocrine organ, releasing a diverse array of compounds affecting vasoactive, immune, growth and coagulation processes [
1,
2]. This makes the endothelium in pulmonary arterial hypertension (PAH) a perfectly positioned critical source of mediators promoting vascular remodeling, e.g., serotonin (5-HT), vasoactive peptides (NO, PGI2, ET-1), cytokines, chemokines, and growth factors [
3]. Endothelial dysfunction is one of the key players in the development of PAH, a multifactorial disorder characterized by a progressive rise in vascular resistance [
3,
4].
Channelopathy in PAH has been traditionally structured around the effect of K
+ and Ca
2+ channel alterations on the phenotype of pulmonary arterial smooth muscle cells (PASMCs) [
5]. In contrast, our knowledge about aberrations in ion channel function in endothelial cells of pulmonary arteries (PAECs) is limited. Posttranslational modifications and decreased K
+ channel expression have been described in idiopathic PAH (IPAH) PASMCs and animal models of PH, linking membrane depolarization to opening of voltage-gated Ca
2+ channels (VGCC), thus promoting the constrictive and proliferative phenotype as seen in PAH [
6,
7,
8,
9]. Moreover, transient receptor potential (TRP) channels were likewise shown to be dysregulated in IPAH PASMCs demonstrating their role in the phenotypic switch [
10]. The focus has recently been extended towards Ca
2+-activated Cl
− channels (CaCC), demonstrating their importance in cell physiology [
11]. Since the discovery of CaCC in 2008 [
12], its most prominent member TMEM16A has been shown to be a key component in maintaining ionic homeostasis of several cells and tissues, including the gut [
13], airway epithelium [
14], kidney [
15] and brain [
16]. On the other hand, its dysfunction has been associated with the pathophysiology of several heterogeneous diseases such as cancer, hypertension, gastrointestinal motility disorders and cystic fibrosis [
17]. Previously we reported the contribution of TMEM16A in the maintenance of PASMC membrane potential [
18]. IPAH-associated increase in the channel activity caused severe dysregulation of the Ca
2+ homeostasis, promoting PASMC constriction and a proliferative phenotype. By using the TMEM16A inhibitor benzbromarone (Bbr) we effectively counteracted the two major hallmarks of PAH; chronic vasoconstriction and remodeling in vivo [
18].
Recently increased TMEM16A activity in the mitochondria has also been shown in PAECs in IPAH [
19], yet studies addressing its functional significance in the endothelial cells remain rare. Here we show the pathological consequences of increased TMEM16A activity in primary human PAECs. We introduce TMEM16A as a player responsible for pathological changes in IPAH PAECs.
2. Materials and Methods
2.1. Human Lung Samples
Human lung tissue samples were obtained from donors or patients with IPAH who underwent lung transplantation at the Department of Surgery, Division of Thoracic Surgery, Medical University of Vienna, Austria. The protocol and tissue usage were approved by the institutional ethics committee (976/2010) and patient consent was obtained before lung transplantation. The patient characteristics included: age at the time of transplantation, weight, height, sex, mean pulmonary arterial pressure (mPAP) measured by right heart catheterization, pulmonary function test, as well as oxygen and medication before transplantation. The chest computed tomography (CT) scans were reviewed and the diagnoses were verified by an independent board including experienced pathologists, radiologists and pulmonologists. Healthy donor lung tissue was obtained from the same source.
Detailed patient characteristics can be found in
Table S1.
2.2. Cell Isolation and Culture
2.2.1. Donor and IPAH PAECs
For the isolation of donor and IPAH pulmonary artery endothelial cells (PAECs), pulmonary arteries (<2 mm in diameter) were isolated and the endothelium incubated with an enzymatic mixture of collagenase, DNAse and dispaze in HBSS at room temperature (RT). Cell suspension was collected, resuspended in VascuLife Complete SMC Medium and cultured in gelatin-coated T25 flasks at 37 °C and 5% CO2. After reaching 70–80% confluency, cells were trypsinized, enriched by 3 consecutive steps of CD31-selective magnetic-activated cell sorting technology and verified via morphological and marker confirmation (smooth muscle actin, fibronectin, vimentin, von Willebrand Factor, smooth muscle myosin heavy chain and CD31). Surplus PAECs were frozen (endothelial cell complete medium containing 12% FCS and 10% DMSO) and stored in liquid nitrogen until further use. Passages 3–9 were used for the experiments.
Human PAECs purchased (Lonza, Basel, Switzerland) or isolated as described above, were cultured in gelatin-(0.1% gelatin solution in PBS) coated cell culture flasks in Lonza endothelial cell growth medium (EBMTM-2 supplemented with EGMTM-2 containing growth factors, cytokines and other supplements, with final 2% FCS concentration), here referred to as complete medium. Whenever the starvation medium was used, EBMTM-2 was supplemented with 0.5% FCS and 0.2% antibiotics.
Detailed patient characteristics of isolated and purchased PAECs can be found in
Tables S1 and S2 respectively.
2.2.2. PASMCs
The isolation and culture of human PASMCs was performed according to Stulnig et al. [
20]. After the removal of the endothelial cell layer, the media was peeled away from the underlying adventitial layer and cut into approximately 1–2 mm
2 sections, centrifuged and resuspended in VascuLife Complete SMC Medium supplemented with 20% FCS and 0.2% antibiotics, then transferred to T75 flasks and cultured at 37 °C and 5% CO
2. After a confluent monolayer of PASMC had formed, the cells were trypsinized and either cultured in VascuLife Complete SMC medium supplemented with 10% FCS and 0.2% antibiotics, or frozen (VascuLife Complete SMC Medium containing 15% FCS and 10% DMSO) and stored in liquid nitrogen until further use. Passages 4–8 were used for the experiments.
Detailed patient characteristics of isolated PASMCs can be found in
Table S1.
2.3. Precision-Cut Lung Slices (PCLS)
Donor lung cuts were filled with 3% low melting agarose and let to solidify at 4 °C for 15 min. Cylindrical cores of 8 mm in diameter were cut and sliced in cutting solution to sections of 250 μm thickness using a Krumdieck live tissue microtome. Freshly cut slices were then transferred to the incubation medium and kept at 37 °C and 5% CO2 in the incubator. The incubation medium was changed 4 times separated by 30 min wash steps and finally left overnight. On the following day the slices were either fixed in 4% formaldehyde for 1 h or kept for further use in culture.
Further information regarding solutions and materials can be found in
Tables S5 and S6 respectively.
2.4. Overexpression of TMEM16A
2.4.1. Cell Preparation
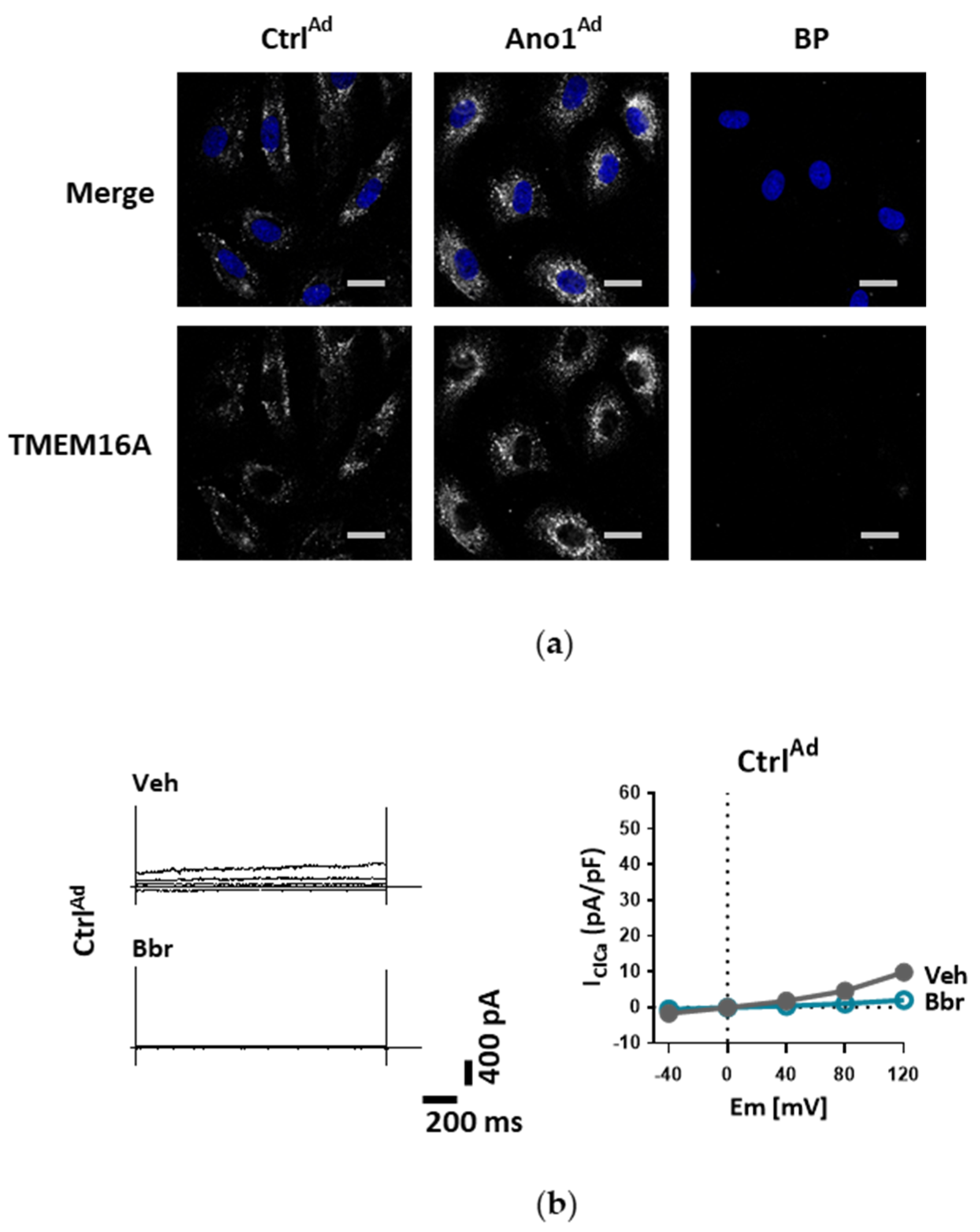
For TMEM16A overexpression, human PAECs or PASMCs were seeded on gelatin-coated chamber slides or in 6-well plates and left to settle in fully supplemented medium overnight. On the next day the medium was replaced by fresh complete medium containing either TMEM16A-encoding adenoviruses Ano1Ad or control CtrlAd (Cyagen Biosciences, Santa Clara, CA, USA) at multiplicity of infection 50 (MOI 50). After 24 h the medium was replaced by fresh complete medium. For further assays, cells were collected either 48 or 72 h after the start of infection.
2.4.2. Isolated Pulmonary Arteries and Precision-Cut Lung Slices (PCLS)
In the case of isolated human pulmonary arteries or PCLS, the vessels or PCLS slices were incubated with the adenovirus in basal VascuLife Medium for 24 h followed by exchange of the viral solution and incubation for further 24 h. After 48 h the vessels were either used for isometric tension measurements, immunofluorescence imaging or collected for protein assessment followed by western blot.
Further information regarding solutions and materials can be found in
Tables S5 and S6 respectively.
2.5. Immunofluorescence Staining
2.5.1. Human Lung sections
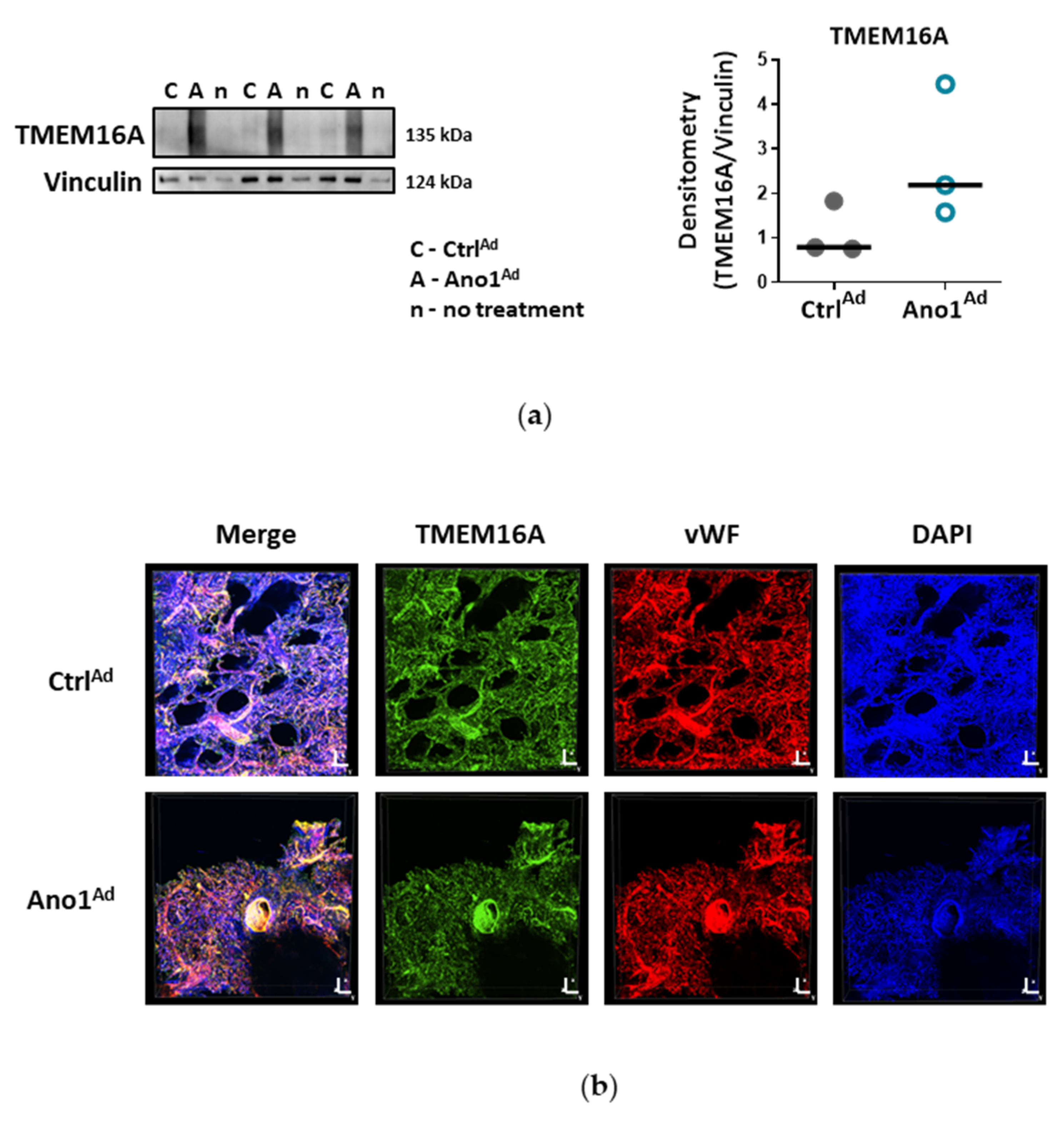
Formalin-fixed paraffin-embedded human lung tissue blocks were cut to 3.5 μm thick slices. Sections were deparaffinized at 60 °C overnight and antigen retrieval was performed with Dako Target Retrieval Solution pH 9.0 at 95 °C. Lung sections were blocked with 10% BSA for 1 h at RT and immunolabelled with antibodies against Von Willebrand factor (vWF) and TMEM16A at 4 °C overnight. On the following day, the sections were washed, then labelled with Alexa Fluor 555 a-rabbit and Alexa Fluor 647 a-mouse secondary antibodies for 1 h at RT. Finally, the slides were preserved using a mounting medium containing 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) and imaged with Zeiss LSM 510 META laser scanning confocal system. Duplicates were processed either without the primary antibody or with the primary antibody against TMEM16A pre-incubated with the immunogen peptide as negative controls.
2.5.2. Precision-Cut Lung Slices (PCLS)
Formalin-fixed pieces were blocked at 4 °C overnight in PBS containing 5% BSA and 0.5% Triton X-100. On the next day, the slices were transferred to primary antibodies against vWF and TMEM16A prepared in the same solution and incubated for 24 h at 4 °C. On the following day, the pieces were washed, then labelled with a mixture of AlexaFluor 555 a-rabbit secondary antibody and DAPI prepared in PBS with 3% BSA and incubated for another 24 h at 4 °C. The pieces were preserved using a DAKO mounting medium and imaged with Nikon’s A1+ confocal laser microscope system.
In the case of TMEM16A overexpression, the staining procedure was similar to the one described above; the pieces were incubated with a mixture of AlexaFluor 488 a-rabbit secondary antibody and DAPI. vWF antibodies were labelled with Mix-n-Stain™ CF™ 633 antibody labeling kit according to the manufacturer’s instructions.
2.5.3. Human PAECs
Cells were seeded onto gelatin-coated chamber slides and formalin-fixed. After blocking with 5% BSA, the cells were incubated overnight at 4 °C with antibodies against Von Willebrand factor and TMEM16A. On the following day, the cells were washed, then labelled with Alexa Fluor 555 a-rabbit and Alexa Fluor 647 a-mouse secondary antibodies for 1 h at RT. Finally, the slides were preserved using a mounting medium containing DAPI and imaged with Nikon’s A1+ confocal laser microscope system. Duplicates were processed either without the primary antibody or with the primary antibody against TMEM16A pre-incubated with the immunogen peptide as negative controls.
For the labelling of TMEM16A-overexpressing human PAECs, cells were infected with a MOI 50 as described above. 72 h after adenoviral infection, the cells were fixed with 4% paraformaldehyde. The following staining protocol was similar as described above; the slides were incubated with Alexa Fluor 647 a-rabbit secondary antibody and imaged with Nikon’s A1+ confocal laser microscope system.
Further information regarding antibodies and materials can be found in
Tables S4 and S6 respectively.
2.6. Analysis of Protein Expression
For the analysis of total protein, cells were lysed in CHAPS [
2] buffer and analyzed with Western blot. Alternatively, pieces of healthy human lung tissue were collected in CHAPS [
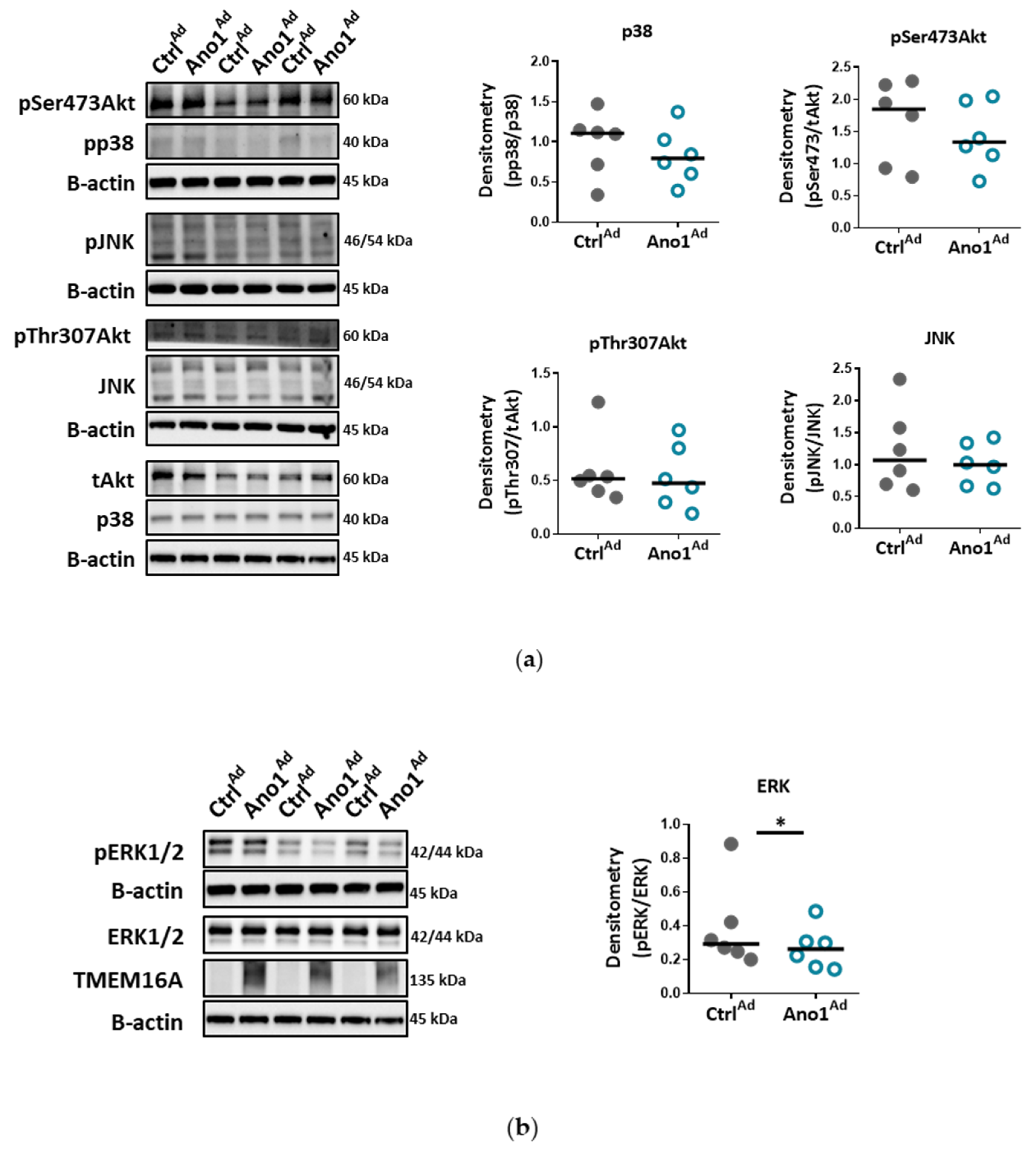
2] buffer followed by homogenization with beads and MagNA Lyser instrument or sonication respectively. After centrifugation (13,000 g, 10 min), the protein concentration of the supernatant was determined with Pierce BCA protein assay kit according to the manufacturer’s instruction. A specific amount of protein from each sample was mixed with 10× Loading buffer and run on 8% or 15% SDS-polyacrylamide gels, followed by electro transfer to a nitrocellulose membrane. After blocking the membrane with 5% BSA in TBS-T, the membrane was probed with one of the following antibodies: TMEM16A, pp38, p38, pERK, ERK, pAkt, pAkt, tAKT, pSAPK/JNK, SAPK/JNK, PCNA, Cl. PARP, Cyclin D1, eNOS, LC3B, pSer1177 eNOS or pThr495 eNOS. Afterwards the membrane was incubated with horseradish peroxidase conjugated secondary antibody and the final detection of the proteins was performed using a SuperSignal West Femto, ECL prime or ECL Start Kit and a ChemiDocTM touch imaging system. Protein density was normalized to the intensity of housekeeping protein. As housekeeping genes B-Actin or Vinculin were used.
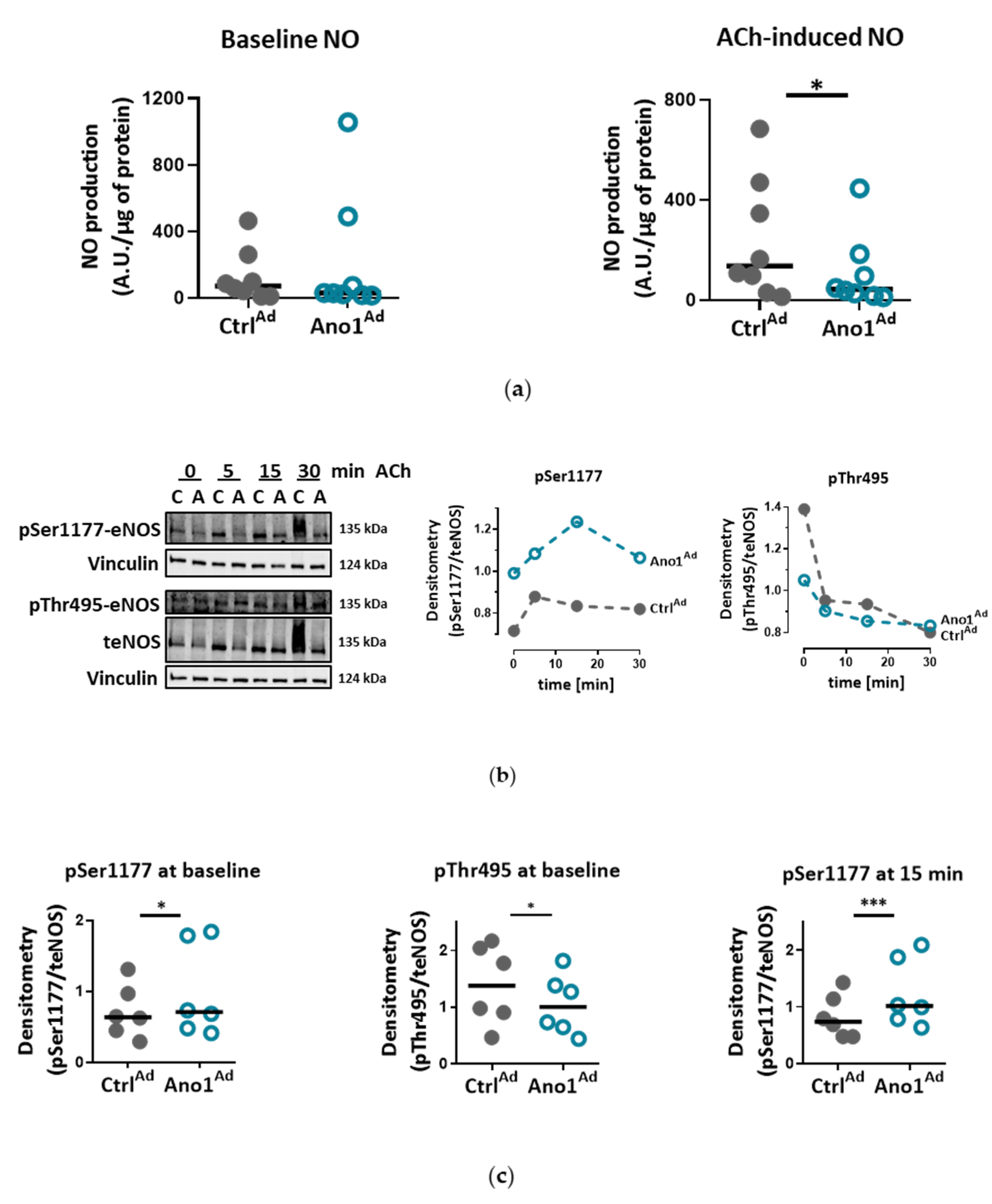
For testing the effect of TMEM16A-overexpression on eNOS phosphorylation, 48 h after adenoviral infection, the cells were 2 h serum-starved and stimulated with 5 µm acetylcholine (ACh). Protein was collected at 0, 5, 15 and 30 min post-stimulation.
For testing the effect of reduced [Cl−]in on eNOS phosphorylation, the cells were incubated for 24 h with either Ringer’s solution containing physiological [Cl−] or solution with KCl and NaCl exchanged with potassium and sodium gluconate to half the amount respectively ([Cl−] was reduced from 129.5 to 67.25 mm). After 2 h serum starvation, the rest of the protocol was performed as described above.
Further information regarding antibodies, solutions and materials can be found in
Tables S4–S6 respectively.
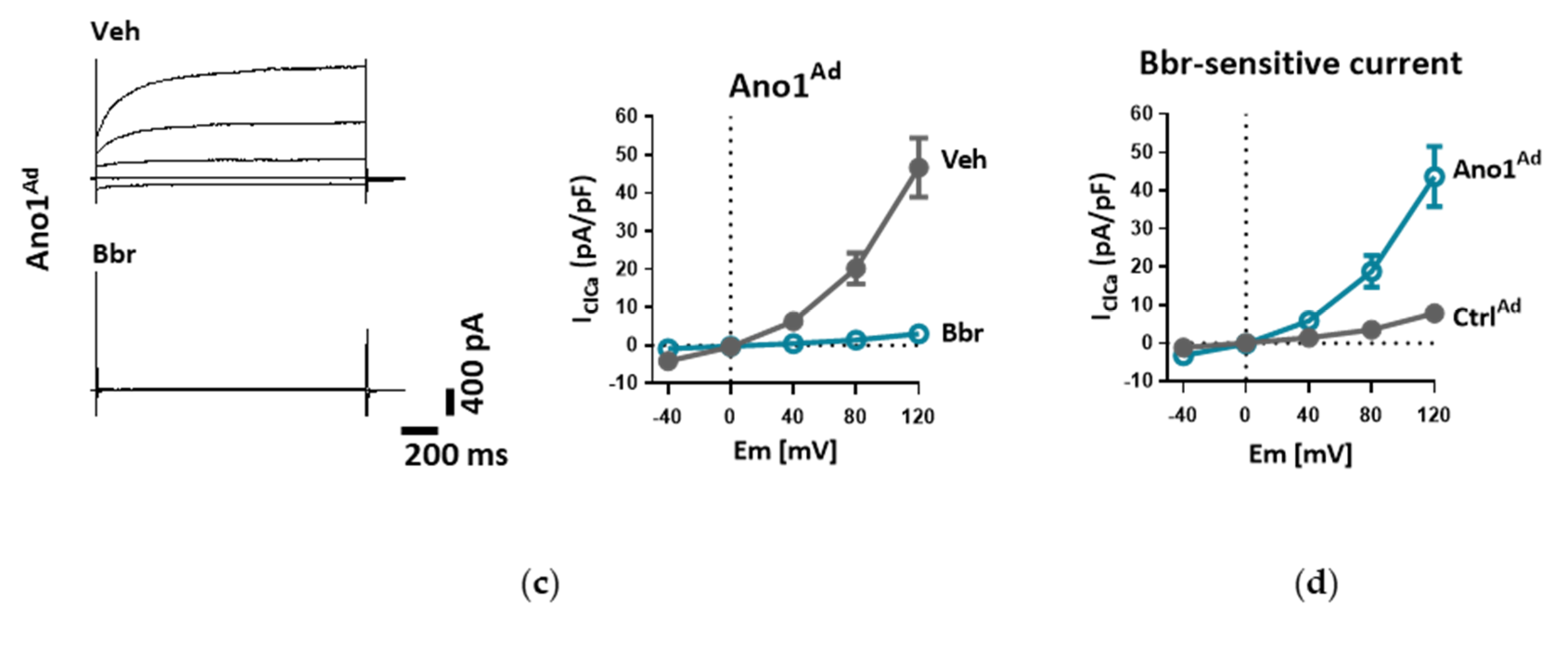
2.7. Measurement of Whole-Cell Ca2+-Activated Cl− Current (IClCa)
Whole-cell Ca
2+-activated Cl
− current was measured as reported previously [
18]. Briefly, donor or IPAH PAECs grown on gelatin-coated dishes were harvested with StemPro Accutase, centrifuged (300 g, 10 min), resuspended in complete medium and incubated at 37 °C for 15 min to allow them to attach before measurements. In the case of adenoviral manipulation of TMEM16A expression, cells were incubated with Ano1
Ad or its control Ctrl
Ad at MOI 50 as described above (see Overexpression of TMEM16A) and used after 48 h.
The cells were incubated in bath solution I until the formation of whole-cell configuration. Once the amplitude of TMEM16A current was stable, the cells were perfused with bath solution II, containing TEA-Cl to minimize K+ current contamination, and vehicle or benzbromarone (Bbr). In order to measure IClCa, the command potential was stepped from a 0 mV holding potential to −40, 0, +40, +80 and +120 mV for 1.5 s, allowing 0.5 s recovery time at the holding potential between each step. The average current measured between 1000 and 1500 ms of each voltage step was plotted against the holding potential. Due to the almost symmetrical Cl− concentration of the bath and pipette solutions, the reversal potential (Erev) for Cl− was expected to be around zero.
Patch pipettes were pulled from thick-walled borosilicate glass by a P-87 puller (Sutter Instrument Co., Novato, CA, USA) and fire-polished. Pipettes were filled with pipette solution (pipette resistance was between 3–5 MΩ) and connected to the headstage of an Axopatch-1D patch clamp amplifier (Axon Instruments, Inc., Foster City, CA, USA). Cut-off frequency of the eight-pole Bessel filter was adjusted to 200 Hz, data were acquired at 2 kHz. Experiments were carried out at RT (21 °C). Solutions were applied using a gravity-driven perfusion system. Data were digitally sampled by Digidata 1550B (Axon Instruments, Inc., Foster City, CA, USA) and analyzed by pCLAMP 10 software.
Further information regarding solutions and materials can be found in
Tables S5 and S6 respectively.
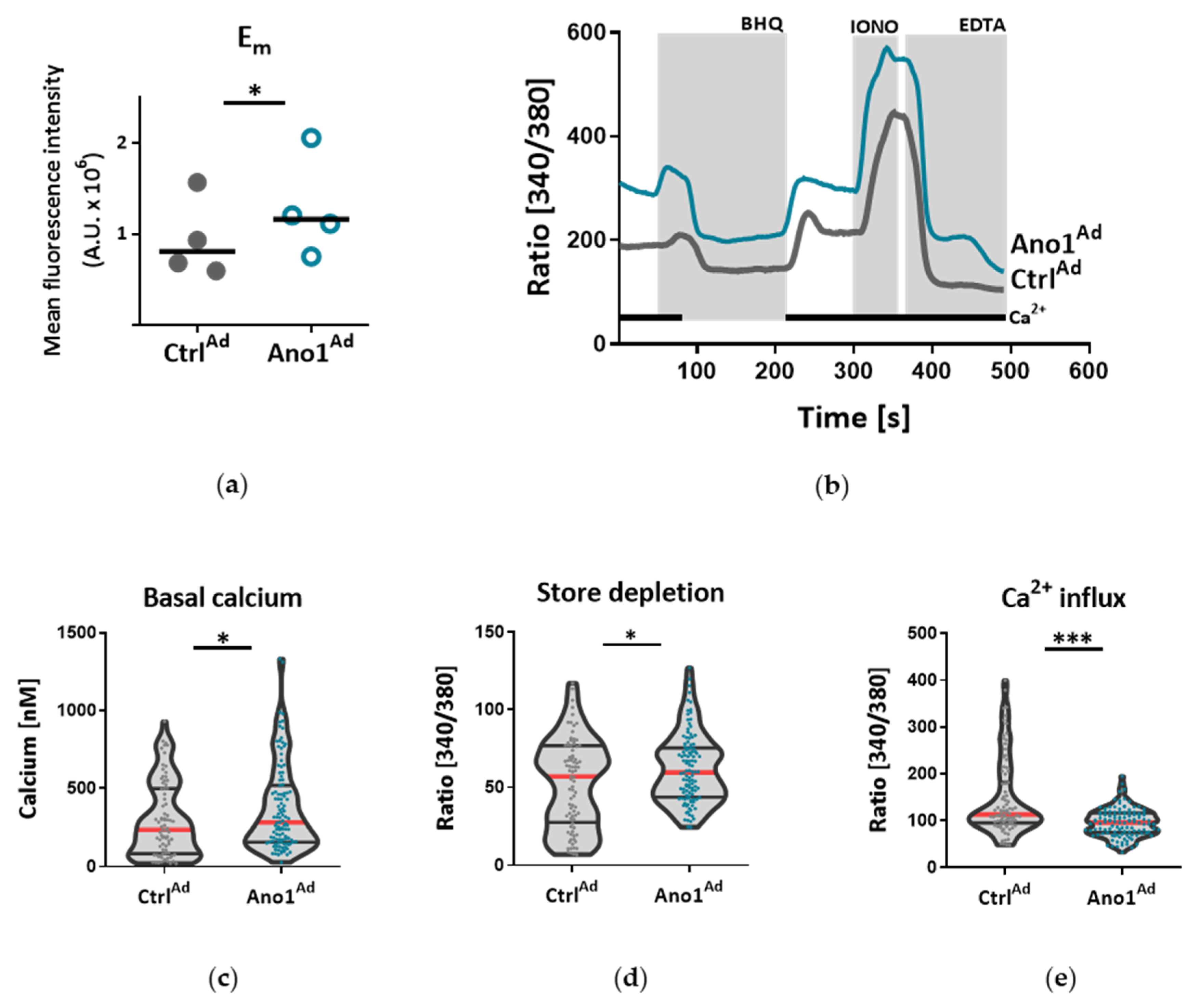
2.8. Live Cell Ca2+ Imaging
Live cell Ca
2+-imaging was done as previously reported [
21]. Human PAECs or PASMCs were seeded on 25 mm glass coverslips and infected with a viral solution of Ano1
Ad or Ctrl
Ad at MOI 50. Forty-eight hours after adenoviral infection, the cells were loaded with 2 µm Fura-2-acetoxymethyl ester (Fura-2AM) for 45 min at 37 °C. The single glass cover slip was mounted on the stage of a Zeiss 200 M inverted epifluorescence microscope coupled to a PolyChrome V monochromator (Till Photonics, Kaufbeuren, Germany) light source in a sealed, temperature-controlled RC-21B imaging chamber (Warner Instruments, Hamden, CT, USA). Fluorescence images were obtained every 3 s with alternate excitation at 340 and 380 nm and the emitted light collected at 510 nm by an air-cooled Andor Ixon camera (Andor Technology, Belfast, Ireland). Background fluorescence was recorded from each cover slip and subtracted before calculation. The acquired images were stored and processed offline with TillVision software (Till Photonics, Germany).
All solutions were prewarmed to 30 °C and cells were stimulated with a 15 µm selective SERCA blocker 2,5-Di-t-butyl-1,4-benzohydroquinone (BHQ) in the presence and absence of extracellular Ca
2+. Maximal and minimal ratio values were determined at the end of each experiment by treating the cells with 5 µm ionomycin (maximal ratio) followed by 20 mm EGTA-mediated chelation of all free Ca
2+ (minimal ratio). Cells that did not respond to ionomycin were discarded. The basal Ca
2+ levels were determined as an average of initial 50 s and [Ca
2+]
i was calculated as previously described [
22]. BHQ-induced Ca
2+ peak and Ca
2+ response following Ca
2+ readmission were calculated as the maximal peak height subtracting the baseline.
Further information regarding solutions and materials can be found in
Table S5 and
Table S6 respectively.
2.9. DAF-DM-Mediated Nitric Oxide Measurement
PAECs were seeded in gelatin-coated dark 96-well plates and treated either with TMEM16A-overexpressing adenovirus or Ringer’s solution with reduced Cl−.
In the case of adenovirus, following infection at MOI 50 and 48 h of overexpression, the cells were starved for 2 h with Ringer’s solution and loaded with 10 µm 4-Amino-5-Methylamino-2′,7′-Difluorofluorescein Diacetate (DAF-FM) for 30 min at 37 °C. The cells were stimulated with 5 µM ACh for 20 min followed by measurement on CLARIOstar Plus (BMG Labtech, Ortenberg, Germany) at Ex/Em = 495/515 nm. All the assays were performed in quadruplicate and normalized to protein content.
Further information regarding solutions and materials can be found in
Tables S5 and S6 respectively.
2.10. Wound Healing Assay
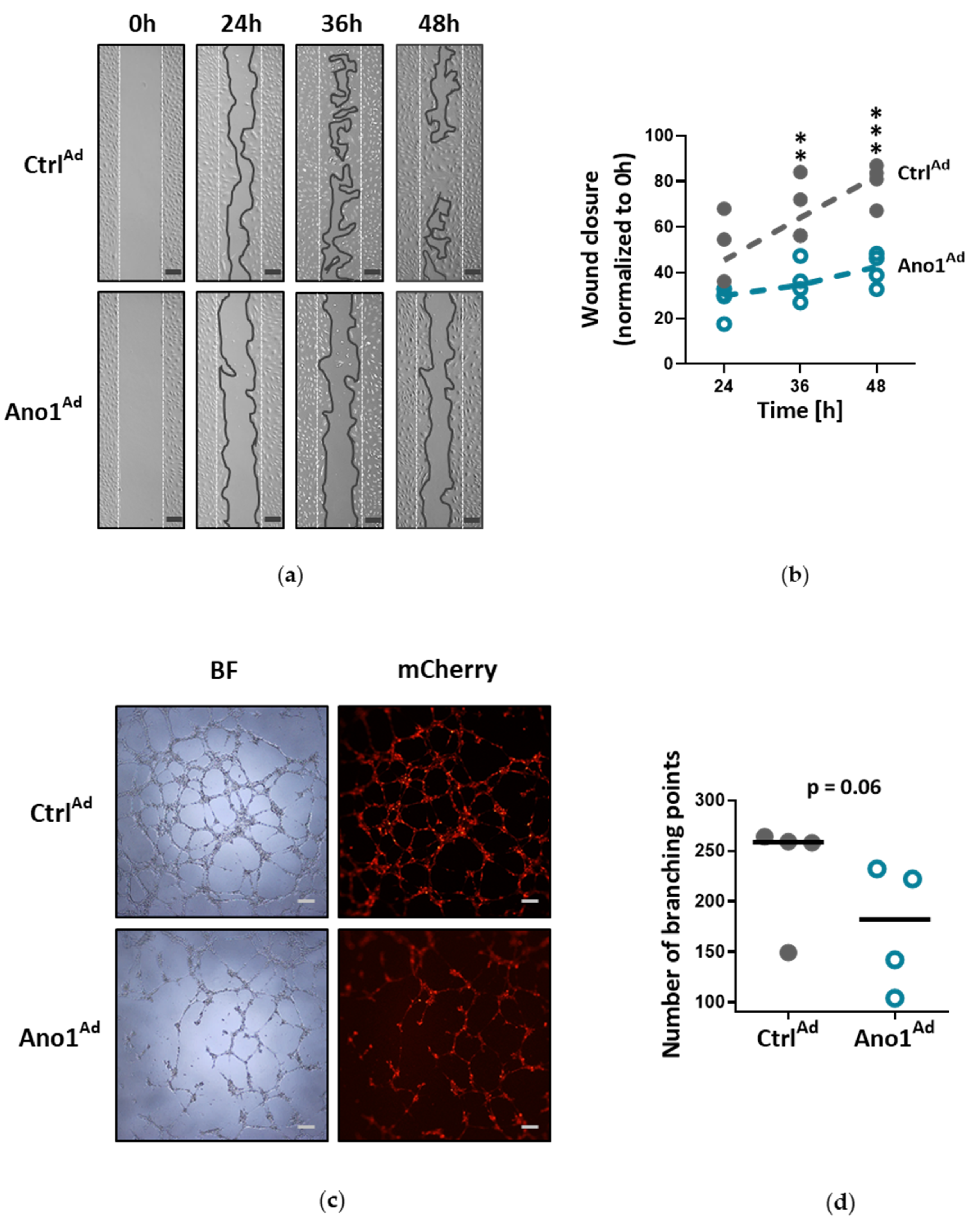
Human PAECs were seeded in 2-well culture-inserts (Ibidi, Planegg, Germany) and infected with either Ano1Ad or CtrlAd at MOI 50. Following overnight starvation and altogether 48 h of TMEM16A overexpression, the inserts were removed to create a gap of approximately 500 µm and the cells were immersed in a complete medium. The closing gaps were photographed at 4x magnification (Olympus CKX41) at 0, 24, 36 and 48 h after removal of the inserts and the photos quantitatively assessed comparing the initial gap with the area of the healing wound using image analyzing software (ImageJ 1.46r).
Further information regarding solutions and materials can be found in
Table S6.
2.11. Pulmonary Arterial Isometric Tension Measurements
All animal studies were approved by the Austrian Ministry of Education, Science and Culture under the license number BMWFW-66.010/0043-WF/V/3b/2016. Ten-to-twelve-week-old wild-type male mice were sacrificed by cervical dislocation and lungs collected for pulmonary artery (PA) isolation. Using a stereo zoom microscope SZX7 (Olympus, Tokyo, Japan) second order PAs of 4 mm in length were isolated and cut in half. One half was always used for a vehicle control. Alternatively, human PAs (4 mm), isolated from human lung tissue obtained from Vienna (976/2010), were isolated, cut in half and incubated with either Ano1Ad or CtrlAd for 24 h, after which the solution was exchanged for VascuLife SMC medium without any growth factors or FCS (LifeLine Cell Technology) for a further 24 h.
In both cases the isolated PAs were mounted on the wire myography system (Danish Myo Technology 620M, Aarhus, Denmark) with the help of tungsten wires as described previously [
23]. Afterwards the PAs were equilibrated for 30 min in physiological salt solution (PSS; pH 7.4, 100% oxygen, 37 °C) followed by an increase of basal tension to 2 mN and stabilization for further 30 min. The vessel viability was tested with 3 sequential steps of depolarization, PSS with 120 mM KCl (KPSS; isotonic replacement of NaCl by KCl) each lasting 15 min. The vessels with mean KPSS response below 2 mN were discarded. The effect of vasoactive agents (high potassium chloride, thromboxane analog U44619 (300 nm), L-NAME (300 µm), Bbr (0.1–30 µm) or ACh (0.1–30 µm)) was tested by directly adding the agents into the chamber and the differences were measured between the vessels incubated with the substances or vehicle, or in the case of human PAs between TMEM16A-overexpressing Ano1
Ad or Ctrl
Ad expressing vessels.
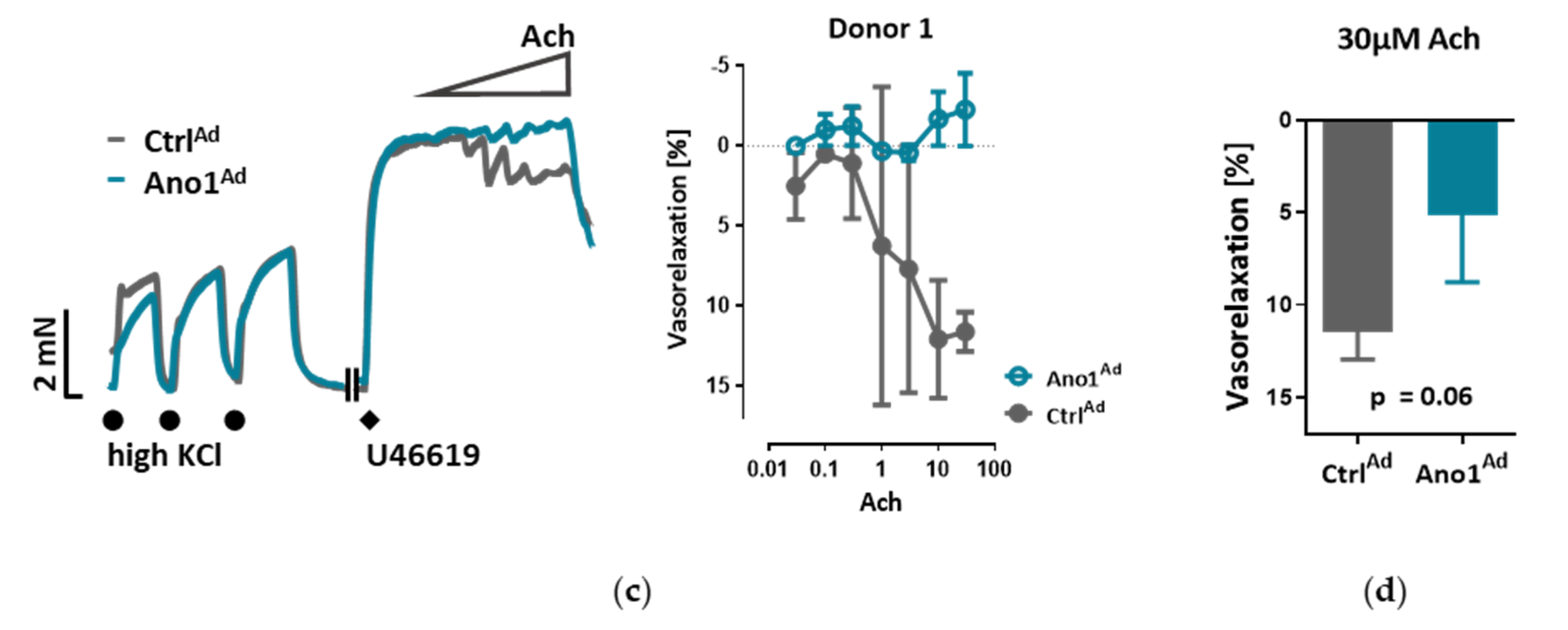
The myography chambers were connected to force transducer units for isometric measurements (PowerLab, ADInstrument, Oxford, UK). The vasorelaxation was calculated as percentage (% relaxation) of the contraction induced by 300 nm U46619. U46619 caused a constriction of 3.88 ± 0.38, 3.99 ± 0.32 or 3.40 ± 0.20 mN in the case of isometric tension measurements of control, endothelium-removed or L-NAME-incubated mouse pulmonary arteries respectively. In the case of isometric tension measurements of human pulmonary arteries U46619 caused a constriction of 5.90 ± 1.63 or 5.90 ± 1.57 mN of control or TMEM16A-overexpressing pulmonary arteries respectively.
Further information regarding solutions and materials can be found in
Tables S5 and S6 respectively.
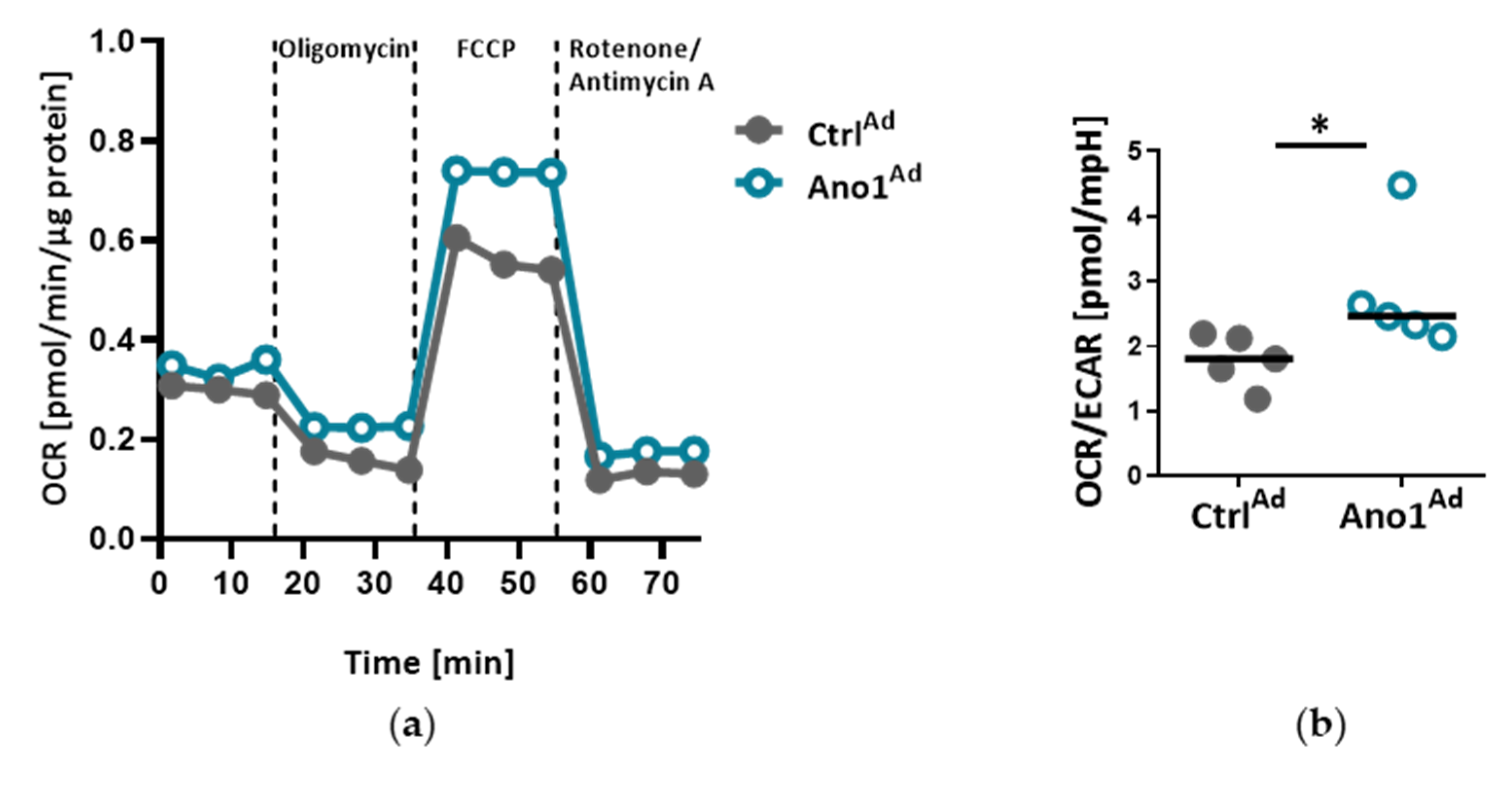
2.12. Measuring Cell Metabolic State
The Agilent Seahorse XFp setup (Agilent, Santa Clara, CA, USA) was used to assess the mitochondrial function of primary human PAECs: 20,000 cells were seeded into 8-well plates and infected with either AnoAd or CtrlAd and incubated for 24 h. Forty-eight hours after adenoviral infection, a Seahorse XFp cell mito stress test kit was used according to the manufacturer’s instructions. Briefly, with the use of Oligomycin, Rotenone and Rotenone/Antimycin A, the mitochondrial state can be assessed. All the assays were performed in triplicate and normalized to protein content.
Further information regarding materials can be found in
Table S6.
2.13. Matrigel Tube-Formation Assay
To test endothelial cell angiogenic potential, Matrigel tube formation assay was used (Merck Millipore, Burlington, MA, USA): 200,000 cells were seeded in a gelatin-covered 6-well plate and infected with either Ano1Ad or CtrlAd at MOI 50 and incubated for 24 h. Following overnight starvation and 48 h after adenoviral infection, the cells were trypsinized, counted and 50,000 cells per 96-well plate were seeded onto prepared matrigel in triplicate according to the manufacturer’s instructions. After 6 h of incubation period at 37 °C, the tubular networks were photographed at 4x magnification (Olympus CKX41) and photos quantitatively assessed comparing the number of branching points using image analyzing software (ImageJ 1.46r).
Further information regarding materials can be found in
Table S6.
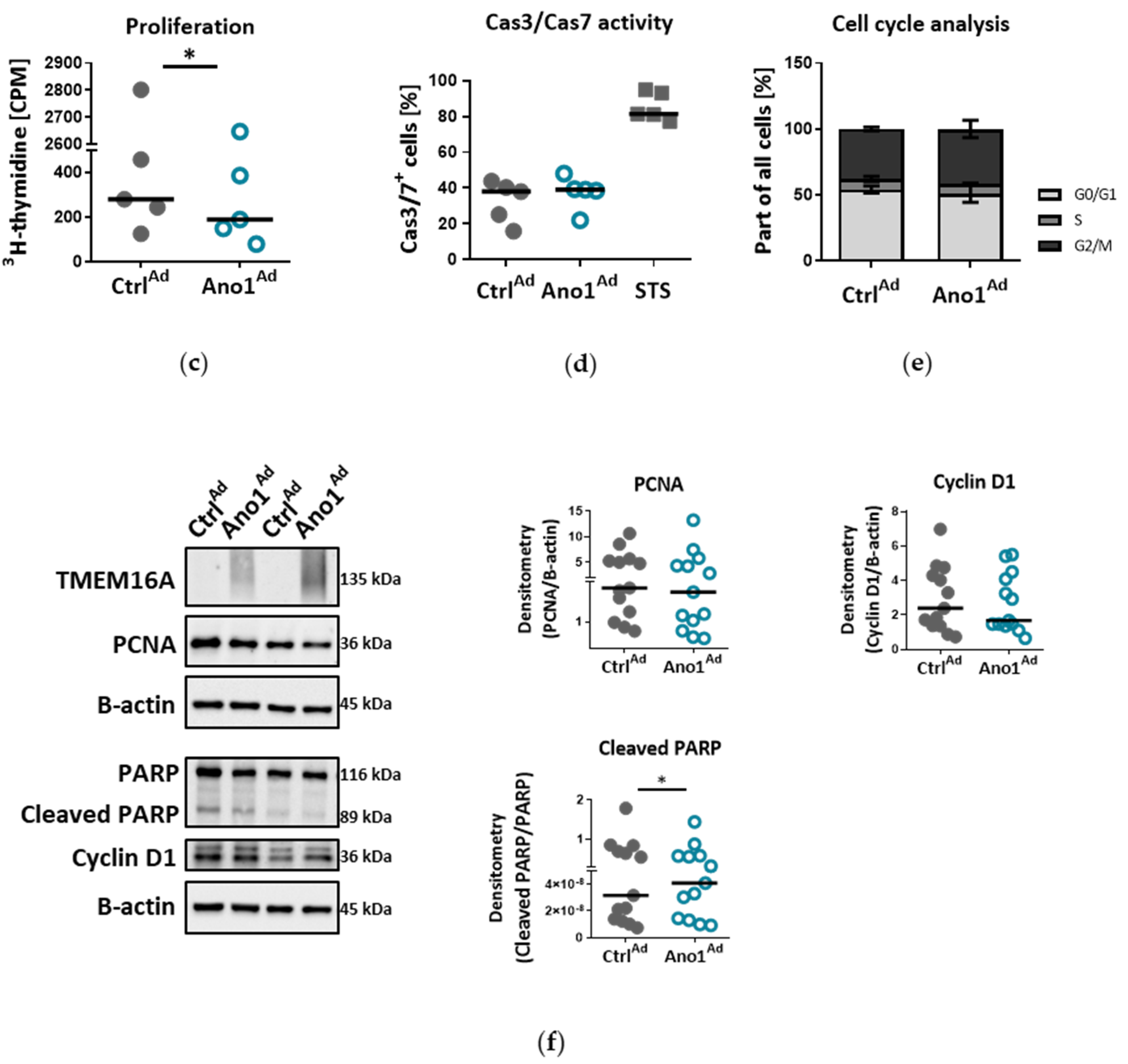
2.14. Assessment of Cell Proliferation In Vitro
To study the influence of TMEM16A on human PAEC proliferation, 10,000 cells per well were seeded in 96-well plates and infected with either Ano1Ad or CtrlAd at MOI 50. After 24 h the medium was replaced by fresh complete medium and the cells serum-starved overnight. On the following day the cell medium was replaced by fresh starvation medium with added 3H-thymidine. The proliferation was determined after 24 h 3H-thymidine incorporation (BIOTREND Chemikalien GmbH, Cologne, Germany) and altogether 72 h after adenoviral infection as an index of DNA synthesis, and detected with a scintillation counter and the results are represented as counts per minute (CPM) (Wallac 1450 MicroBeta TriLux liquid scintillation counter and luminometer, PerkinElmer, Waltham, MA, USA). Each independent experiment was performed in quintuplicate.
Further information regarding materials can be found in
Table S6.
2.15. Assessment of Cell Resting Membrane Potential In Vitro
To study the influence of TMEM16A on human PAEC resting membrane potential, 80,000 cells per well in a 6-well plate were seeded and transfected with either CtrlAd or Ano1Ad at MOI 50. After 24 h the medium was replaced by fresh complete medium and the cells were serum-starved overnight. 48 h after adenoviral infection, the medium was replaced with starvation medium supplemented with 10 µm DiBAC4(3) dye for 30 min at 37 °C after which the cells were collected with cell-scratcher in PSS. Fluorescence signal intensity was measured on CytoFLEX S flow cytometer (Beckman Coulter, Brea, CA, USA) at Ex/Em = 490/516 nm. Increased depolarization results in additional influx of the anionic dye and an increase in fluorescence.
Further information regarding solutions and materials can be found in
Tables S5 and S6 respectively.
2.16. Assessment of Cell Cas3/Cas7 Activation In Vitro
To study the influence of TMEM16A on human PAEC Cas3/Cas7 activation, we seeded 150,000 cells per well in a 6-well plate and transfected the next day with either CtrlAd or Ano1Ad at MOI 50. After 24 h the medium was replaced by fresh complete medium and the cells serum-starved overnight. The next day the medium was exchanged by fresh starvation medium. As a positive control, we incubated the cells with 10 µm Staurosporin (STS) for 24 h. Seventy-two hours after adenoviral infection, CellEventTM Caspase-3/7 green detection reagent was used according to the manufacturer’s instruction. The fluorescent signal shift was measured with a CytoFLEX S flow cytometer (Beckman Coulter, Brea, CA, USA) at Ex/Em = 503/530 nm.
Further information regarding solutions and materials can be found in
Table S6.
2.17. Cell-Cycle Analysis
To study the effect of TMEM16A overexpression on cell cycle progression of primary human PAECs we seeded 200,000 cells per well in a 6-well plate and transfected with either CtrlAd or Ano1Ad at MOI 50. After 24 h the medium was replaced by fresh complete medium and the cells serum-starved overnight. The next day the medium was exchanged by fresh starvation medium for a further 24 h. Seventy-two hours after adenoviral infection, the cells were trypsinized, resuspended in PBS, then transferred into cold EtOH under continuous vortexing and incubated at 4 °C for at least 30 min. After washing using PBS supplemented with 0.5% FCS, the cell pellet was resuspended in a solution containing 1 µg/mL DAPI and 0.1% Triton x-100 in PBS and incubated at RT for 10 min, then transferred on ice until measurements were done. Fluorescence signal intensity was measured on a CytoFLEX LX flow cytometer (Beckman Coulter, Brea, CA, USA) at Ex/Em = 355/461 (bandpass filter 450/45). The data was analyzed using ModFit software.
Further information regarding materials can be found in
Table S6.
2.18. qRT-PCR
qRT-PCR was performed as described previously [
18]. Briefly, PAECs were grown until confluence and serum-starved overnight. RNA was collected using the PeqGOLD Total RNA kit (PeqLab, Erlangen, Germany) and transcribed into cDNA with the iScript reagent mix (Bio-Rad, Hercules CA, USA). To assess ANO1 expression, exon-exon junction spanning primers targeting the boundaries of exons 1 and 2 were acquired from Eurofins, Graz, Austria (see
Table S3). The primers covered the region without reported alternative splicing, therefore they amplified all splice variants. qRT-PCR was performed using Blue S’Green qPCR kit.
Further information regarding primer sequences and materials can be found in
Tables S3 and S6 respectively.
2.19. Statistical Analysis
Data is shown either as individual data plots with median, as mean ± s.e.m or as floating bars plot (min-to-max). In all cases, n numbers are representing number of replicates, N numbers are representing number of patients. Numbers are given in the corresponding figure legend. Statistical analyses were performed using Prism 8.0 (GraphPad Software, La Jolla, CA, USA) and are identified in the corresponding figure legend. All datasets met the assumptions of the statistical test used, statistical analyses were two-sided and p values < 0.05 were considered significant.
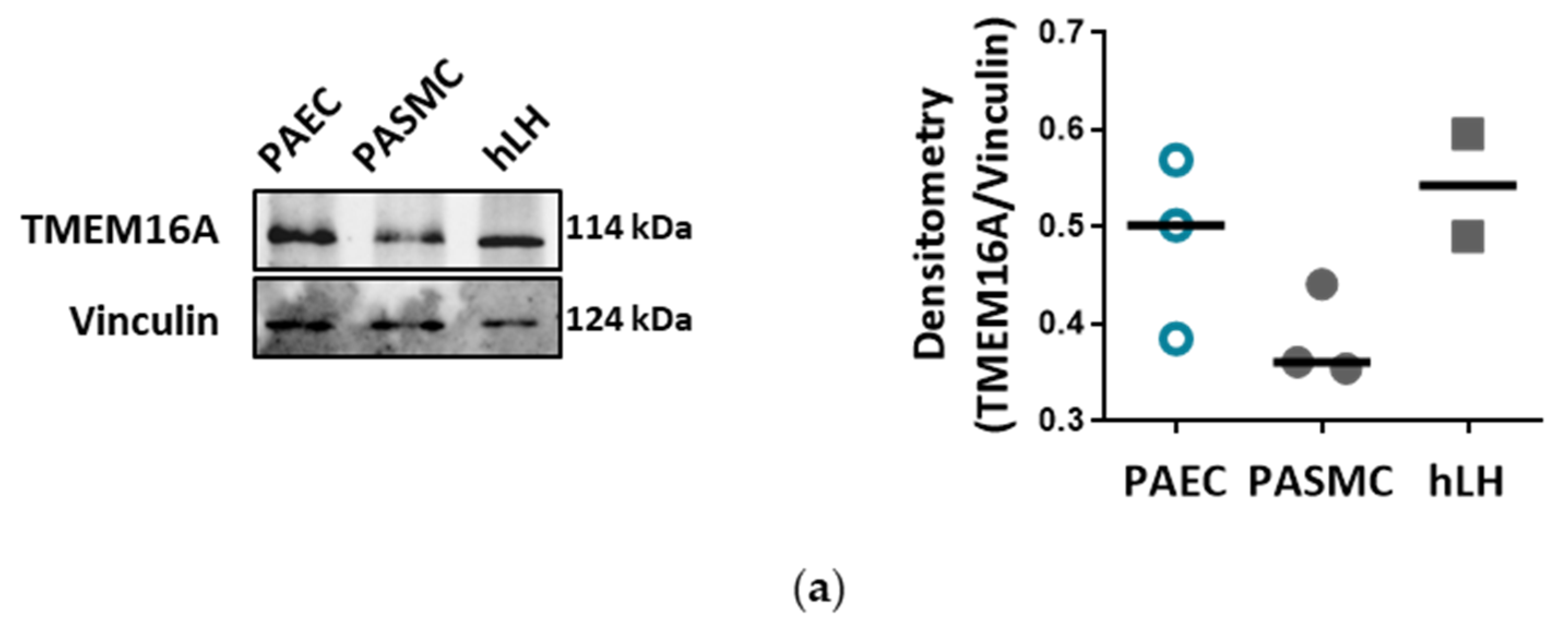
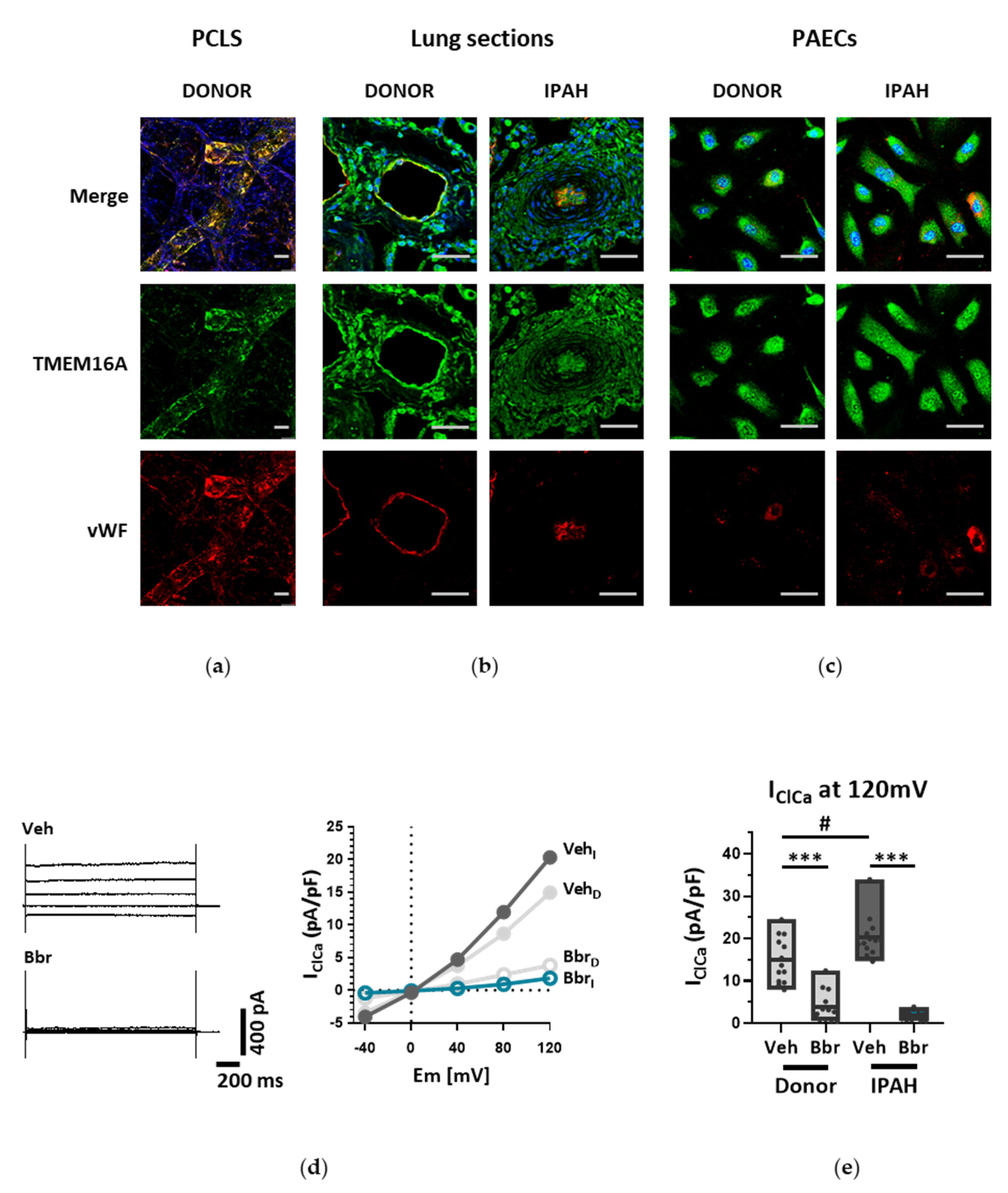
4. Discussion
Endothelial cells (ECs) express a bewildering diversity of different chloride channels responsible for a variety of cellular functions [
24]. The Ca
2+-activated Cl
− channel (CaCC) TMEM16A has been previously detected in the mitochondria in human pulmonary artery endothelial cells (PAECs) [
19]. Our investigation is the first to report the presence of active TMEM16A in the plasma membrane of primary human PAECs. Previously, we have demonstrated the importance of TMEM16A in PASMCs and revealed the therapeutic potential of targeting TMEM16A to reverse pathophysiological signatures of PH in animal models [
18]. With our current results we provide further insights into the role of TMEM16A for the physiologic function of the endothelium and the potential impact of elevated TMEM16A activity for pulmonary endothelial dysfunction, a hallmark of PH.
The Ca
2+-activated Cl
− channel TMEM16A, encoded by the gene ANO1, was found in many excitable tissues such as smooth muscle, cardiac muscle and sensory neurons. The channel is activated by relatively low intracellular Ca
2+ and, upon Ca
2+ binding, an allosteric change induces an opening of the channel pore leading to conduction of an outwardly rectifying current. Although Ca
2+-activated Cl
− current has been studied at the functional level for more than 30 years in different tissues, only a few studies have investigated its role in endothelium. Almost two decades ago, by employing 3D, precision-cut lung slices, we reported the postnatal development of the chloride current contribution to the membrane potential in rat PAECs in situ [
25]. A recent report indicated a relationship between TMEM16A activity and oxidative stress in endothelial cells in the systemic circulation. Transgenic endothelium-specific overexpression of TMEM16A facilitated Nox2/p22phox expression and reactive oxygen species production leading to endothelial dysfunction in a mouse model [
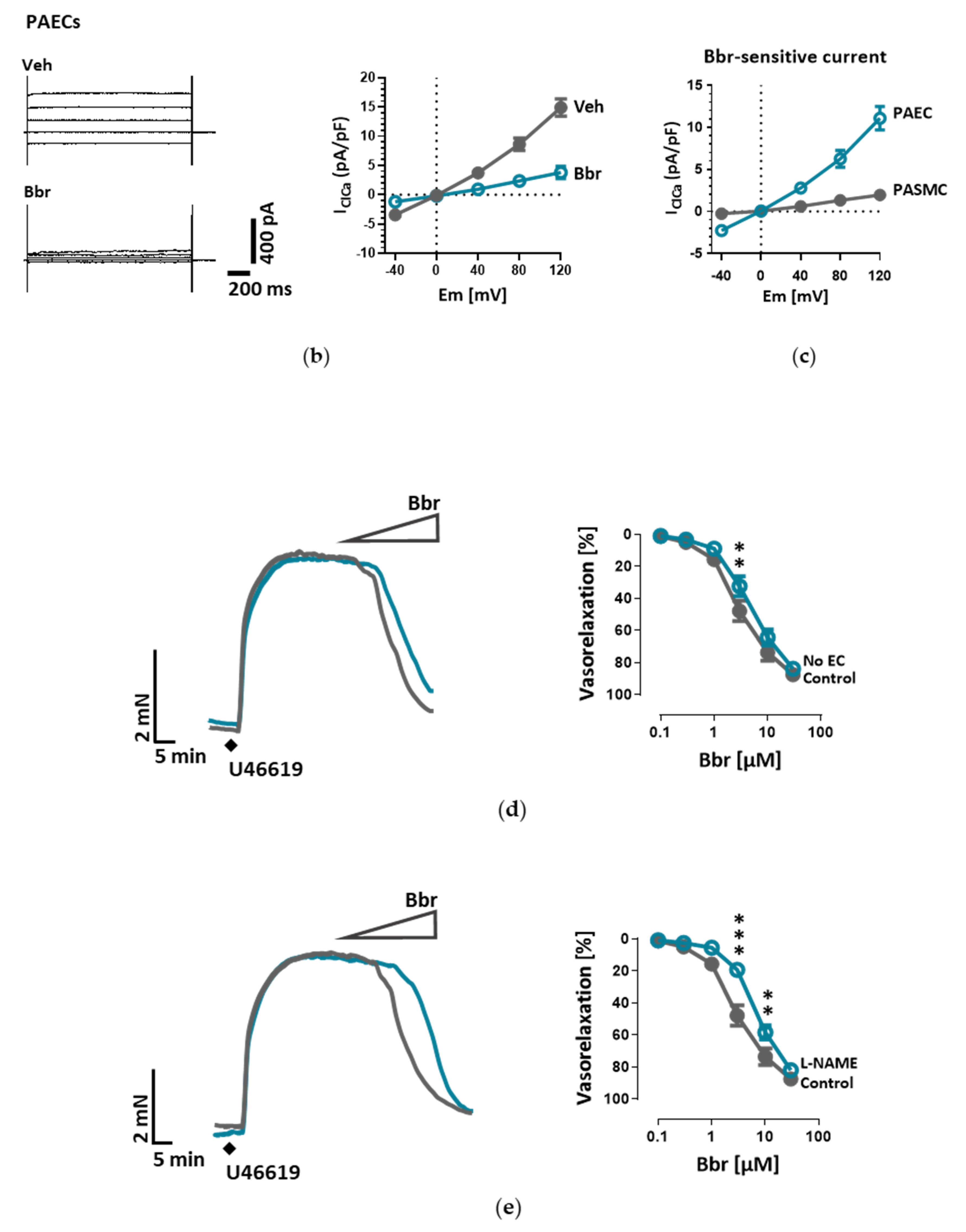
26]. Our study is however the first to show the functional presence of TMEM16A in the cell membrane of human PAECs using patch-clamp recordings. We demonstrate the relevance of this channel for the endothelial function ex vivo and establish TMEM16A as an important player in human pulmonary endothelium under physiological and pathophysiological conditions.
Under disease conditions, elevated TMEM16A in endothelial cells of the systemic circulation was linked to hypertension by showing that TMEM16A plays a specific and critical role in mediating the hemodynamic response to angiotensin II [
26]. Our investigations provide evidence for the functional consequences of increased TMEM16A activity and demonstrate enhanced chloride current in the cell membrane of PAECs from IPAH patients. In order to examine the functional consequences of the elevated TMEM16A expression in endothelial cells, we successfully established a model system for subsequent investigations. Using an IPAH-mimicking enhancement of TMEM16A activity achieved by adenoviral overexpression, we detected depolarization of resting membrane potential and altered Ca
2+ homeostasis in primary human PAECs. Depolarization was also observed in PASMCs, as outlined in the current and the previous study [
18]. However, the depolarization in both of the cell-types have different physiological consequences. After the removal and reintroduction of extracellular Ca
2+, the Ca
2+ influx decreased in PAECs overexpressing TMEM16A and increased in PASMCs. The difference between the two cell types might be the result of a cell-type specific channelome. The absence of L-type Ca
2+ channels in PAECs [
27,
28,
29], which participate mainly in voltage-dependent Ca
2+ influx in many other cell types including PASMCs, indicates an alternative mechanism in PAECs. Although we did not perform additional experiments, according to previous studies it is possible that store-operated Ca
2+ entry (SOCE) plays a key role here [
30,
31]. The reduced Ca
2+ influx in TMEM16A-overexpressing PAECs can be a hint of mitochondrial dysfunction in these cells [
31]. By emptying the endoplasmic reticulum (ER) with BHQ, we detected that stores hold higher amounts of Ca
2+ in TMEM16A-overexpressing PAECs. Increased cytosolic Ca
2+ concentration due to Ca
2+ leak from the ER reticulum has been described in the coronary ECs of diabetic mice [
32]. Thus, with regard to the elevated intracellular Ca
2+ concentration we speculate that TMEM16A-overexpressing PAECs may develop a similar ER Ca
2+ leak contributing to the observed increased Ca
2+ level. Enhanced ER Ca
2+ leak activates SOCE, a newly reported activator of calcium-dependent chloride channels [
33,
34]. Furthermore, a recent study has shown that TMEM16A is directly activated by Ca
2+ release from the ER via the IP
3 receptors [
35]. Thus, the increased intracellular Ca
2+ level together with depolarization might provide a re-entrant loop for the activation of the TMEM16A in IPAH PAECs.
Mitochondria are recognized at the crossroads of many cellular functions. Our investigations reinforced our hypothesis that mitochondrial function is altered in TMEM16A-overexpressing PAECs, highlighted by the predominance of oxidative over glycolytic metabolism. As previously reported, endothelial cells rely primarily on glycolysis with in vitro glycolysis rates comparable to or even higher than in cancer cells [
36]. One of the advantages of aerobic glycolysis in ECs is that lower oxidative phosphorylation generates less reactive oxygen species (ROS) [
37]. Pulmonary hypertension is characterized by perturbations in metabolic pathways affecting NO generation, smooth muscle proliferation, migration and oxidative stress [
3,
38]. Our data points to a TMEM16A-overexpression-driven shift toward oxidative phosphorylation in PAEC. However, further work is needed to determine whether this metabolic alteration, intimately related to mitochondrial function, represents a functional feature of a PAEC subtype or an early step in the disease development.
Chloride movement across the cell plasma membrane has been suggested to play an important role in regulating a variety of physiological processes, including cell proliferation, apoptosis, migration or mucus secretion. Elevated TMEM16A activity results in increased chloride efflux in endothelial cells and thus causes the decrease of the intracellular chloride concentration [
39]. Our data show that TMEM16A overexpression in primary human PAECs led to decreased proliferation, wound-healing and tubulogenesis, while apoptosis remained unaffected. The TMEM16A-induced endothelial dysfunction, shown as aberrant proliferation and tube formation, was ERK1/2-dependent as TMEM16A overexpression led to decreased ERK1/2 but not JNK, p38 or Akt phosphorylation. These findings are in line with recent studies indicating that the cell-specific role of elevated TMEM16A for migration, proliferation or angiogenesis depends on the cellular environment and it is specific for a particular cell type [
34]. Accordingly, our results very likely reflect that the ERK1/2 pathway in human PAECs was specifically affected by TMEM16A overexpression, leading to reduced wound-healing and vessel formation.
We show that IPAH-mimicking TMEM16A overexpression alters eNOS function in human PAECs. eNOS acts as a master regulator of vascular tone through the generation of NO and dysregulation of eNOS is a hallmark of many cardiovascular diseases. We noticed that baseline as well as ACh-induced activatory eNOS phosphorylation were increased, however, NO bioavailability was decreased under stimulated conditions in TMEM16A overexpressing cells. Increase in the intracellular concentration of free calcium has been proposed to regulate eNOS phosphorylation and activity [
40]. Consistent with this, the enhanced TMEM16A activity-mediated increase in cytosolic Ca
2+ levels in our study could lead to increased phosphorylation at eNOS activation sites, including Ser1177 [
41,
42]. The discordance between eNOS activatory phosphorylation and NO availability as observed in the present study was also found in endothelial cells incubated with insulin or VEGF [
41]. Several potential mechanisms such as eNOS uncoupling, or dimer disruption as well as NO scavenging by ROS, (possibly raised by augmented oxidative metabolism), may contribute to the decreased NO availability in TMEM16A-overexpressing PAECs [
41,
42,
43]. Our findings imply that increased TMEM16A activity in PAECs interferes with background eNOS activity and the stable production of nitric oxide, essential components for maintaining low vascular tone in normoxic conditions [
44].
There are some discrepancies between our findings and the previously published reports on TMEM16A in IPAH PAECs, as we did not detect an increased apoptosis or a channel activator-mediated attenuated p38 phosphorylation. However, most of the results in the previous study have been generated on rat lung microvascular ECs based on activated mitochondrial TMEM16A. Thus, the regional and species-specific heterogeneity might explain the differences. Furthermore, previous studies postulated several stages in PAEC transformation in PAH [
45,
46,
47]. Accordingly, our data suggest that increased TMEM16A activity in the cell membrane represents one stage of the IPAH PAEC phenotype [
48]. Moreover, similar to our findings, IPAH patients may have disrupted eNOS pathways [
49,
50]. Although eNOS is considered a beneficial factor in the pulmonary arteries, persistent eNOS activation may also induce PH in mice and humans through PKG nitration causing disturbed vasodilation [
51]. Chronic hypoxic and monocrotaline rat models of PH are characterized by impaired ACh-mediated vasorelaxation, further supporting our hypothesis [
52].
Our study has some limitations: applying a broad range of functional read-outs made it necessary to utilize freshly isolated pulmonary arteries, fresh precision-cut lung slices as well as primary human endothelial cells in cell culture in one study. It is possible that culturing causes phenotypical changes of primary cells and this may impair the conclusions drawn from experiments in intact vessels. However, this type of combination of different methods still provides the backbone for the majority of preclinical studies in the vascular system. The other drawback is, that we can’t exclude changes in the biophysical properties of the TMEM16A channel in IPAH PAECs. Further investigations on activation and on the Ca
2+-binding of the channel are needed [
53]. Finally, we found a marked overexpression of TMEM16A in IPAH PAEC and the respective whole-cell Ca
2+-activated Cl
− current as well as the Bbr-responsive current but we did not investigate other readouts in the IPAH PAECs because the number of available cells was very much limited for high throughput read-outs. Therefore, we cannot provide direct evidence that silencing of TMEM16A would normalize the IPAH phenotype.
Within the scope of this work, we were able to extend the pathological footprint of TMEM16A overexpression beyond its effect on pulmonary arterial smooth muscle cell and show its role in endothelial dysfunction. We traced the vasorelaxation and angiogenic defects of TMEM16A overexpression and the decrease in NO production in PAECs and established the disease-associated increased TMEM16A activity in PAH.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












