The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications—Update in Pathogenesis, Treatment and Monitoring


Abstract
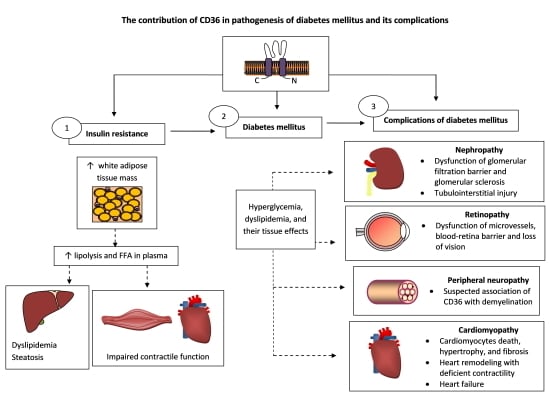
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Characterization of CD36
3. Diabetes Mellitus—Is It an Epidemic?
4. Association of sCD36 with Diabetes Mellitus
5. The Role of CD36 in the Pathogenesis of DM
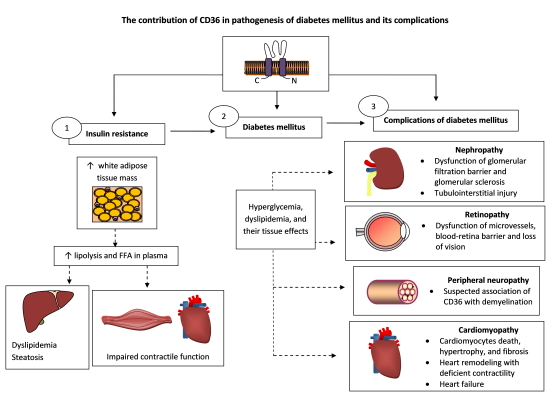
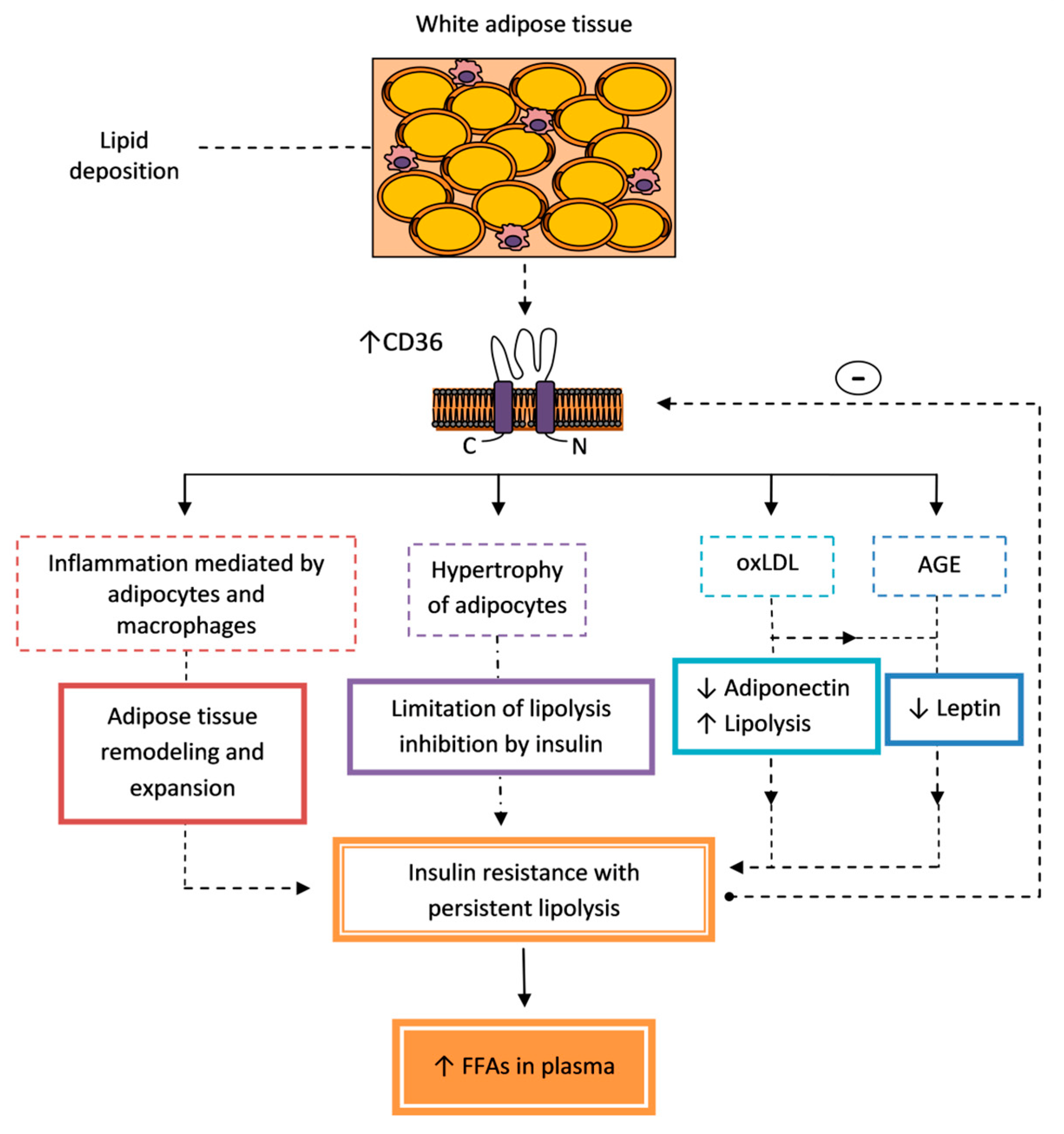
5.1. Insulin Resistance
5.1.1. Adipose Tissue
5.1.2. Liver
5.1.3. Skeletal and Cardiac Muscles
5.2. Pancreatic β-Cell Dysfunction and Damage
6. Diabetic Complications
6.1. Hyperglycemia
6.2. Alterations in Lipid Metabolism
7. The Role of CD36 in Diabetic Complications
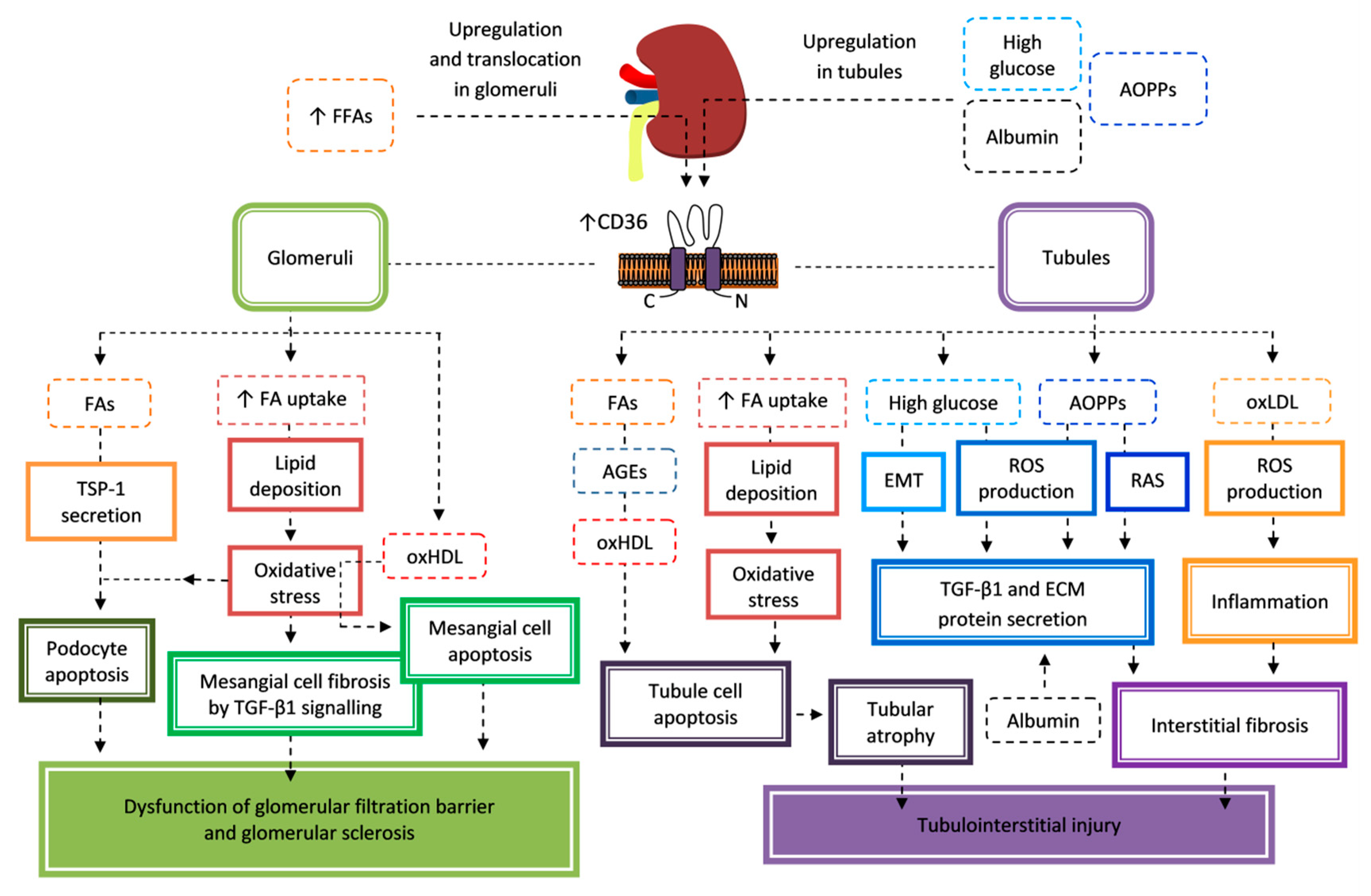
7.1. Nephropathy
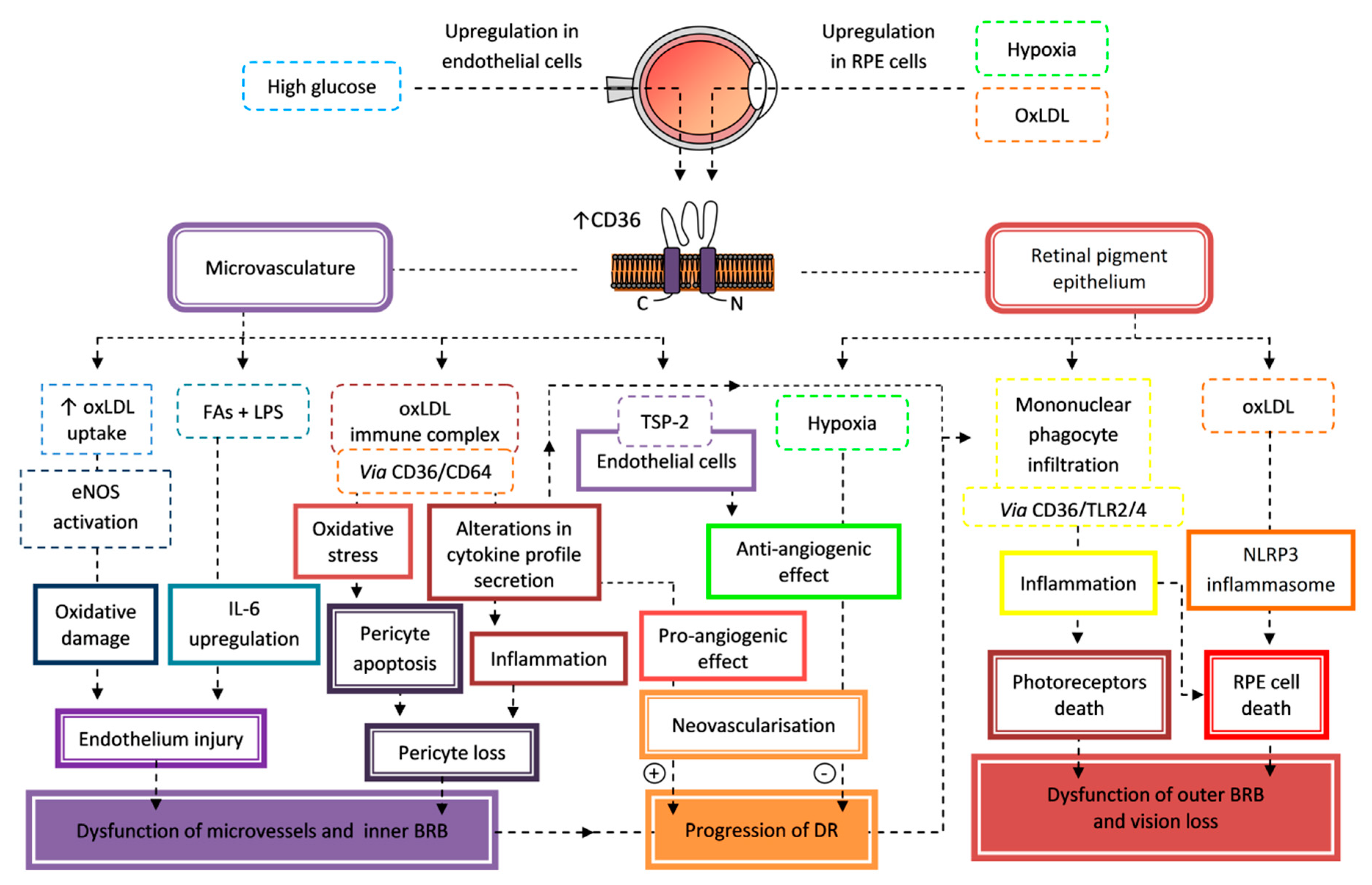
7.2. Retinopathy
7.3. Peripheral Neuropathy
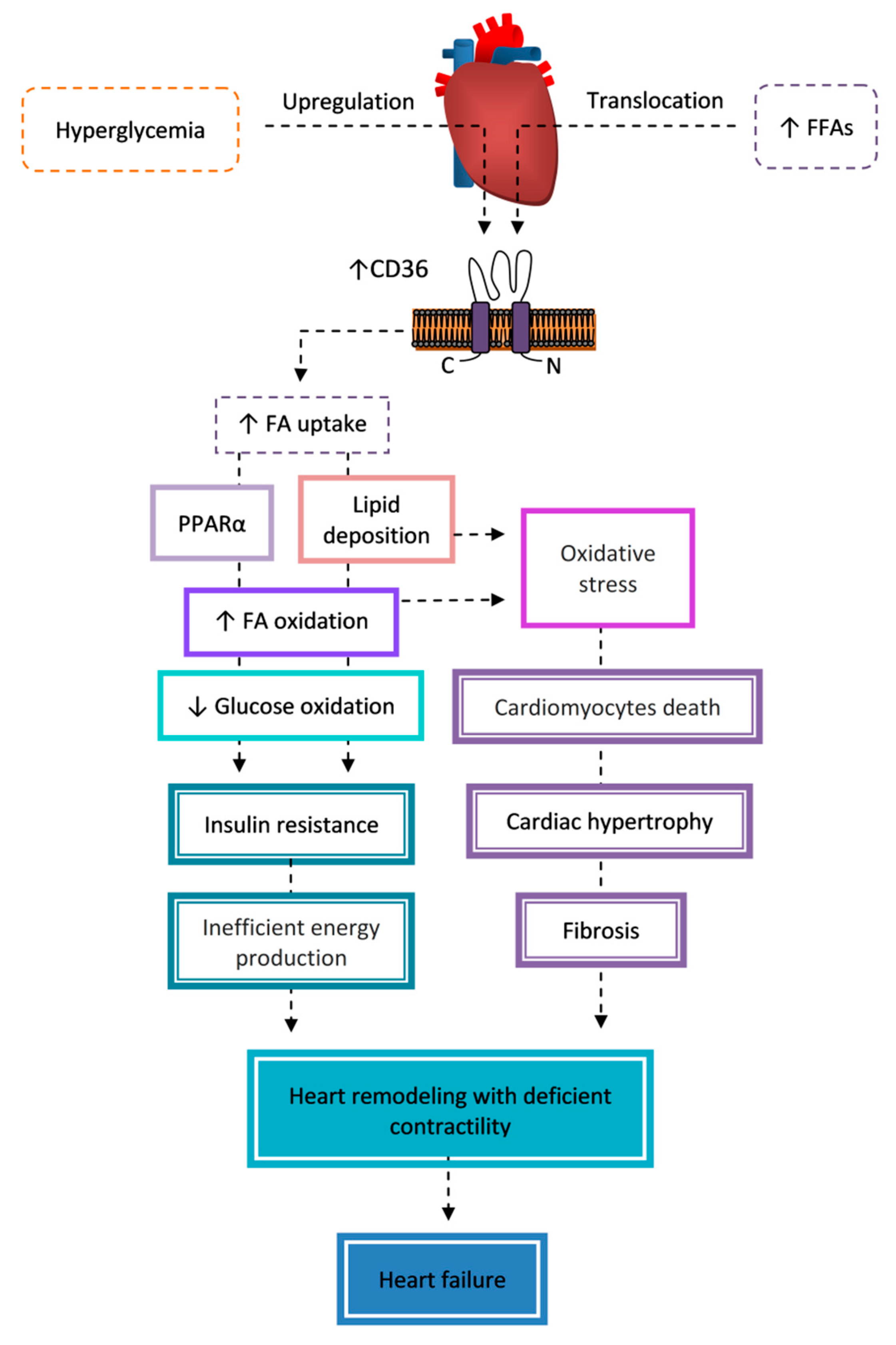
7.4. Cardiomyopathy
8. Summary and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hsieh, F.L.; Turner, L.; Bolla, J.R.; Robinson, C.V.; Lavstsen, T.; Higgins, M.K. The structural basis for CD36 binding by the malaria parasite. Nat. Commun. 2016, 7, 12837. [Google Scholar] [CrossRef]
- Zhao, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; Ruan, X.Z. CD36 and lipid metabolism in the evolution of atherosclerosis. Br. Med. Bull. 2018, 126, 101–112. [Google Scholar] [CrossRef]
- World Health Organization. Global Report on Diabetes. Available online: http://apps.who.int/iris/bitstream/10665/204871/1/9789241565257_eng.pdf?ua=1 (accessed on 21 April 2020).
- Eid, S.; Sas, K.M.; Abcouwer, S.F.; Feldman, E.L.; Gardner, T.W.; Pennathur, S.; Fort, P.E. New insights into the mechanisms of diabetic complications: Role of lipids and lipid metabolism. Diabetologia 2019, 62, 1539–1549. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Kuchibhotla, S.; Westfall, K.M.; Silverstein, R.L.; Morton, R.E.; Febbraio, M. A CD36-dependent pathway enhances macrophage and adipose tissue inflammation and impairs insulin signalling. Cardiovasc. Res. 2011, 89, 604–613. [Google Scholar] [CrossRef]
- Cai, L.; Wang, Z.; Ji, A.; Meyer, J.M.; van der Westhuyzen, D.R. Scavenger receptor CD36 expression contributes to adipose tissue inflammation and cell death in diet-induced obesity. PLoS ONE 2012, 7, e36785. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef]
- Chen, X.F.; Wang, L.; Wu, Y.Z.; Song, S.Y.; Min, H.Y.; Yang, Y.; He, X.; Liang, Q.; Yi, L.; Wang, Y.; et al. Effect of puerarin in promoting fatty acid oxidation by increasing mitochondrial oxidative capacity and biogenesis in skeletal muscle in diabetic rats. Nutr. Diabetes 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Yao, L.; Lin, X.; Gu, T.; Rong, X.; Batey, R.; Yamahara, J.; Wang, J.; Li, Y. A mixture of apple pomace and rosemary extract improves fructose consumption-induced insulin resistance in rats: Modulation of sarcolemmal CD36 and glucose transporter-4. Am. J. Transl. Res. 2016, 8, 3791–3801. [Google Scholar] [PubMed]
- Mansor, L.S.; Sousa Fialho, M.D.L.; Yea, G.; Coumans, W.A.; West, J.A.; Kerr, M.; Carr, C.A.; Luiken, J.J.; Glatz, J.F.C.; Evans, R.D.; et al. Inhibition of sarcolemmal FAT/CD36 by sulfo-N-succinimidyl oleate rapidly corrects metabolism and restores function in the diabetic heart following hypoxia/reoxygenation. Cardiovasc. Res. 2017, 113, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 dependent redoxosomes promotes ceramide-mediated pancreatic β-cell failure via p66Shc activation. Free Radic. Biol. Med. 2019, 134, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential protection against type 2 diabetes in obesity through lower CD36 expression and improved exocytosis in β-cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 mediated fatty acid-induced podocyte apoptosis via oxidative stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gu, C.; Li, Y.; Huang, J. High glucose promotes CD36 expression by upregulating peroxisome proliferator-activated receptor γ levels to exacerbate lipid deposition in renal tubular cells. Biomed. Res. Int. 2017, 2017, 1414070. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chen, Q.; Ma, K.; Ju, Y.; Ji, T.; Wang, Z.; Li, W.; Li, W. Astragaloside IV inhibits palmitate-mediated oxidative stress and fibrosis in human glomerular mesangial cells via downregulation of CD36 expression. Pharmacol. Rep. 2019, 71, 319–329. [Google Scholar] [CrossRef]
- Li, X.; Zhang, T.; Geng, J.; Wu, Z.; Xu, L.; Liu, J.; Tian, J.; Zhou, Z.; Nie, J.; Bai, X. Advanced oxidation protein products promote lipotoxicity and tubulointerstitial fibrosis via CD36/β-catenin pathway in diabetic nephropathy. Antioxid. Redox Signal. 2019, 31, 521–538. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Interaction of palmitate and LPS regulates cytokine expression and apoptosis through sphingolipids in human retinal microvascular endothelial cells. Exp. Eye Res. 2019, 178, 61–71. [Google Scholar] [CrossRef]
- Fu, D.; Yu, J.Y.; Wu, M.; Du, M.; Chen, Y.; Abdelsamie, S.A.; Li, Y.; Chen, J.; Boulton, M.E.; Ma, J.X.; et al. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J. Lipid Res. 2014, 55, 860–869. [Google Scholar] [CrossRef]
- Mellal, K.; Omri, S.; Mulumba, M.; Tahiri, H.; Fortin, C.; Dorion, M.F.; Pham, H.; Garcia Ramos, Y.; Zhang, J.; Pundir, S.; et al. Immunometabolic modulation of retinal inflammation by CD36 ligand. Sci. Rep. 2019, 9, 12903. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, W. Gene expression profiling reveals candidate biomarkers and probable molecular mechanism in diabetic peripheral neuropathy. Diabetes Metab. Syndr. Obes. 2019, 12, 1213–1223. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Guo, K.; Eid, S.A.; Rumora, A.E.; Hinder, L.M.; Hayes, J.M.; Mendelson, F.E.; Hur, J.; Feldman, E.L. Integrated lipidomic and transcriptomic analyses identify altered nerve triglycerides in mouse models of prediabetes and type 2 diabetes. Dis. Model. Mech. 2020, 13, dmm042101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bao, M.; Dai, M.; Wang, X.; He, W.; Tan, T.; Lin, D.; Wang, W.; Wen, Y.; Zhang, R. Cardiospecific CD36 suppression by lentivirus-mediated RNA interference prevents cardiac hypertrophy and systolic dysfunction in high-fat-diet induced obese mice. Cardiovasc. Diabetol. 2015, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Czarnowska, E.; Domal-Kwiatkowska, D.; Reichman-Warmusz, E.; Bierla, J.B.; Sowinska, A.; Ratajska, A.; Goral-Radziszewska, K.; Wojnicz, R. The correlation of PPARα activity and cardiomyocyte metabolism and structure in idiopathic dilated cardiomyopathy during heart failure progression. PPAR Res. 2016, 2016, 7508026. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Fan, J.; Zhao, Y.; Zhang, X.; Dai, B.; Zhan, J.; Yin, Z.; Nie, X.; Fu, X.D.; Chen, C.; et al. Nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ. Res. 2019, 125, 1106–1120. [Google Scholar] [CrossRef]
- Wang, S.; Wong, L.Y.; Neumann, D.; Liu, Y.; Sun, A.; Antoons, G.; Strzelecka, A.; Glatz, J.F.C.; Nabben, M.; Luiken, J.J. Augmenting vacuolar H(+)-ATPase function prevents cardiomyocytes from lipid-overload induced dysfunction. Int. J. Mol. Sci. 2020, 21, 1520. [Google Scholar] [CrossRef]
- Hur, J.; Sullivan, K.A.; Pande, M.; Hong, Y.; Sima, A.A.; Jagadish, H.V.; Kretzler, M.; Feldman, E.L. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain 2011, 134, 3222–3235. [Google Scholar] [CrossRef]
- Bonen, A.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Heigenhauser, G.J. The fatty acid transporter FAT/CD36 is upregulated in subcutaneous and visceral adipose tissues in human obesity and type 2 diabetes. Int. J. Obes. (Lond.) 2006, 30, 877–883. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.L.; Ong, H.; Vance, D.E.; Dyck, J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar] [CrossRef]
- Steneberg, P.; Sykaras, A.G.; Backlund, F.; Straseviciene, J.; Söderström, I.; Edlund, H. Hyperinsulinemia enhances hepatic expression of the fatty acid transporter Cd36 and provokes hepatosteatosis and hepatic insulin resistance. J. Biol. Chem. 2015, 290, 19034–19043. [Google Scholar] [CrossRef]
- Lou, P.H.; Lucchinetti, E.; Scott, K.Y.; Huang, Y.; Gandhi, M.; Hersberger, M.; Clanachan, A.S.; Lemieux, H.; Zaugg, M. Alterations in fatty acid metabolism and sirtuin signaling characterize early type-2 diabetic hearts of fructose-fed rats. Physiol. Rep. 2017, 5, e13388. [Google Scholar] [CrossRef]
- Farhangkhoee, H.; Khan, Z.A.; Barbin, Y.; Chakrabarti, S. Glucose-induced up-regulation of CD36 mediates oxidative stress and microvascular endothelial cell dysfunction. Diabetologia 2005, 48, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Man, CD36 (173510). Available online: https://omim.org/entry/173510 (accessed on 29 April 2020).
- Kobylka, D.; Carraway, K.L. Proteolytic digestion of proteins of the milk fat globule membrane. Biochim. Biophys. Acta 1973, 307, 133–140. [Google Scholar] [CrossRef]
- Clemetson, K.J.; Pfueller, S.L.; Luscher, E.F.; Jenkins, C.S. Isolation of the membrane glycoproteins of human blood platelets by lectin affinity chromatography. Biochim. Biophys. Acta 1977, 464, 493–508. [Google Scholar] [CrossRef]
- Tandon, N.N.; Kralisz, U.; Jamieson, G.A. Identification of glycoprotein IV (CD36) as a primary receptor for platelet-collagen adhesion. J. Biol. Chem. 1989, 264, 7576–7583. [Google Scholar] [PubMed]
- Tandon, N.N.; Lipsky, R.H.; Burgess, W.H.; Jamieson, G.A. Isolation and characterization of platelet glycoprotein IV (CD36). J. Biol. Chem. 1989, 264, 7570–7575. [Google Scholar]
- Fernández-Ruiz, E.; Armesilla, A.L.; Sánchez-Madrid, F.; Vega, M.A. Gene encoding the collagen type I and thrombospondin receptor CD36 is located on chromosome 7q11.2. Genomics 1993, 17, 759–761. [Google Scholar] [CrossRef]
- Armesilla, A.L.; Vega, M.A. Structural organization of the gene for human CD36 glycoprotein. J. Biol. Chem. 1994, 269, 18985–18991. [Google Scholar]
- Armesilla, A.L.; Calvo, D.; Vega, M.A. Structural and functional characterization of the human CD36 gene promoter: Identification of a proximal PEBP2/CBF site. J. Biol. Chem. 1996, 271, 7781–7787. [Google Scholar] [CrossRef]
- Rać, M.E.; Safranow, K.; Poncyljusz, W. Molecular basis of human CD36 gene mutations. Mol. Med. 2007, 13, 288–296. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef]
- Andersen, M.; Lenhard, B.; Whatling, C.; Eriksson, P.; Odeberg, J. Alternative promoter usage of the membrane glycoprotein CD36. BMC Mol. Biol. 2006, 7, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Glatz, J.F.; Luiken, J.J. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Niculite, C.M.; Enciu, A.M.; Hinescu, M.E. CD36: Focus on epigenetic and post-transcriptional regulation. Front. Genet. 2019, 10, 680. [Google Scholar] [CrossRef]
- Qiao, L.; Zou, C.; Shao, P.; Schaack, J.; Johnson, P.F.; Shao, J. Transcriptional regulation of fatty acid translocase/CD36 expression by CCAAT/enhancer-binding protein alpha. J. Biol. Chem. 2008, 283, 8788–8795. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, L.; Laviolette, M.; Rodrigue-Way, A.; Sow, B.; Brochu, M.; Caron, V.; Tremblay, A. The CD36-PPARγ pathway in metabolic disorders. Int. J. Mol. Sci. 2018, 19, 1529. [Google Scholar] [CrossRef]
- Raghavan, S.; Singh, N.K.; Gali, S.; Mani, A.M.; Rao, G.N. Protein kinase Cθ via activating transcription factor 2-mediated CD36 expression and foam cell formation of Ly6Chi cells contributes to atherosclerosis. Circulation 2018, 138, 2395–2412. [Google Scholar] [CrossRef]
- Neculai, D.; Schwake, M.; Ravichandran, M.; Zunke, F.; Collins, R.F.; Peters, J.; Neculai, M.; Plumb, J.; Loppnau, P.; Pizarro, J.C.; et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013, 504, 172–176. [Google Scholar] [CrossRef]
- Van Schravendijk, M.R.; Handunnetti, S.M.; Barnwell, J.W.; Howard, R.J. Normal human erythrocytes express CD36, an adhesion molecule of monocytes, platelets, and endothelial cells. Blood 1992, 80, 2105–2114. [Google Scholar] [CrossRef]
- Savill, J.; Hogg, N.; Ren, Y.; Haslett, C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Investig. 1992, 90, 1513–1522. [Google Scholar] [CrossRef]
- Berendt, A.R.; Simmons, D.L.; Tansey, J.; Newbold, C.I.; Marsh, K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature 1989, 341, 57–59. [Google Scholar] [CrossRef]
- Abumrad, N.A.; el-Maghrabi, M.R.; Amri, E.Z.; Lopez, E.; Grimaldi, P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 1993, 268, 17665–17668. [Google Scholar] [PubMed]
- Van Nieuwenhoven, F.A.; Verstijnen, C.P.; Abumrad, N.A.; Willemsen, P.H.; van Eys, G.J.; van der Vusse, G.J.; Glatz, J.F. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem. Biophys. Res. Commun. 1995, 207, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bao, Y.; Yang, Y.; Wu, Y.; Chen, X.; Si, S.; Hong, B. Discovery of antagonists for human scavenger receptor CD36 via an ELISA-like high-throughput screening assay. J. Biomol. Screen. 2010, 15, 239–250. [Google Scholar] [CrossRef]
- Alkhatatbeh, M.J.; Mhaidat, N.M.; Enjeti, A.K.; Lincz, L.F.; Thorne, R.F. The putative diabetic plasma marker, soluble CD36, is non-cleaved, non-soluble and entirely associated with microparticles. J. Thromb. Haemost. 2011, 9, 844–851. [Google Scholar] [CrossRef]
- Li, S.; Wei, J.; Zhang, C.; Li, X.; Meng, W.; Mo, X.; Zhang, Q.; Liu, Q.; Ren, K.; Du, R.; et al. Cell-derived microparticles in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. Cell. Physiol. Biochem. 2016, 39, 2439–2450. [Google Scholar] [CrossRef]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Kuniyasu, A.; Horiuchi, S.; Nakayama, H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann. N. Y. Acad. Sci. 2001, 947, 350–355. [Google Scholar] [CrossRef]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar]
- Hoebe, K.; Georgel, P.; Rutschmann, S.; Du, X.; Mudd, S.; Crozat, K.; Sovath, S.; Shamel, L.; Hartung, T.; Zähringer, U.; et al. CD36 is a sensor of diacylglycerides. Nature 2005, 433, 523–527. [Google Scholar] [CrossRef]
- Albert, M.L.; Pearce, S.F.; Francisco, L.M.; Sauter, B.; Roy, P.; Silverstein, R.L.; Bhardwaj, N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998, 188, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar] [CrossRef] [PubMed]
- Khawandanah, J. Double or hybrid diabetes: A systematic review on disease prevalence, characteristics and risk factors. Nutr. Diabetes 2019, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Lopez-Carmona, M.D.; Plaza-Seron, M.C.; Vargas-Candela, A.; Tinahones, F.J.; Gomez-Huelgas, R.; Bernal-Lopez, M.R. CD36 overexpression: A possible etiopathogenic mechanism of atherosclerosis in patients with prediabetes and diabetes. Diabetol. Metab. Syndr. 2017, 9, 55. [Google Scholar] [CrossRef]
- Handberg, A.; Levin, K.; Højlund, K.; Beck-Nielsen, H. Identification of the oxidized low-density lipoprotein scavenger receptor CD36 in plasma: A novel marker of insulin resistance. Circulation 2006, 114, 1169–1176. [Google Scholar] [CrossRef]
- Ekici, M.; Kisa, U.; Durmaz, S.A.; Ugur, E.; Nergiz-Unal, R. Fatty acid transport receptor soluble CD36 and dietary fatty acid pattern in type 2 diabetic patients: A comparative study. Br. J. Nutr. 2018, 119, 153–162. [Google Scholar] [CrossRef]
- Kulkarni, N.B.; Ganu, M.U.; Godbole, S.G.; Deo, S.S. Assessment of potential biomarkers of atherosclerosis in Indian patients with type 2 diabetes mellitus. Indian J. Med. Res. 2018, 147, 169–176. [Google Scholar] [CrossRef]
- Koonen, D.P.; Jensen, M.K.; Handberg, A. Soluble CD36- a marker of the (pathophysiological) role of CD36 in the metabolic syndrome? Arch. Physiol. Biochem. 2011, 117, 57–63. [Google Scholar] [CrossRef]
- Liani, R.; Halvorsen, B.; Sestili, S.; Handberg, A.; Santilli, F.; Vazzana, N.; Formoso, G.; Aukrust, P.; Davì, G. Plasma levels of soluble CD36, platelet activation, inflammation, and oxidative stress are increased in type 2 diabetic patients. Free Radic. Biol. Med. 2012, 52, 1318–1324. [Google Scholar] [CrossRef] [PubMed]
- Castelblanco, E.; Sanjurjo, L.; Falguera, M.; Hernández, M.; Fernandez-Real, J.M.; Sarrias, M.R.; Alonso, N.; Mauricio, D. Circulating soluble CD36 is similar in type 1 and type 2 diabetes mellitus versus non-diabetic subjects. J. Clin. Med. 2019, 8, 710. [Google Scholar] [CrossRef] [PubMed]
- Handberg, A.; Lopez-Bermejo, A.; Bassols, J.; Vendrell, J.; Ricart, W.; Fernandez-Real, J.M. Circulating soluble CD36 is associated with glucose metabolism and interleukin-6 in glucose-intolerant men. Diabetes Vasc. Dis. Res. 2009, 6, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Handberg, A.; Norberg, M.; Stenlund, H.; Hallmans, G.; Attermann, J.; Eriksson, J.W. Soluble CD36 (sCD36) clusters with markers of insulin resistance, and high sCD36 is associated with increased type 2 diabetes risk. J. Clin. Endocrinol. Metab. 2010, 95, 1939–1946. [Google Scholar] [CrossRef]
- Handberg, A.; Højlund, K.; Gastaldelli, A.; Flyvbjerg, A.; Dekker, J.M.; Petrie, J.; Piatti, P.; Beck-Nielsen, H. RISC Investigators. Plasma sCD36 is associated with markers of atherosclerosis, insulin resistance and fatty liver in a nondiabetic healthy population. J. Intern. Med. 2012, 271, 294–304. [Google Scholar] [CrossRef]
- Alkhatatbeh, M.J.; Ayoub, N.M.; Mhaidat, N.M.; Saadeh, N.A.; Lincz, L.F. Soluble cluster of differentiation 36 concentrations are not associated with cardiovascular risk factors in middle-aged subjects. Biomed. Rep. 2016, 4, 642–648. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; Aroner, S.; Overvad, K.; Cai, T.; Yang, M.; Tjønneland, A.; Handberg, A.; Jensen, M.K. Plasma CD36 and incident diabetes: A case-cohort study in danish men and women. Diabetes Metab. J. 2020, 44, 134–142. [Google Scholar] [CrossRef]
- Kim, H.J.; Moon, J.S.; Park, I.R.; Kim, J.H.; Yoon, J.S.; Won, K.C.; Lee, H.W. A novel index using soluble CD36 is associated with the prevalence of type 2 diabetes mellitus: Comparison study with triglyceride-glucose index. Endocrinol. Metab. 2017, 32, 375–382. [Google Scholar] [CrossRef]
- Chmielewski, M.; Bragfors-Helin, A.C.; Stenvinkel, P.; Lindholm, B.; Anderstam, B. Serum soluble CD36, assessed by a novel monoclonal antibody-based sandwich ELISA, predicts cardiovascular mortality in dialysis patients. Clin. Chim. Acta 2010, 411, 2079–2082. [Google Scholar] [CrossRef]
- Alkhatatbeh, M.J.; Enjeti, A.K.; Acharya, S.; Thorne, R.F.; Lincz, L.F. The origin of circulating CD36 in type 2 diabetes. Nutr. Diabetes 2013, 3, e59. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; Handberg, A.; Ortega, F.; Højlund, K.; Vendrell, J.; Ricart, W. Circulating soluble CD36 is a novel marker of liver injury in subjects with altered glucose tolerance. J. Nutr. Biochem. 2009, 20, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Glintborg, D.; Højlund, K.; Andersen, M.; Henriksen, J.E.; Beck-Nielsen, H.; Handberg, A. Soluble CD36 and risk markers of insulin resistance and atherosclerosis are elevated in polycystic ovary syndrome and significantly reduced during pioglitazone treatment. Diabetes Care 2008, 31, 328–334. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Palmer, A.K.; Tchkonia, T.; LeBrasseur, N.K.; Chini, E.N.; Xu, M.; Kirkland, J.L. Cellular senescence in type 2 diabetes: A therapeutic opportunity. Diabetes 2015, 64, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Regulski, M. Understanding diabetic induction of cellular senescence: A concise review. Wounds 2018, 30, 96–101. [Google Scholar] [PubMed]
- Takasugi, M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging Cell 2018, 17, e12734. [Google Scholar] [CrossRef]
- Knøsgaard, L.; Kazankov, K.; Birkebæk, N.H.; Holland-Fischer, P.; Lange, A.; Solvig, J.; Hørlyck, A.; Kristensen, K.; Rittig, S.; Vilstrup, H.; et al. Reduced sCD36 following weight loss corresponds to improved insulin sensitivity, dyslipidemia and liver fat in obese children. Eur. J. Clin. Nutr. 2016, 70, 1073–1077. [Google Scholar] [CrossRef]
- Knøsgaard, L.; Thomsen, S.B.; Støckel, M.; Vestergaard, H.; Handberg, A. Circulating sCD36 is associated with unhealthy fat distribution and elevated circulating triglycerides in morbidly obese individuals. Nutr. Diabetes 2014, 4, e114. [Google Scholar] [CrossRef]
- Rehman, K.; Akash, M.S. Mechanisms of inflammatory responses and development of insulin resistance: How are they interlinked? J. Biomed. Sci. 2016, 23, 87. [Google Scholar] [CrossRef]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.S.; et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Samovski, D.; Okunade, A.L.; Stahl, P.D.; Abumrad, N.A.; Su, X. CD36 level and trafficking are determinants of lipolysis in adipocytes. FASEB J. 2012, 26, 4733–4742. [Google Scholar] [CrossRef]
- Wan, Z.; Matravadia, S.; Holloway, G.P.; Wright, D.C. FAT/CD36 regulates PEPCK expression in adipose tissue. Am. J. Physiol. Cell Physiol. 2013, 304, C478–C484. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Arumugam, Y.; Dyck, D.J.; Bell, R.C.; Pelsers, M.M.; Turcotte, L.P.; Tandon, N.N.; Glatz, J.F.; Bonen, A. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J. Biol. Chem. 2001, 276, 40567–40573. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Park, K.W.; Cho, S. Inhibition of the CD36 receptor reduces visceral fat accumulation and improves insulin resistance in obese mice carrying the BDNF-Val66Met variant. J. Biol. Chem. 2018, 293, 13338–13348. [Google Scholar] [CrossRef] [PubMed]
- Pietka, T.A.; Schappe, T.; Conte, C.; Fabbrini, E.; Patterson, B.W.; Klein, S.; Abumrad, N.A.; Love-Gregory, L. Adipose and muscle tissue profile of CD36 transcripts in obese subjects highlights the role of CD36 in fatty acid homeostasis and insulin resistance. Diabetes Care 2014, 37, 1990–1997. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Kim, S.H.; Chung, J.H.; Song, S.W.; Jung, W.S.; Lee, Y.A.; Kim, H.N. Relationship between deep subcutaneous abdominal adipose tissue and metabolic syndrome: A case control study. Diabetol. Metab. Syndr. 2016, 8, 10. [Google Scholar] [CrossRef]
- Vroegrijk, I.O.; van Klinken, J.B.; van Diepen, J.A.; van den Berg, S.A.; Febbraio, M.; Steinbusch, L.K.; Glatz, J.F.; Havekes, L.M.; Voshol, P.J.; Rensen, P.C.; et al. CD36 is important for adipocyte recruitment and affects lipolysis. Obesity (Silver Spring) 2013, 21, 2037–2045. [Google Scholar] [CrossRef]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA. 2009, 106, 15430–15435. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Wang, H.; Liang, F. Mechanisms linking inflammation to insulin resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Strieder-Barboza, C.; Baker, N.A.; Flesher, C.G.; Karmakar, M.; Neeley, C.K.; Polsinelli, D.; Dimick, J.B.; Finks, J.F.; Ghaferi, A.A.; Varban, O.A.; et al. Advanced glycation end-products regulate extracellular matrix-adipocyte metabolic crosstalk in diabetes. Sci. Rep. 2019, 9, 19748. [Google Scholar] [CrossRef]
- Unno, Y.; Sakai, M.; Sakamoto, Y.; Kuniyasu, A.; Nakayama, H.; Nagai, R.; Horiuchi, S. Advanced glycation end products-modified proteins and oxidized LDL mediate down-regulation of leptin in mouse adipocytes via CD36. Biochem. Biophys. Res. Commun. 2004, 325, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Unno, Y.; Sakai, M.; Sakamoto, Y.; Kuniyasu, A.; Nagai, R.; Nakayama, H.; Horiuchi, S. Glycolaldehyde-modified bovine serum albumin downregulates leptin expression in mouse adipocytes via a CD36-mediated pathway. Ann. N. Y. Acad. Sci. 2005, 1043, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.H.; Ginsberg, H.N. Adipocyte signaling and lipid homeostasis: Sequelae of insulin-resistant adipose tissue. Circ. Res. 2005, 96, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Luiken, J.J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid. Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef]
- Jedidi, I.; Couturier, M.; Thérond, P.; Gardès-Albert, M.; Legrand, A.; Barouki, R.; Bonnefont-Rousselot, D.; Aggerbeck, M. Cholesteryl ester hydroperoxides increase macrophage CD36 gene expression via PPARalpha. Biochem. Biophys. Res. Commun. 2006, 351, 733–738. [Google Scholar] [CrossRef]
- Kotla, S.; Rao, G.N. Reactive oxygen species (ROS) mediate p300-dependent STAT1 protein interaction with peroxisome proliferator-activated receptor (PPAR)-γ in CD36 protein expression and foam cell formation. J. Biol. Chem. 2015, 290, 30306–30320. [Google Scholar] [CrossRef]
- Goto, K.; Iso, T.; Hanaoka, H.; Yamaguchi, A.; Suga, T.; Hattori, A.; Irie, Y.; Shinagawa, Y.; Matsui, H.; Syamsunarno, M.R.; et al. Peroxisome proliferator-activated receptor-γ in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J. Am. Heart Assoc. 2013, 2, e004861. [Google Scholar] [CrossRef]
- Lim, H.J.; Lee, S.; Lee, K.S.; Park, J.H.; Jang, Y.; Lee, E.J.; Park, H.Y. PPARgamma activation induces CD36 expression and stimulates foam cell like changes in rVSMCs. Prostaglandins Other Lipid Mediat. 2006, 80, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Caspar-Bauguil, S.; Kolditz, C.I.; Lefort, C.; Vila, I.; Mouisel, E.; Beuzelin, D.; Tavernier, G.; Marques, M.A.; Zakaroff-Girard, A.; Pecher, C.; et al. Fatty acids from fat cell lipolysis do not activate an inflammatory response but are stored as triacylglycerols in adipose tissue macrophages. Diabetologia 2015, 58, 2627–2636. [Google Scholar] [CrossRef] [PubMed]
- Coats, B.R.; Schoenfelt, K.Q.; Barbosa-Lorenzi, V.C.; Peris, E.; Cui, C.; Hoffman, A.; Zhou, G.; Fernandez, S.; Zhai, L.; Hall, B.A.; et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep. 2017, 20, 3149–3161. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Castro, R.; Soto-Rodriguez, I.; Deschamps-Lago, R.A.; Gruber-Pagola, P.; Rodriguez Antolin, J.; Peres Quintal, A.; Rivera, J.R.; Alexander-Aguilera, A. Dietary sucrose regulates the expression of the Cd36 gene in hepatic tissue of rats with obesity and Non Alcoholic Fatty Liver Disease (NAFLD). Biomed. Pap. 2018, 162, 99–106. [Google Scholar] [CrossRef]
- Quintana-Castro, R.; Aguirre-Maldonado, I.; Soto-Rodríguez, I.; Deschamps-Lago, R.A.; Gruber-Pagola, P.; Urbina de Larrea, Y.K.; Juárez-Rivera, V.E.; Ramos-Manuel, L.E.; Alexander-Aguilera, A. Cd36 gene expression in adipose and hepatic tissue mediates the lipids accumulation in liver of obese rats with sucrose-induced hepatic steatosis. Prostaglandins Other Lipid Mediat. 2020, 147, 106404. [Google Scholar] [CrossRef]
- Park, W.J.; Park, J.W.; Merrill, A.H.; Storch, J.; Pewzner-Jung, Y.; Futerman, A.H. Hepatic fatty acid uptake is regulated by the sphingolipid acyl chain length. Biochim. Biophys. Acta 2014, 1841, 1754–1766. [Google Scholar] [CrossRef]
- Buqué, X.; Cano, A.; Miquilena-Colina, M.E.; García-Monzón, C.; Ochoa, B.; Aspichueta, P. High insulin levels are required for FAT/CD36 plasma membrane translocation and enhanced fatty acid uptake in obese Zucker rat hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E504–E514. [Google Scholar] [CrossRef]
- Nassir, F.; Adewole, O.L.; Brunt, E.M.; Abumrad, N.A. CD36 deletion reduces VLDL secretion, modulates liver prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. J. Lipid Res. 2013, 54, 2988–2997. [Google Scholar] [CrossRef]
- Heebøll, S.; Poulsen, M.K.; Ornstrup, M.J.; Kjær, T.N.; Pedersen, S.B.; Nielsen, S.; Grønbæk, H.; Handberg, A. Circulating sCD36 levels in patients with non-alcoholic fatty liver disease and controls. Int. J. Obes. (Lond.) 2017, 41, 262–267. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.W.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Sokolowska, E.; Blachnio-Zabielska, A. The role of ceramides in insulin resistance. Front. Endocrinol. (Lausanne) 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A. Ceramides: Nutrient signals that drive hepatosteatosis. J. Lipid Atheroscler. 2020, 9, 50–65. [Google Scholar] [CrossRef]
- Xia, J.Y.; Holland, W.L.; Kusminski, C.M.; Sun, K.; Sharma, A.X.; Pearson, M.J.; Sifuentes, A.J.; McDonald, J.G.; Gordillo, R.; Scherer, P.E. Targeted induction of ceramide degradation leads to improved systemic metabolism and reduced hepatic steatosis. Cell Metab. 2015, 22, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Irimia, J.M.; Meyer, C.M.; Segvich, D.M.; Surendran, S.; DePaoli-Roach, A.A.; Morral, N.; Roach, P.J. Lack of liver glycogen causes hepatic insulin resistance and steatosis in mice. J. Biol. Chem. 2017, 292, 10455–10464. [Google Scholar] [CrossRef]
- Garbacz, W.G.; Lu, P.; Miller, T.M.; Poloyac, S.M.; Eyre, N.S.; Mayrhofer, G.; Xu, M.; Ren, S.; Xie, W. Hepatic overexpression of CD36 improves glycogen homeostasis and attenuates high-fat diet-induced hepatic steatosis and insulin resistance. Mol. Cell. Biol. 2016, 36, 2715–2727. [Google Scholar] [CrossRef]
- Samovski, D.; Sun, J.; Pietka, T.; Gross, R.W.; Ecke, R.H.; Su, X.; Stahl, P.D.; Abumrad, N.A. Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes 2015, 64, 353–359. [Google Scholar] [CrossRef]
- Momken, I.; Chabowski, A.; Dirkx, E.; Nabben, M.; Jain, S.S.; McFarlan, J.T.; Glatz, J.F.; Luiken, J.J.; Bonen, A. A new leptin-mediated mechanism for stimulating fatty acid oxidation: A pivotal role for sarcolemmal FAT/CD36. Biochem. J. 2017, 474, 149–162. [Google Scholar] [CrossRef]
- Holloway, G.P.; Schwenk, R.W.; Luiken, J.J.; Glatz, J.F.; Bonen, A. Fatty acid transport in skeletal muscle: Role in energy provision and insulin resistance. Clin. Lipidol. 2010, 5, 731–745. [Google Scholar] [CrossRef]
- Samovski, D.; Dhule, P.; Pietka, T.; Jacome-Sosa, M.; Penrose, E.; Son, N.H.; Flynn, C.R.; Shoghi, K.I.; Hyrc, K.L.; Goldberg, I.J.; et al. Regulation of insulin receptor pathway and glucose metabolism by CD36 signaling. Diabetes 2018, 67, 1272–1284. [Google Scholar] [CrossRef]
- Mullen, K.L.; Pritchard, J.; Ritchie, I.; Snook, L.A.; Chabowski, A.; Bonen, A.; Wright, D.; Dyck, D.J. Adiponectin resistance precedes the accumulation of skeletal muscle lipids and insulin resistance in high-fat-fed rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R243–R251. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Parolin, M.L.; Steinberg, G.R.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Heigenhauser, G.J.; Dyck, D.J. Triacylglycerol accumulation in human obesity and type 2 diabetes is associated with increased rates of skeletal muscle fatty acid transport and increased sarcolemmal FAT/CD36. FASEB J. 2004, 18, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; Mercier, J.; Man, C.Y.; Metz, L.; Bordenave, S.; Lambert, K.; Jean, E.; Lantier, L.; Bounoua, L.; Brun, J.F.; et al. Intramyocellular lipid accumulation is associated with permanent relocation ex vivo and in vitro of fatty acid translocase (FAT)/CD36 in obese patients. Diabetologia 2010, 53, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; Foretz, M.; Lantier, L.; Hebrard, S.; Viollet, B.; Mercier, J.; Kitzmann, M. Increased FAT/CD36 cycling and lipid accumulation in myotubes derived from obese type 2 diabetic patients. PLoS ONE 2011, 6, e28981. [Google Scholar] [CrossRef] [PubMed]
- Chabowski, A.; Coort, S.L.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Insulin stimulates fatty acid transport by regulating expression of FAT/CD36 but not FABPpm. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E781–E789. [Google Scholar] [CrossRef]
- Liu, Y.; Steinbusch, L.K.M.; Nabben, M.; Kapsokalyvas, D.; van Zandvoort, M.; Schönleitner, P.; Antoons, G.; Simons, P.J.; Coumans, W.A.; Geomini, A.; et al. Palmitate-induced vacuolar-type H(+)-ATPase inhibition feeds forward into insulin resistance and contractile dysfunction. Diabetes 2017, 66, 1521–1534. [Google Scholar] [CrossRef]
- Zhu, B.; Li, M.Y.; Lin, Q.; Liang, Z.; Xin, Q.; Wang, M.; He, Z.; Wang, X.; Wu, X.; Chen, G.G.; et al. Lipid oversupply induces CD36 sarcolemmal translocation via dual modulation of PKCζ and TBC1D1: An early event prior to insulin resistance. Theranostics 2020, 10, 1332–1354. [Google Scholar] [CrossRef]
- Luiken, J.J.; Nabben, M.; Neumann, D.; Glatz, J.F. Understanding the distinct subcellular trafficking of CD36 and GLUT4 during the development of myocardial insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165775. [Google Scholar] [CrossRef]
- Glatz, J.F.; Angin, Y.; Steinbusch, L.K.; Schwenk, R.W.; Luiken, J.J. CD36 as a target to prevent cardiac lipotoxicity and insulin resistance. Prostaglandins Leukot. Essent. Fatty Acids 2013, 88, 71–77. [Google Scholar] [CrossRef]
- Summers, S.A.; Goodpaster, B.H. CrossTalk proposal: Intramyocellular ceramide accumulation does modulate insulin resistance. J. Physiol. 2016, 594, 3167–3170. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Jurczak, M.J. CrossTalk opposing view: Intramyocellular ceramide accumulation does not modulate insulin resistance. J. Physiol. 2016, 594, 3171–3174. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Cooney, G.J.; Kraegen, E.W.; Bruce, C.R. Fatty acid metabolism, energy expenditure and insulin resistance in muscle. J. Endocrinol. 2014, 220, T61–T79. [Google Scholar] [CrossRef]
- Abel, E.D. Free fatty acid oxidation in insulin resistance and obesity. Heart Metab. 2010, 48, 5–10. [Google Scholar]
- Lopaschuk, G.D. Fatty acid oxidation and its relation with insulin resistance and associated disorders. Ann. Nutr. Metab. 2016, 68, 15–20. [Google Scholar] [CrossRef]
- Perona, J.S. Membrane lipid alterations in the metabolic syndrome and the role of dietary oils. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1690–1703. [Google Scholar] [CrossRef]
- Weir, G.C. Glucolipotoxicity, β-cells, and diabetes: The emperor has no clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Yoon, J.S.; Moon, J.S.; Kim, Y.W.; Won, K.C.; Lee, H.W. The glucotoxicity protecting effect of ezetimibe in pancreatic beta cells via inhibition of CD36. J. Korean Med. Sci. 2016, 31, 547–552. [Google Scholar] [CrossRef]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. Diabetes mellitus and the β cell: The last ten years. Cell 2012, 148, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Karunakaran, U.; Lee, I.K.; Moon, J.S.; Won, K.C. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017, 11, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Moon, J.S.; Seo, Y.J.; Park, S.Y.; Kim, J.Y.; Yoon, J.S.; Lee, I.K.; Lee, H.W.; Won, K.C. Inhibition of fatty acid translocase cluster determinant 36 (CD36), stimulated by hyperglycemia, prevents glucotoxicity in INS-1 cells. Biochem. Biophys. Res. Commun. 2012, 420, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, C.S.; Collins, S.; Bengtsson, M.; Eliasson, L.; Salehi, A.; Shimomura, K.; Tarasov, A.; Holm, C.; Ashcroft, F.; Rorsman, P. Long-term exposure to glucose and lipids inhibits glucose-induced insulin secretion downstream of granule fusion with plasma membrane. Diabetes 2007, 56, 1888–1897. [Google Scholar] [CrossRef][Green Version]
- Hoppa, M.B.; Collins, S.; Ramracheya, R.; Hodson, L.; Amisten, S.; Zhang, Q.; Johnson, P.; Ashcroft, F.M.; Rorsman, P. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca2+ channels from secretory granules. Cell Metab. 2011, 13, 487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karunakaran, U.; Moon, J.S.; Lee, H.W.; Won, K.C. CD36 initiated signaling mediates ceramide-induced TXNIP expression in pancreatic beta-cells. Biochim. Biophys. Acta 2015, 1852, 2414–2422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Igoillo-Esteve, M.; Marselli, L.; Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Yuan, T.; Maedler, K. Macrophage-associated pro-inflammatory state in human islets from obese individuals. Nutr. Diabetes 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.H.; Wollam, J.; Riopel, M.; et al. Expansion of islet-resident macrophages leads to inflammation affecting β cell proliferation and function in obesity. Cell Metab. 2019, 29, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef] [PubMed]
- Dalgaard, L.T.; Thams, P.; Gaarn, L.W.; Jensen, J.; Lee, Y.C.; Nielsen, J.H. Suppression of FAT/CD36 mRNA by human growth hormone in pancreatic β-cells. Biochem. Biophys. Res. Commun. 2011, 410, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kowluru, A. CD36 mediates lipid accumulation in pancreatic beta cells under the duress of glucolipotoxic conditions: Novel roles of lysine deacetylases. Biochem. Biophys. Res. Commun. 2018, 495, 2221–2226. [Google Scholar] [CrossRef]
- Zheng, S.; Ren, X.; Han, T.; Chen, Y.; Qiu, H.; Liu, W.; Hu, Y. Fenofibrate attenuates fatty acid-induced islet β-cell dysfunction and apoptosis via inhibiting the NF-κB/MIF dependent inflammatory pathway. Metabolism 2017, 77, 23–38. [Google Scholar] [CrossRef]
- Bigagli, E.; Lodovici, M. Circulating oxidative stress biomarkers in clinical studies on type 2 diabetes and its complications. Oxid. Med. Cell. Longev. 2019, 2019, 5953685. [Google Scholar] [CrossRef]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef]
- Shi, X.Y.; Hou, F.F.; Niu, H.X.; Wang, G.B.; Xie, D.; Guo, Z.J.; Zhou, Z.M.; Yang, F.; Tian, J.W.; Zhang, X. Advanced oxidation protein products promote inflammation in diabetic kidney through activation of renal nicotinamide adenine dinucleotide phosphate oxidase. Endocrinology 2008, 149, 1829–1839. [Google Scholar] [CrossRef]
- Zhou, L.L.; Cao, W.; Xie, C.; Tian, J.; Zhou, Z.; Zhou, Q.; Zhu, P.; Li, A.; Liu, Y.; Miyata, T.; et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int. 2012, 82, 759–770. [Google Scholar] [CrossRef]
- Iwao, Y.; Nakajou, K.; Nagai, R.; Kitamura, K.; Anraku, M.; Maruyama, T.; Otagiri, M. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am. J. Physiol. Renal. Physiol. 2008, 295, F1871–F1880. [Google Scholar] [CrossRef]
- Cao, W.; Hou, F.F.; Nie, J. AOPPs and the progression of kidney disease. Kidney Int. Suppl. 2014, 4, 102–106. [Google Scholar] [CrossRef]
- Rivas-Urbina, A.; Benitez, S.; Perez, A.; Sanchez-Quesada, J.L. Modified low-density lipoproteins as biomarkers in diabetes and metabolic syndrome. Front. Biosci. (Landmark Ed.) 2018, 23, 1220–1240. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wu, J.; Qian, Y.; Fu, L.; Wu, G.; Xu, C.; Mei, C. Oxidized high-density lipoprotein impairs the function of human renal proximal tubule epithelial cells through CD36. Int. J. Mol. Med. 2014, 34, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Gutwein, P.; Abdel-Bakky, M.S.; Doberstein, K.; Schramme, A.; Beckmann, J.; Schaefer, L.; Amann, K.; Doller, A.; Kämpfer-Kolb, N.; Abdel-Aziz, A.A.; et al. CXCL16 and oxLDL are induced in the onset of diabetic nephropathy. J. Cell. Mol. Med. 2009, 13, 3809–3825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gao, X.; Wu, J.; Liu, D.; Cai, H.; Fu, L.; Mei, C. Oxidized high-density lipoprotein enhances inflammatory activity in rat mesangial cells. Diabetes Metab. Res. Rev. 2010, 26, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Kuppan, K.; Mohanlal, J.; Mohammad, A.M.; Babu, K.A.; Sen, P.; Undurti, N.D.; Natarajan, V.; Narayanasamy, A. Elevated serum OxLDL is associated with progression of type 2 Diabetes Mellitus to diabetic retinopathy. Exp. Eye Res. 2019, 186, 107668. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T. Abnormal lipoprotein metabolism in diabetic nephropathy. Clin. Exp. Nephrol. 2014, 18, 206–209. [Google Scholar] [CrossRef]
- Schofield, J.D.; Liu, Y.; Rao-Balakrishna, P.; Malik, R.A.; Soran, H. Diabetes dyslipidemia. Diabetes Ther. 2016, 7, 203–219. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Diabetes and Dyslipidemia. Available online: https://www.ncbi.nlm.nih.gov/books/NBK305900/ (accessed on 15 May 2020).
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Chen, Y.; Huang, W.; Viterna, J.; Liu, J.; Westfall, K.; Tian, J.; Bartlett, D.J.; Tang, W.H.; Xie, Z.; et al. CD36 and Na/K-ATPase-α1 form a proinflammatory signaling loop in kidney. Hypertension 2013, 61, 216–224. [Google Scholar] [CrossRef]
- Furuichi, K.; Shimizu, M.; Okada, H.; Narita, I.; Wada, T. Clinico-pathological features of kidney disease in diabetic cases. Clin. Exp. Nephrol. 2018, 22, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.C.; Tang, S.C. Diabetic nephropathy: Landmark clinical trials and tribulations. Nephrol. Dial. Transplant. 2016, 31, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.; Aditya, B.S. An overview of diabetic nephropathy: Epidemiology, pathophysiology and treatment. J. Diabetes Nurs. 2015, 19, 61–67. [Google Scholar]
- Lim, A.K. Diabetic nephropathy—Complications and treatment. Int. J. Nephrol. Renovasc. Dis. 2014, 7, 361–381. [Google Scholar] [CrossRef] [PubMed]
- Magee, C.; Grieve, D.J.; Watson, C.J.; Brazil, D.P. Diabetic nephropathy: A tangled web to unweave. Cardiovasc. Drugs Ther. 2017, 31, 579–592. [Google Scholar] [CrossRef]
- Sagoo, M.K.; Gnudi, L. Diabetic nephropathy: An overview. Methods Mol. Biol. 2020, 2067, 3–7. [Google Scholar] [CrossRef]
- Herman-Edelstein, M.; Scherzer, P.; Tobar, A.; Levi, M.; Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 2014, 55, 561–572. [Google Scholar] [CrossRef]
- Susztak, K.; Ciccone, E.; McCue, P.; Sharma, K.; Böttinger, E.P. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. 2005, 2, e45. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Lee, E.; Choi, J.; Lee, H.S. Palmitate induces mitochondrial superoxide generation and activates AMPK in podocytes. J. Cell. Physiol. 2017, 232, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, X.M.; Sun, J.Y.; Jiang, X.S.; Wu, Y.; Yang, S.; Huang, H.Z.; Ruan, X.Z.; Du, X.G. Palmitic acid-induced podocyte apoptosis via the reactive oxygen species-dependent mitochondrial pathway. Kidney Blood Press. Res. 2018, 43, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Maimaitiyiming, H.; Zhou, Q.; Norman, H.; Zhou, C.; Wang, S. Interaction of thrombospondin1 and CD36 contributes to obesity-associated podocytopathy. Biochim. Biophys. Acta 2015, 1852, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Shi, Y.; Han, B.; Liu, X.; Qiao, X.; Qi, Y.; Wang, L. The antioxidant peptide SS31 prevents oxidative stress, downregulates CD36 and improves renal function in diabetic nephropathy. Nephrol. Dial. Transplant. 2018, 33, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Wintergerst, E.S.; Jelk, J.; Rahner, C.; Asmis, R. Apoptosis induced by oxidized low density lipoprotein in human monocyte-derived macrophages involves CD36 and activation of caspase-3. Eur. J. Biochem. 2000, 267, 6050–6059. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Morcos, M.; Sayed, A.A.; Bierhaus, A.; Yard, B.; Waldherr, R.; Merz, W.; Kloeting, I.; Schleicher, E.; Mentz, S.; Abd el Baki, R.F.; et al. Activation of tubular epithelial cells in diabetic nephropathy. Diabetes 2002, 51, 3532–3544. [Google Scholar] [CrossRef]
- Chang, C.C.; Chen, C.Y.; Huang, C.H.; Wu, C.L.; Wu, H.M.; Chiu, P.F.; Kor, C.T.; Chen, T.H.; Chang, G.D.; Kuo, C.C.; et al. Urinary glycated uromodulin in diabetic kidney disease. Clin. Sci. (Lond.) 2017, 131, 1815–1829. [Google Scholar] [CrossRef]
- Sasaki, H.; Kamijo-Ikemori, A.; Sugaya, T.; Yamashita, K.; Yokoyama, T.; Koike, J.; Sato, T.; Yasuda, T.; Kimura, K. Urinary fatty acids and liver-type fatty acid binding protein in diabetic nephropathy. Nephron Clin. Pract. 2009, 12, c148–c156. [Google Scholar] [CrossRef]
- Hou, Y.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Li, Y.; Shi, Y. CD36 is involved in high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2015, 468, 281–286. [Google Scholar] [CrossRef]
- Loeffler, I.; Wolf, G. Epithelial-to-mesenchymal transition in diabetic nephropathy: Fact or fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef]
- Yang, Y.L.; Lin, S.H.; Chuang, L.Y.; Guh, J.Y.; Liao, T.N.; Lee, T.C.; Chang, W.T.; Chang, F.R.; Hung, M.Y.; Chiang, T.A.; et al. CD36 is a novel and potential anti-fibrogenic target in albumin-induced renal proximal tubule fibrosis. J. Cell. Biochem. 2007, 101, 735–744. [Google Scholar] [CrossRef]
- Cao, W.; Xu, J.; Zhou, Z.M.; Wang, G.B.; Hou, F.F.; Nie, J. Advanced oxidation protein products activate intrarenal renin-angiotensin system via a CD36-mediated, redox-dependent pathway. Antioxid. Redox Signal. 2013, 18, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Shiju, T.M.; Mohan, V.; Balasubramanyam, M.; Viswanathan, P. Soluble CD36 in plasma and urine: A plausible prognostic marker for diabetic nephropathy. J. Diabetes Complicat. 2015, 29, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Sabanayagam, C. Strategies to tackle the global burden of diabetic retinopathy: From epidemiology to artificial intelligence. Ophthalmologica 2020, 243, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kollias, A.N.; Ulbig, M.W. Diabetic retinopathy: Early diagnosis and effective treatment. Dtsch. Arztebl. Int. 2010, 107, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fernandez-Loaiza, P.; Sauma, J.; Hernandez-Bogantes, E.; Masis, M. Classification of diabetic retinopathy and diabetic macular edema. World J. Diabetes 2013, 4, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, K.B.; Frydkjaer-Olsen, U.; Grauslund, J. Vascular changes and neurodegeneration in the early stages of diabetic retinopathy: Which comes first? Ophthalmic Res. 2016, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rossino, M.G.; Dal Monte, M.; Casini, G. Relationships between neurodegeneration and vascular damage in diabetic retinopathy. Front. Neurosci. 2019, 13, 1172. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Rizzolo, L.J. Effects of diabetic retinopathy on the barrier functions of the retinal pigment epithelium. Vis. Res. 2017, 139, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Ponnalagu, M.; Subramani, M.; Jayadev, C.; Shetty, R.; Das, D. Retinal pigment epithelium-secretome: A diabetic retinopathy perspective. Cytokine 2017, 95, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Courtois, Y. The role of CD36 receptor in the phagocytosis of oxidized lipids and AMD. Aging (Albany NY) 2010, 2, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Nawaz, M.I.; Ola, M.S.; De Hertogh, G.; Opdenakker, G.; Geboes, K. Expression of thrombospondin-2 as a marker in proliferative diabetic retinopathy. Acta Ophthalmol. 2013, 91, e169–e177. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Houssier, M.; Bujold, K.; Sapieha, P.; Lubell, W.; Dorfman, A.; Racine, J.; Hardy, P.; Febbraio, M.; Lachapelle, P.; et al. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging (Albany NY) 2010, 2, 981–989. [Google Scholar] [CrossRef]
- Gnanaguru, G.; Choi, A.R.; Amarnani, D.; D’Amore, P.A. Oxidized lipoprotein uptake through the CD36 receptor activates the NLRP3 inflammasome in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4704–4712. [Google Scholar] [CrossRef]
- Farhangkhoee, H.; Khan, Z.A.; Chakrabarti, S. CD36 regulates glucose-induced endothelial nitric oxide synthase and oxidant injury in microvascular endothelial cells. In Proceedings of the 66th Annual Scientific Sessions of the American Diabetes Association, Washington, DC, USA, 9–13 June 2006; p. 1910. [Google Scholar]
- Antonetti, D.A.; Klein, R.; Gardner, T.W. Diabetic retinopathy. N. Engl. J. Med. 2012, 366, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Nishi, K.; Itabe, H.; Uno, M.; Kitazato, K.T.; Horiguchi, H.; Shinno, K.; Nagahiro, S. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.Y.; Du, M.; Elliott, M.H.; Wu, M.; Fu, D.; Yang, S.; Basu, A.; Gu, X.; Ma, J.X.; Aston, C.E.; et al. Extravascular modified lipoproteins: A role in the propagation of diabetic retinopathy in a mouse model of type 1 diabetes. Diabetologia 2016, 59, 2026–2035. [Google Scholar] [CrossRef]
- Du, M.; Wu, M.; Fu, D.; Yang, S.; Chen, J.; Wilson, K.; Lyons, T.J. Effects of modified LDL and HDL on retinal pigment epithelial cells: A role in diabetic retinopathy? Diabetologia 2013, 56, 2318–2328. [Google Scholar] [CrossRef]
- Wu, M.; Chen, Y.; Wilson, K.; Chirindel, A.; Ihnat, M.A.; Yu, Y.; Boulton, M.E.; Szweda, L.I.; Ma, J.-X.; Lyons, T.J. Intraretinal leakage and oxidation of LDL in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2679–2685. [Google Scholar] [CrossRef]
- Lopes-Virella, M.F.; Baker, N.L.; Hunt, K.J.; Lyons, T.J.; Jenkins, A.J.; Virella, G. High concentrations of AGE-LDL and oxidized LDL in circulating immune complexes are associated with progression of retinopathy in type 1 diabetes. Diabetes Care 2012, 35, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.J.; Li, W.; Wells-Knecht, M.C.; Jokl, R. Toxicity of mildly modified low-density lipoproteins to cultured retinal capillary endothelial cells and pericytes. Diabetes 1994, 43, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Wu, M.; Zhang, J.; Du, M.; Yang, S.; Hammad, S.M.; Wilson, K.; Chen, J.; Lyons, T.J. Mechanisms of modified LDL-induced pericyte loss and retinal injury in diabetic retinopathy. Diabetologia 2012, 55, 3128–3140. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Yu, J.Y.; Yang, S.; Wu, M.; Hammad, S.M.; Connell, A.R.; Du, M.; Chen, J.; Lyons, T.J. Survival or death: A dual role for autophagy in stress-induced pericyte loss in diabetic retinopathy. Diabetologia 2016, 59, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2009, 2, re3. [Google Scholar] [CrossRef]
- Piano, I.; Novelli, E.; Della Santina, L.; Strettoi, E.; Cervetto, L.; Gargini, C. Involvement of autophagic pathway in the progression of retinal degeneration in a mouse model of diabetes. Front. Cell. Neurosci. 2016, 10, 42. [Google Scholar] [CrossRef]
- Truman, J.P.; Al Gadban, M.M.; Smith, K.J.; Jenkins, R.W.; Mayroo, N.; Virella, G.; Lopes-Virella, M.F.; Bielawska, A.; Hannun, Y.A.; Hammad, S.M. Differential regulation of acid sphingomyelinase in macrophages stimulated with oxidized low-density lipoprotein (LDL) and oxidized LDL immune complexes: Role in phagocytosis and cytokine release. Immunology 2012, 136, 30–45. [Google Scholar] [CrossRef]
- Kamei, M.; Yoneda, K.; Kume, N.; Suzuki, M.; Itabe, H.; Matsuda, K.; Shimaoka, T.; Minami, M.; Yonehara, S.; Kita, T.; et al. Scavenger receptors for oxidized lipoprotein in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1801–1807. [Google Scholar] [CrossRef]
- Bessho, H.; Honda, S.; Kondo, N.; Kusuhara, S.; Tsukahara, Y.; Negi, A. The association of CD36 variants with polypoidal choroidal vasculopathy compared to typical neovascular age-related macular degeneration. Mol. Vis. 2012, 18, 121–127. [Google Scholar]
- Kondo, N.; Honda, S.; Kuno, S.; Negi, A. Positive association of common variants in CD36 with neovascular age-related macular degeneration. Aging (Albany NY) 2009, 1, 266–274. [Google Scholar] [CrossRef][Green Version]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef] [PubMed]
- Lavalette, S.; Conart, J.B.; Touhami, S.; Roubeix, C.; Houssier, M.; Augustin, S.; Raoul, W.; Combadière, C.; Febbraio, M.; Ong, H.; et al. CD36 deficiency inhibits retinal inflammation and retinal degeneration in Cx3cr1 knockout mice. Front. Immunol. 2020, 10, 3032. [Google Scholar] [CrossRef] [PubMed]
- Lois, N.; McCarter, R.V.; O’Neill, C.; Medina, R.J.; Stitt, A.W. Endothelial progenitor cells in diabetic retinopathy. Front. Endocrinol. (Lausanne) 2014, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Mwaikambo, B.R.; Yang, C.; Chemtob, S.; Hardy, P. Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1- and phosphatidylinositol 3-kinase-dependent mechanisms. J. Biol. Chem. 2009, 284, 26695–26707. [Google Scholar] [CrossRef]
- Mwaikambo, B.R.; Sennlaub, F.; Ong, H.; Chemtob, S.; Hardy, P. Activation of CD36 inhibits and induces regression of inflammatory corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4356–4364. [Google Scholar] [CrossRef]
- Matušková, V.; Balcar, V.J.; Khan, N.A.; Bonczek, O.; Ewerlingová, L.; Zeman, T.; Kolář, P.; Vysloužilová, D.; Vlková, E.; Šerý, O. CD36 gene is associated with intraocular pressure elevation after intravitreal application of anti-VEGF agents in patients with age-related macular degeneration: Implications for the safety of the therapy. Ophthalmic Genet. 2018, 39, 4–10. [Google Scholar] [CrossRef]
- Koch, M.; Hussein, F.; Woeste, A.; Gründker, C.; Frontzek, K.; Emons, G.; Hawighorst, T. CD36-mediated activation of endothelial cell apoptosis by an N-terminal recombinant fragment of thrombospondin-2 inhibits breast cancer growth and metastasis in vivo. Breast Cancer Res. Treat. 2011, 128, 337–346. [Google Scholar] [CrossRef]
- Yang, H.; Sloan, G.; Ye, Y.; Wang, S.; Duan, B.; Tesfaye, S.; Gao, L. New perspective in diabetic neuropathy: From the periphery to the brain, a call for early detection, and precision medicine. Front. Endocrinol. (Lausanne) 2020, 10, 929. [Google Scholar] [CrossRef]
- Juster-Switlyk, K.; Smith, A.G. Updates in diabetic peripheral neuropathy. F1000Res. 2016, 5, 738. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Tesfaye, S.; Boulton, A.J.; Dyck, P.J.; Freeman, R.; Horowitz, M.; Kempler, P.; Lauria, G.; Malik, R.A.; Spallone, V.; Vinik, A.; et al. Diabetic Neuropathy Expert Group. Diabetic neuropathies: Update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010, 33, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Pande, M.; Hur, J.; Hong, Y.; Backus, C.; Hayes, J.M.; Oh, S.S.; Kretzler, M.; Feldman, E.L. Transcriptional profiling of diabetic neuropathy in the BKS db/db mouse: A model of type 2 diabetes. Diabetes 2011, 60, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, W.; Xie, C.; Zhang, M.; Yin, X.; Wang, F.; Xu, J.; Shi, B. Identification of genes and signaling pathways associated with diabetic neuropathy using a weighted correlation network analysis: A consort study. Medicine (Baltimore) 2016, 95, e5443. [Google Scholar] [CrossRef] [PubMed]
- Padilla, A.; Descorbeth, M.; Almeyda, A.L.; Payne, K.; De Leon, M. Hyperglycemia magnifies Schwann cell dysfunction and cell death triggered by PA-induced lipotoxicity. Brain Res. 2011, 1370, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Isoda, F.; Kurland, I.; Mobbs, C.V. Glucose-induced metabolic memory in Schwann cells: Prevention by PPAR agonists. Endocrinology 2013, 154, 3054–3066. [Google Scholar] [CrossRef] [PubMed]
- Montani, L.; Pereira, J.A.; Norrmén, C.; Pohl, H.B.F.; Tinelli, E.; Trötzmüller, M.; Figlia, G.; Dimas, P.; von Niederhäusern, B.; Schwager, R.; et al. De novo fatty acid synthesis by Schwann cells is essential for peripheral nervous system myelination. J. Cell Biol. 2018, 217, 1353–1368. [Google Scholar] [CrossRef]
- Gulsin, G.S.; Athithan, L.; McCann, G.P. Diabetic cardiomyopathy: Prevalence, determinants and potential treatments. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018819834869. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic cardiomyopathy: An update of mechanisms contributing to this clinical entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Gilca, G.E.; Stefanescu, G.; Badulescu, O.; Tanase, D.M.; Bararu, I.; Ciocoiu, M. Diabetic cardiomyopathy: Current approach and potential diagnostic and therapeutic targets. J. Diabetes Res. 2017, 2017, 1310265. [Google Scholar] [CrossRef]
- Ying, Y.; Zhu, H.; Liang, Z.; Ma, X.; Li, S. GLP1 protects cardiomyocytes from palmitate-induced apoptosis via Akt/GSK3b/b-catenin pathway. J. Mol. Endocrinol. 2015, 55, 245–262. [Google Scholar] [CrossRef]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Yang, J.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Kovacs, A.; Febbraio, M.; Finck, B.N.; Kelly, D.P. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ. Res. 2007, 100, 1208–1217. [Google Scholar] [CrossRef]
- Wu, L.; Wang, K.; Wang, W.; Wen, Z.; Wang, P.; Liu, L.; Wang, D.W. Glucagon-like peptide-1 ameliorates cardiac lipotoxicity in diabetic cardiomyopathy via the PPARα pathway. Aging Cell 2018, 17, e12763. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhao, Y.; He, M.; Li, H.; Fan, J.; Nie, X.; Yan, M.; Chen, C.; Wang, D.W. MiR-30c/PGC-1β protects against diabetic cardiomyopathy via PPARα. Cardiovasc. Diabetol. 2019, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA. 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-C.; Kovacs, A.; Ford, D.A.; Hsu, F.-F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef]
- Koonen, D.P.; Febbraio, M.; Bonnet, S.; Nagendran, J.; Young, M.E.; Michelakis, E.D.; Dyck, J.R. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation 2007, 116, 2139–2147. [Google Scholar] [CrossRef]
- Sung, M.M.; Byrne, N.J.; Kim, T.T.; Levasseur, J.; Masson, G.; Boisvenue, J.J.; Febbraio, M.; Dyck, J.R. Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H552–H560. [Google Scholar] [CrossRef]
- Umbarawan, Y.; Syamsunarno, M.R.A.A.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Sci. Rep. 2018, 8, 12035. [Google Scholar] [CrossRef]
- Angin, Y.; Steinbusch, L.K.; Simons, P.J.; Greulich, S.; Hoebers, N.T.; Douma, K.; van Zandvoort, M.A.; Coumans, W.A.; Wijnen, W.; Diamant, M.; et al. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem. J. 2012, 448, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Meng, Z.; Zheng, Y.; Hu, B.; Shen, E. Fibroblast growth factor 21 inhibition aggravates cardiac dysfunction in diabetic cardiomyopathy by improving lipid accumulation. Exp. Ther. Med. 2018, 15, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhao, H.; Du, M.; Wu, X. The effect of apelin-13 on pancreatic islet beta cell mass and myocardial fatty acid and glucose metabolism of experimental type 2 diabetic rats. Peptides 2019, 114, 1–7. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puchałowicz, K.; Rać, M.E. The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications—Update in Pathogenesis, Treatment and Monitoring. Cells 2020, 9, 1877. https://doi.org/10.3390/cells9081877
Puchałowicz K, Rać ME. The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications—Update in Pathogenesis, Treatment and Monitoring. Cells. 2020; 9(8):1877. https://doi.org/10.3390/cells9081877
Chicago/Turabian StylePuchałowicz, Kamila, and Monika Ewa Rać. 2020. "The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications—Update in Pathogenesis, Treatment and Monitoring" Cells 9, no. 8: 1877. https://doi.org/10.3390/cells9081877
APA StylePuchałowicz, K., & Rać, M. E. (2020). The Multifunctionality of CD36 in Diabetes Mellitus and Its Complications—Update in Pathogenesis, Treatment and Monitoring. Cells, 9(8), 1877. https://doi.org/10.3390/cells9081877