Anti-NASH Drug Development Hitches a Lift on PPAR Agonism

 ,
,  ,
,
Abstract
:1. Introduction
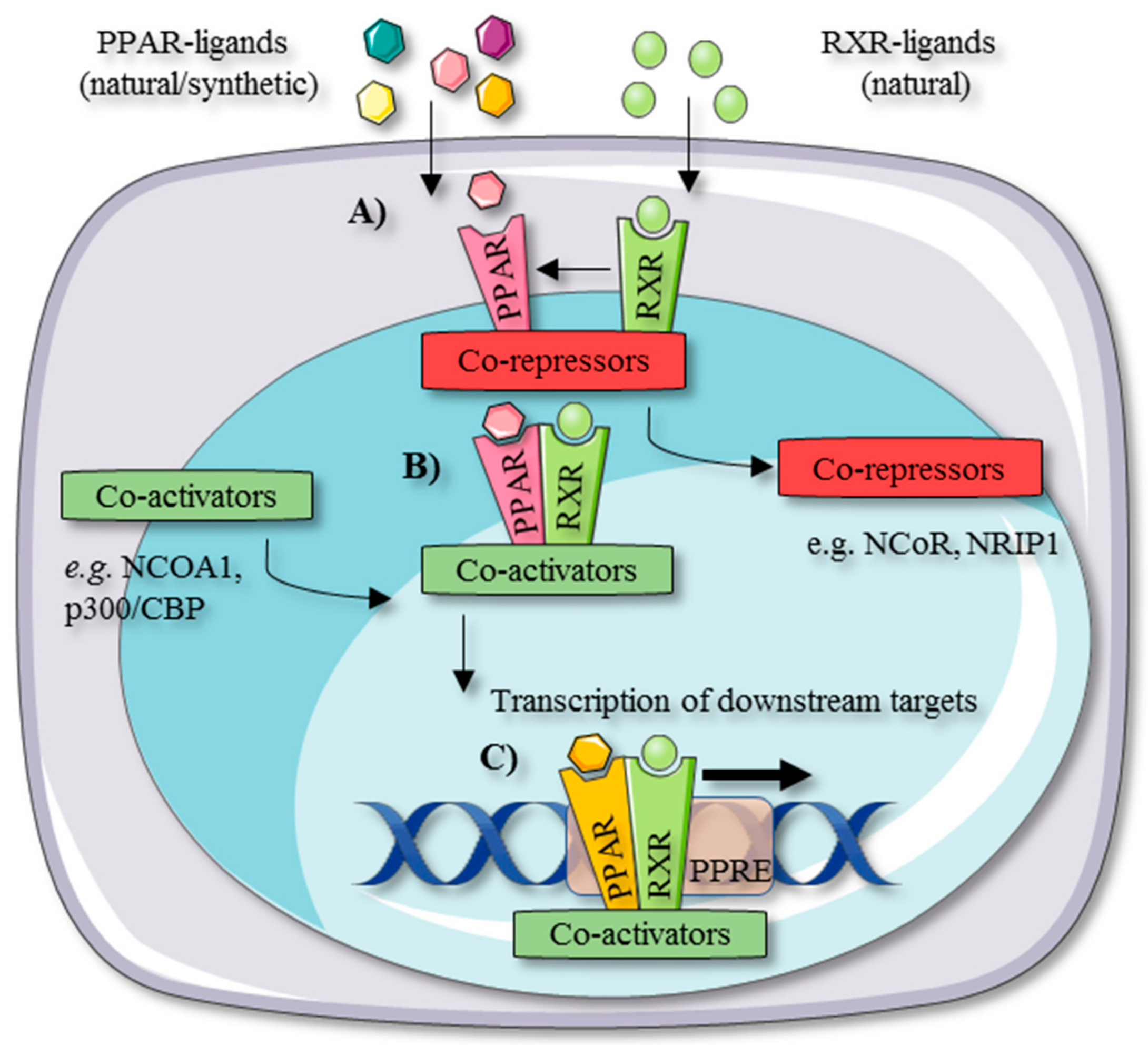
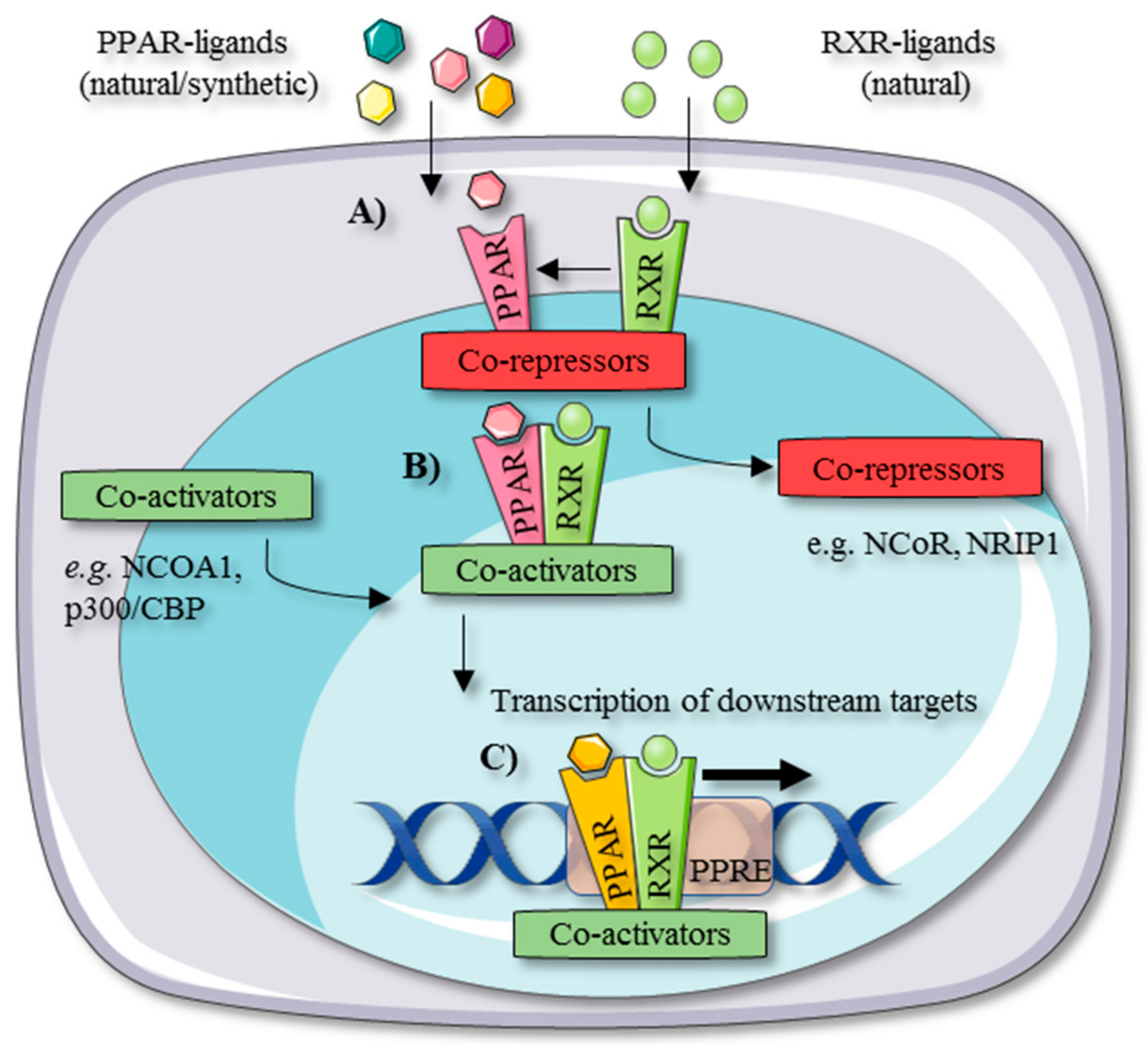
2. PPAR Tissue Distribution and Working Mechanism
3. Dysregulation of PPARs during NASH
4. PPAR Agonists as Potential Anti-NASH Treatment
4.1. PPAR-α Agonists
4.2. PPAR-δ Agonists
4.3. PPAR-γ Agonists
4.4. PPAR-α/δ Agonists
4.5. PPAR-α/γ Agonists
4.6. PPAR-pan Agonists
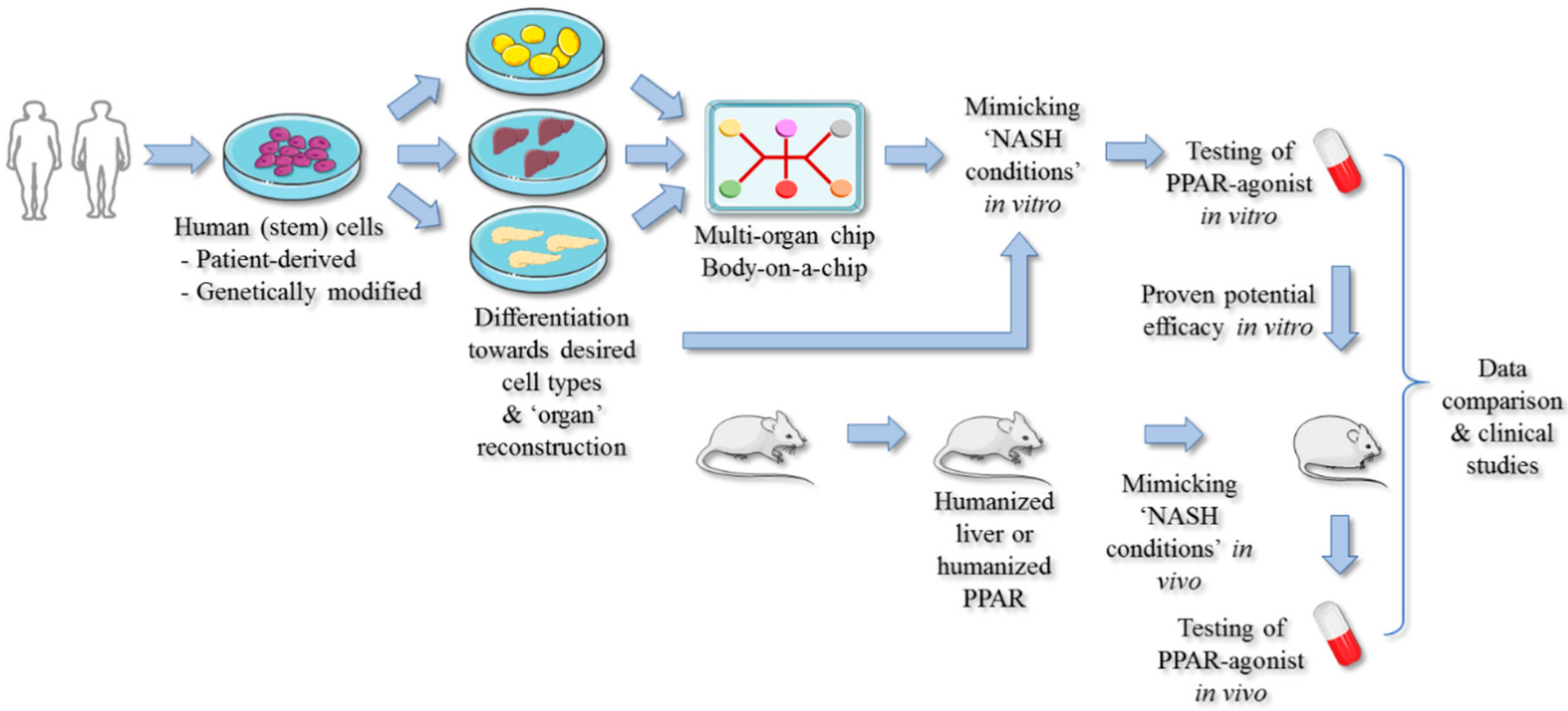
5. Strategies for Improving PPAR-Targeted Anti-NASH Drug Testing and Therapy
PPAR-Targeted Preclinical Drug Testing
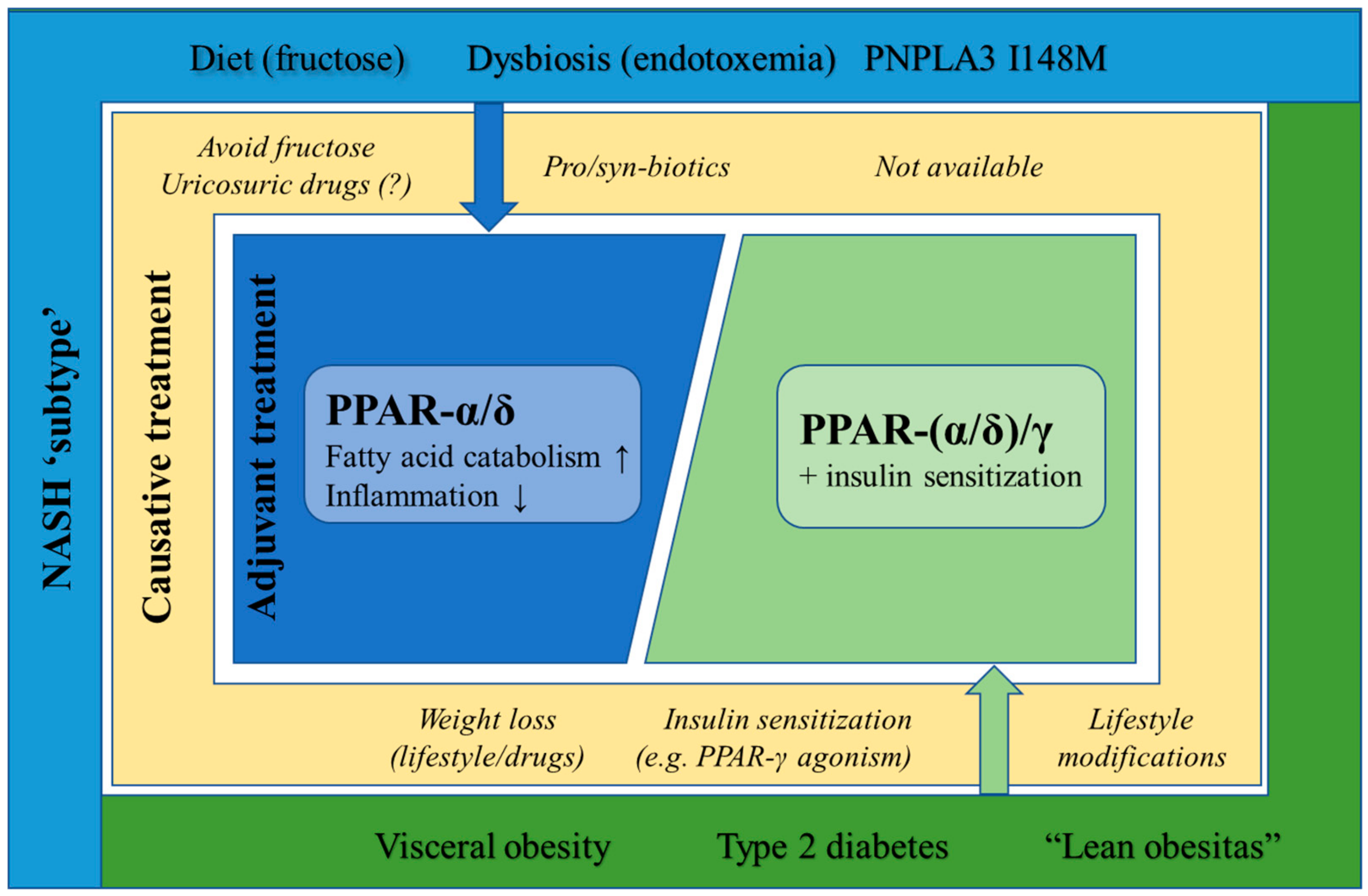
6. Targeted PPAR Agonism as NASH-Specific Therapy
6.1. Diet-Induced NASH
6.2. Obesity and Type 2 Diabetes-Induced NASH
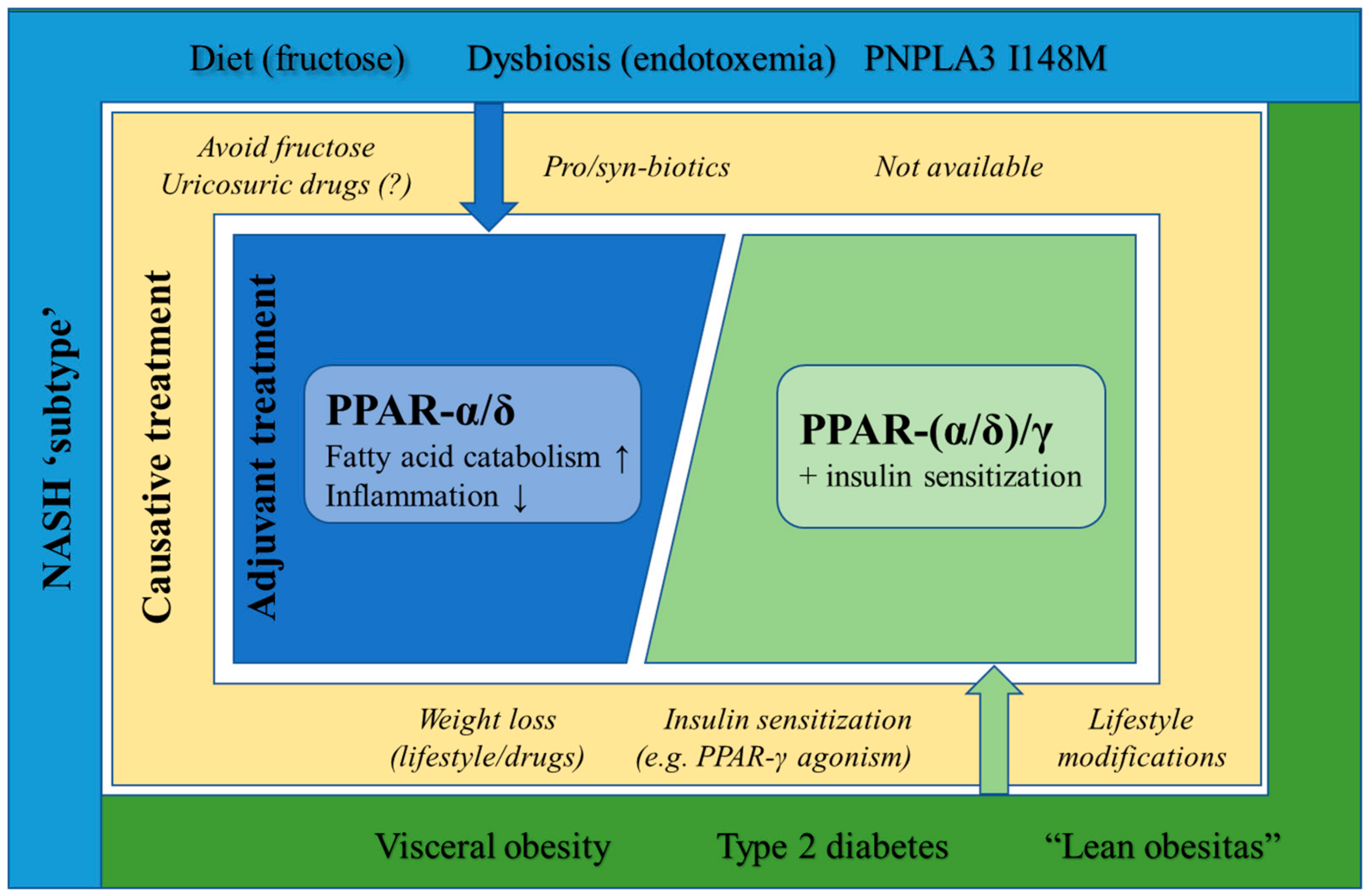
6.3. Lean NASH
6.4. Microbiome-Induced NASH
7. Outlook and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.M.; Marchesini, G.; Pinto-Cortez, H.; Petta, S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation 2019, 103, 22–27. [Google Scholar] [CrossRef]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the Epidemic of Nonalcoholic Fatty Liver Disease Demonstrates an Exponential Increase in Burden of Disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric Surgery Reduces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kota, B.P.; Huang, T.H.-W.; Roufogalis, B.D. An Overview on Biological Mechanisms of PPARs. Pharmacol. Res. 2005, 51, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.I.; Siersbæk, M.; Mandrup, S. PPARs: Fatty Acid Sensors Controlling Metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef]
- Feng, X.; Gao, X.; Jia, Y.; Zhang, H.; Xu, Y.; Wang, G. PPAR-α Agonist Fenofibrate Decreased RANTES Levels in Type 2 Diabetes Patients with Hypertriglyceridemia. Med. Sci. Monit. 2016, 22, 743–751. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Connolly, J.J.; Ooka, K.; Lim, J.K. Future Pharmacotherapy for Non-Alcoholic Steatohepatitis (NASH): Review of Phase 2 and 3 Trials. J. Clin. Transl. Hepatol. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, N.; Aoyama, T.; Kimura, S.; Gonzalez, F.J. Targeting Nuclear Receptors for the Treatment of Fatty Liver Disease. Pharmacol. Ther. 2017, 179, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Burn, K.A.; Vanden Heuvel, J.P. Modulation of PPAR Activity via Phosphorylation. Biochim Biophys Acta 2009, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugge, A.; Mandrup, S. Molecular Mechanisms and Genome-Wide Aspects of PPAR Subtype Specific Transactivation. PPAR Res. 2010, 2010, 169506. [Google Scholar] [CrossRef] [Green Version]
- Liss, K.H.H.; Finck, B.N. PPARs and Nonalcoholic Fatty Liver Disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Oberfield, J.L.; Collins, J.L.; Holmes, C.P.; Goreham, D.M.; Cooper, J.P.; Cobb, J.E.; Lenhards, J.M.; Hull-Ryde, E.A.; Mohr, C.P.; Blanchard, S.G.; et al. A Peroxisome Proliferator-Activated Receptor γ Ligand Inhibits Adipocyte Differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 6102–6106. [Google Scholar] [CrossRef] [Green Version]
- Berger, J.; Moller, D.E. The Mechanisms of Action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [Green Version]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in Obesity-Induced T2DM, Dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Giby, V.G.; Ajith, T.A. Role of Adipokines and Peroxisome Proliferator-Activated Receptors in Nonalcoholic Fatty Liver Disease. World J. Hepatol. 2014, 6, 570–579. [Google Scholar] [CrossRef]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural Basis for PPAR Partial or Full Activation Revealed by a Novel Ligand Binding Mode. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Ricote, M.; Glass, C.K. PPARs and Molecular Mechanisms of Transrepression. Biochim. Biophys. Acta 2007, 1171, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schupp, M.; Lazar, M.A. Endogenous Ligands for Nuclear Receptors: Digging Deeper. J. Biol. Chem. 2010, 285, 40409–40415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome Proliferator-Activated Receptor Alpha Target Genes. PPAR Res. 2010, 2010, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinin, E.; Moschettal, A. Hepatic-Specific PPARa-FGF21 Action in NAFLD. Gut 2016, 65, 1075–1076. [Google Scholar] [CrossRef]
- Zhang, N.; Chu, E.S.H.; Zhang, J.; Li, X.; Liang, Q.; Chen, J.; Chen, M.; Teoh, N.; Farrell, G.; Sung, J.J.Y.; et al. Peroxisome Proliferator Activated Receptor Alpha Inhibits Hepatocarcinogenesis through Mediating NF-KB Signaling Pathway. Oncotarget 2014, 5, 8330–8340. [Google Scholar] [CrossRef]
- Iroz, A.; Montagner, A.; Benhamed, F.; Levavasseur, F.; Polizzi, A.; Anthony, E.; Régnier, M.; Fouché, E.; Lukowicz, C.; Cauzac, M.; et al. A Specific ChREBP and PPARα Cross-Talk Is Required for the Glucose-Mediated FGF21 Response. Cell Rep. 2017, 21, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.J. Fenofibrate Simultaneously Induces Hepatic Fatty Acid Oxidation, Synthesis, and Elongation in Mice. J. Biol. Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [Green Version]
- Palomer, X.; Barroso, E.; Pizarro-Delgado, J.; Peña, L.; Botteri, G.; Zarei, M.; Aguilar, D.; Montori-Grau, M.; Vázquez-Carrera, M. PPARβ/δ: A Key Therapeutic Target in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 913. [Google Scholar] [CrossRef] [Green Version]
- Zarei, M.; Barroso, E.; Palomer, X.; Dai, J.; Rada, P.; Quesada-López, T.; Escolà-Gil, J.C.; Cedó, L.; Zali, M.R.; Molaei, M.; et al. Hepatic Regulation of VLDL Receptor by PPARβ/δ and FGF21 Modulates Non-Alcoholic Fatty Liver Disease. Mol. Metab. 2018, 8, 117–131. [Google Scholar] [CrossRef]
- Heikkinen, S.; Auwerx, J.; Argmann, C.A. PPARγ in Human and Mouse Physiology. Biochim. Biophys. Acta 2007, 1771, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Ahn, Y. Role of Peroxisome Proliferator–Activated Receptor-γ in the Glucose-Sensing Apparatus of Liver and β-Cells. Biochemistry 2004, 53, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pais, R.; Moraru, I.; Ratziu, V. Glitazones for Human Nonalcoholic Steatohepatitis. Therap. Adv. Gastroenterol. 2011, 4, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The Peroxisome Proliferator-Activated Receptor-γ Is a Negative Regulator of Macrophage Activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Chengyu, J.; Ting, A.T.; Seed, B. PPAR-γ Agonists Inhibit Production Of monocyte Inflammatory Cytokine. Nature 1999, 391, 82–86. [Google Scholar]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPAR-α Gene Expression Correlates with Severity and Histological Treatment Response in Patients with Non-Alcoholic Steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K.; Horiuchi, A.; Komatsu, M.; Nakajima, T.; Nishizawa, T.; Joshita, S.; Umemura, T.; et al. Down-Regulation of SREBP-1c Is Associated with the Development of Burned-out NASH. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Z.; Klaunig, J.E. Modulation of Xenobiotic Nuclear Receptors in High-Fat Diet Induced Non-Alcoholic Fatty Liver Disease. Toxicology 2018, 1, 199–213. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [Green Version]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A Controlled Trial of Gemfibrozil in the Treatment of Patients with Nonalcoholic Steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A Pilot Trial of Fenofibrate for the Treatment of Non-Alcoholic Fatty Liver Disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, B.M. Ursodeoxycholic Acid or Clofibrate in the Treatment of Non-Alcohol- Induced Steatohepatitis: A Pilot Study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Arai, H.; Yokote, K.; Araki, E.; Suganami, H.; Yamashita, S. Efficacy and Safety of Pemafibrate (K-877), a Selective Peroxisome Proliferator-Activated Receptor α Modulator, in Patients with Dyslipidemia: Results from a 24-Week, Randomized, Double Blind, Active-Controlled, Phase 3 Trial. J. Clin. Lipidol. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, Y.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; Takizawa, T.; Saito, S.; Nagashima, Y.; et al. Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor Alpha Modulator, Improves the Pathogenesis in a Rodent Model of Nonalcoholic Steatohepatitis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E.; Schwartz, S.; Littlejohn, T.; Kerzner, B.; Krauss, R.M.; Karpf, D.B.; Choi, Y.J.; Wang, X.; Naim, S.; Roberts, B.K. MBX-8025, a Novel Peroxisome Proliferator Receptor-Δagonist: Lipid and Other Metabolic Effects in Dyslipidemic Overweight Patients Treated with and without Atorvastatin. J. Clin. Endocrinol. Metab. 2011, 96, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Roberts, B.K.; Wang, X.; Geaney, J.C.; Naim, S.; Wojnoonski, K.; Karpf, D.B.; Krauss, R.M. Effects of the PPAR-δ Agonist MBX-8025 on Atherogenic Dyslipidemia. Atherosclerosis 2012, 220, 470–476. [Google Scholar] [CrossRef]
- Ooi, E.M.M.; Watts, G.F.; Sprecher, D.L.; Chan, D.C.; Barrett, P.H.R. Mechanism of Action of a Peroxisome Proliferator-Activated Receptor (PPAR)-δ Agonist on Lipoprotein Metabolism in Dyslipidemic Subjects with Central Obesity. J. Clin. Endocrinol. Metab. 2011, 96, 1568–1576. [Google Scholar] [CrossRef] [Green Version]
- Risérerus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of Peroxisome Proliferator–δ Activated Receptor (PPAR) Promotes Reversal of Multiple Metabolic Abnormalities, Reduces Oxidative Stress, and Increases Fatty Acid Oxidation in Moderately Obese Men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.-J.; McWherter, C.A.; Teoh, N.C.-H.; et al. The Selective Peroxisome Proliferator-Activated Receptor-Delta Agonist Seladelpar Reverses Nonalcoholic Steatohepatitis Pathology by Abrogating Lipotoxicity in Diabetic Obese Mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [Green Version]
- Oseini, A.M.; Sanyal, A.J. Therapies in Non-Alcoholic Steatohepatitis (NASH). Liver Int. 2017, 37, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Ozturk, Z.A.; Kadayifci, A. Insulin Sensitizers for the Treatment of Non-Alcoholic Fatty Liver Disease. World J. Hepatol. 2014, 6, 199–206. [Google Scholar] [CrossRef]
- Yong-ho, L.; Jae, H.K.; So, R.K.; Heung, J.Y.; Eun-Jung, R.; Young, M.C.; Byung-Wan, L. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness Study Patients. Endocrinol. Nutr. Metab. 2017, 32, 60–69. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, Vitamin E, or Placebo for Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.M.; Xia, M.F.; Wang, Y.; Chang, X.X.; Yao, X.Z.; Rao, S.X.; Zeng, M.S.; Tu, Y.F.; Feng, R.; Jia, W.P.; et al. Efficacy of Berberine in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J.; Mofrad, P.S.; Contos, M.J.; Sargeant, C.; Luketic, V.A.; Sterling, R.K.; Stravitz, R.T.; Shiffman, M.L.; Clore, J.; Mills, A.S. A Pilot Study of Vitamin E Versus Vitamin E and Pioglitazone for the Treatment of Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2004, 2, 1107–1115. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus a Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, Placebo-Controlled Trial of Pioglitazone in Nondiabetic Subjects With Nonalcoholic Steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The Diagnosis and Management of Non-Alcoholic Fatty Liver Disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Marchesini, G.; Day, C.P.; Dufour, J.F.; Canbay, A.; Nobili, V.; Ratziu, V.; Tilg, H.; Roden, M.; Gastaldelli, A.; Yki-Jarvinen, H.; et al. EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [Green Version]
- Mahady, S.E.; Webster, A.C.; Walker, S.; Sanyal, A.; George, J. The Role of Thiazolidinediones in Non-Alcoholic Steatohepatitis—A Systematic Review and Meta Analysis. J. Hepatol. 2011, 55, 1383–1390. [Google Scholar] [CrossRef]
- Tuccori, M.; Filion, K.B.; Yin, H.; Yu, O.H.; Platt, R.W.; Azoulay, L. Pioglitazone Use and Risk of Bladder Cancer: Population Based Cohort Study. Br. Med. J. 2016, 352, i1541. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T. Long-Term Efficacy of Rosiglitazone in Nonalcoholic Steatohepatitis: Results of the Fatty Liver Improvement by Rosiglitazone Therapy (FLIRT 2) Extension Trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for Nonalcoholic Steatohepatitis: One-Year Results of the Randomized Placebo-Controlled Fatty Liver Improvement With Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Loke, Y.K.; Furberg, C.D. Long-Term Risk of Cardiovascular Events With Rosiglitazone. Am. Med. Assoc. 2007, 298, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Pladevall, M.; Riera-Guardia, N.; Margulis, A.V.; Varas-Lorenzo, C.; Calingaert, B.; Perez-Gutthann, S. Cardiovascular Risk Associated with the Use of Glitazones, Metformin and Sufonylureas: Meta-Analysis of Published Observational Studies. BMC Cardiovasc. Disord. 2016, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Sin, G.K.; Doo, M.K.; Jeong-Teak, W.; Hak, C.J.; Choon, H.C.; Kyung, S.K.; Jeong, H.P.; Yong, S.P.; Sang, J.K.; Dong, S.C. Efficacy and Safety of Lobeglitazone Monotherapy in Patients with Type 2 Diabetes Mellitus over 24-Weeks: A Multicenter, Randomized, Double-Blind, Parallel-Group, Placebo Controlled Trial. PLoS ONE 2014, 9, e92843. [Google Scholar]
- Sun, H.K.; Sin, G.K.; Doo, M.K.; Jeong-Teak, W.; Hak, C.J.; Choon, H.C.; Kyung, S.K.; Jeong, H.P.; Yong, S.P.; Sang, J.K.; et al. Safety and Efficacy of Lobeglitazone Monotherapy in Patients with Type 2 Diabetes Mellitus over 52 Weeks: An Open-Label Extension Study. Diabetes Res. Clin. Pract. 2015, 110, e27–e30. [Google Scholar]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-α and -δ, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Hanf, R.; Millatt, L.J.; Cariou, B.; Noel, B.; Rigou, G.; Delataille, P.; Daix, V.; Hum, D.W.; Staels, B. The Dual Peroxisome Proliferator-Activated Receptor Alpha/Delta Agonist GFT505 Exerts Anti-Diabetic Effects in Db/Db Mice without Peroxisome Proliferator-Activated Receptor Gamma-Associated Adverse Cardiac Effects. Diabetes Vasc. Dis. Res. 2014, 11, 440–447. [Google Scholar] [CrossRef]
- Fiévet, C.; Fruchart, J.C.; Staels, B. PPARα and PPARγ Dual Agonists for the Treatment of Type 2 Diabetes and the Metabolic Syndrome. Curr. Opin. Pharmacol. 2006, 6, 606–614. [Google Scholar] [CrossRef]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New Dual Peroxisome Proliferator Activated Receptor Agonist—Saroglitazar in Diabetic Dyslipidemia and Non-Alcoholic Fatty Liver Disease: Integrated Analysis of the Real World Evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARα/γ Agonist Saroglitazar Improves Liver Histopathology and Biochemistry in Experimental NASH Models. Liver Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, C.J.; Viraswami-Appanna, K.; Fiedorek, F.T. Efficacy and Safety of Muraglitazar: A Double-Blind, 24-Week, Dose-Ranging Study in Patients with Type 2 Diabetes. Diabetes Vasc. Dis. Res. 2009, 6, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, S.; Banshi, S.; Bhavana, S. Saroglitazar for the Treatment of Hypertriglyceridemia in Patients with Type 2 Diabetes: Current Evidence. DiabetesMetab. Syndr. Obes. Targets Ther. 2015, 8, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettstein, G.; Luccarini, J.-M.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrene, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The New-Generation Pan-Peroxisome Proliferator-Activated Receptor Agonist IVA337 Protects the Liver From Metabolic Disorders and Fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef]
- Holden, P.R.; Tugwood, J.D. Peroxisome Proliferator-Activated Receptor Alpha: Role in Rodent Liver Cancer and Species Differences. J. Mol. Endocrinol. 1999, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M. The Role of PPARα in Lipid Metabolism and Obesity: Focusing on the Effects of Estrogen on PPARα Actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef]
- Mukherjee, R.; Jow, L.; McDonnell, D.P. Human and Rat Peroxisome Proliferator Activated Receptors (PPARs) Demonstrate Similar Tissue Distribution but Different Responsiveness to PPAR Activators. J. Steroid Biochem. Mol. Biol. 1994, 51, 157–166. [Google Scholar] [CrossRef]
- Peters, J.M.; Cheung, C.; Gonzalez, F.J. Peroxisome Proliferator-Activated Receptor- α and Liver Cancer: Where Do We Stand? J. Mol. Med. 2005, 83, 774–785. [Google Scholar] [CrossRef]
- Yang, Q.; Nagano, T.; Shah, Y.; Cheung, C.; Ito, S.; Gonzalez, F.J. The PPARα-Humanized Mouse: A Model to Investigate Species Differences in Liver Toxicity Mediated by PPARα. Toxicol. Sci. 2008, 101, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Tateno, C.; Miya, F.; Wake, K.; Kataoka, M.; Ishida, Y.; Yamasaki, C.; Yanagi, A.; Kakuni, M.; Wisse, E.; Verheyen, F.; et al. Morphological and Microarray Analyses of Human Hepatocytes from Xenogeneic Host Livers. Lab. Investig. 2013, 93, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Tateno, C.; Yamamoto, T.; Utoh, R.; Yamasaki, C.; Ishida, Y.; Myoken, Y.; Oofusa, K.; Okada, M.; Tsutsui, N.; Yoshizato, K. Chimeric Mice with Hepatocyte-Humanized Liver as an Appropriate Model to Study Human Peroxisome Proliferator-Activated Receptor-α. Toxicol. Pathol. 2015, 43, 233–248. [Google Scholar] [CrossRef] [PubMed]
- de la Rosa Rodriguez, M.A.; Sugahara, G.; Hooiveld, G.J.E.J.; Ishida, Y.; Tateno, C.; Kersten, S. The Whole Transcriptome Effects of the PPARα Agonist Fenofibrate on Livers of Hepatocyte Humanized Mice. BMC Genom. 2018, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rogue, A.; Anthérieu, S.; Vluggens, A.; Umbdenstock, T.; Claude, N.; De la Moureyre-Spire, C.; Weaver, R.J.; Guillouzo, A. PPAR Agonists Reduce Steatosis in Oleic Acid-Overloaded HepaRG Cells. Toxicol. Appl. Pharmacol. 2014, 276, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in Vitro Human Liver System for Interrogating Nonalcoholic Steatohepatitis. J. Clin. Invest. 2016, 1, e90954. [Google Scholar] [CrossRef]
- Feaver, R.E.; Cole, B.K. Comparison of Obeticholic Acid and GFT505 (Elafibranor) for Treatment of Non-Alcoholic Steatohepatitis (NASH) in a Human in Vitro Surrogate System. Poster presented at ‘The Liver Meeting’, Boston, MA, USA, 11–15 November 2016. [Google Scholar]
- Boeckmans, J.; Buyl, K.; Natale, A.; Vandenbempt, V.; Branson, S.; De Boe, V.; Rogiers, V.; De Kock, J.; Rodrigues, R.M.; Vanhaecke, T. Elafibranor Restricts Lipogenic and Inflammatory Responses in a Human Skin Stem Cell-Derived Model of NASH. Pharmacol. Res. 2019, 144, 377–389. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical Models of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef]
- Parafati, M.; Kirby, R.J.; Khorasanizadeh, S.; Rastinejad, F.; Malany, S. A Nonalcoholic Fatty Liver Disease Model in Human Induced Pluripotent Stem Cell-Derived Hepatocytes, Created by Endoplasmic Reticulum Stress-Induced Steatosis. Dis. Model. Mech. 2018, 11, dmm033530. [Google Scholar] [CrossRef] [Green Version]
- Sung, J.H.; Wang, Y.I.; Narasimhan Sriram, N.; Jackson, M.; Long, C.; Hickman, J.J.; Shuler, M.L. Recent Advances in Body-on-a-Chip Systems. Anal. Chem. 2019, 91, 330–351. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/Body-on-a-Chip Based on Microfluidic Technology for Drug Discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]
- Skardal, A.; Shupe, T.; Atala, A. Organoid-on-a-Chip and Body-on-a-Chip Systems for Drug Screening and Disease Modeling. Drug Discov. Today 2016, 21, 1399–1411. [Google Scholar] [CrossRef]
- Della Corte, C.; Ferrari, F.; Villani, A.; Nobili, V. Epidemiology and Natural History of NAFLD. J. Med. Biochem. 2014, 34, 13–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism. 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and Sugar: A Major Mediator of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, F.W.B.; Griffin, J.L. De Novo Lipogenesis in the Liver in Health and Disease: More than Just a Shunting Yard for Glucose. Biol. Rev. 2016, 91, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Yonemitsu, S.; Erion, D.M.; Iwasaki, T.; Stark, R.; Weismann, D.; Dong, J.; Zhang, D.; Jurczak, M.J.; Löffler, M.G.; et al. The Role of Peroxisome Proliferator-Activated Receptor γ Coactivator-1 β in the Pathogenesis of Fructose-Induced Insulin Resistance. Cell Metab. 2009, 9, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Dekker, M.J.; Su, Q.; Baker, C.; Rutledge, A.C.; Adeli, K. Fructose: A Highly Lipogenic Nutrient Implicated in Insulin Resistance, Hepatic Steatosis, and the Metabolic Syndrome. Am. J. Physiol. Metab. 2010, 299, E685–E694. [Google Scholar] [CrossRef] [Green Version]
- Roglans, N.; Vilà, L.; Farré, M.; Alegret, M.; Sánchez, R.M.; Vázquez-Carrera, M.; Laguna, J.C. Impairment of Hepatic STAT-3 Activation and Reduction of PPARα Activity in Fructose-Fed Rats. Hepatology 2007, 45, 778–788. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome Activation and Function in Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Lombardi, R.; Pisano, G.; Fargion, S. Role of Serum Uric Acid and Ferritin in the Development and Progression of NAFLD. Int. J. Mol. Sci. 2016, 17, 548. [Google Scholar] [CrossRef] [Green Version]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric Acid Induces Hepatic Steatosis by Generation of Mitochondrial Oxidative Stress. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poiley, J.; Steinberg, A.S.; Choi, Y.J.; Davis, C.S.; Martin, R.L.; McWherter, C.A.; Boudes, P.F.; Bays, H.; Bolshoun, D.; Bolster, E.; et al. A Randomized, Double-Blind, Active- and Placebo-Controlled Efficacy and Safety Study of Arhalofenate for Reducing Flare in Patients With Gout. Arthritis Rheumatol. 2016, 68, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.; Le, K.; Davis, J.; Alderete, T.; Cherry, R.; Lebel, S.; Goran, M. High Rates of Fructose Malabsorption Are Associated with Reduced Liver Fat in Obese African Americans. J. Am. Coll. Nutr. 2012, 369–374. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-Alcoholic Fatty Liver Disease—A Global Public Health Perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, Stress, and Diabetes. J. Clin. Invest. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Kitade, H.; Ni, Y.; Ota, T. Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules 2015, 5, 1563–1579. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.k.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-Mediated Inhibition of Insulin Receptor Tyrosine Kinase Activity in TNF-Alpha- and Obesity-Induced Insulin Resistance. Sci. (80-. ). 1996, 271, 665–670. [Google Scholar] [CrossRef]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The Expression of Tumor Necrosis Factor in Human Adipose Tissue. J. Clin. Invest. 1995, 95, 2111–2119. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Weinstock, R.; Thusu, K.; Abdel-rahman, E.; Aljada, A.; Wadden, T. Tumor Necrosis Factor-Alpha in Sera of Obese Patients: Fall with Weight Loss. J. Clin. Endocrinol. Metab. 1998, 83, 2907–2910. [Google Scholar]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennachio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and Epigenetics of NAFLD and NASH: Clinical Impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. Combined Effects of the TM6SF2 Rs58542926, PNPLA3 Rs738409 and MBOAT7 Rs641738 Variants on NAFLD Severity: Multicentre Biopsy-Based Study. J. Lipid Res. 2017, 58, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A Sequence Variation (I148M) in PNPLA3 Associated with Nonalcoholic Fatty Liver Disease Disrupts Triglyceride Hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotronen, A.; Johansson, L.E.; Johansson, L.M.; Roos, C.; Westerbacka, J.; Hamsten, A.; Bergholm, R.; Arkkila, P.; Arola, J.; Kiviluoto, T.; et al. A Common Variant in PNPLA3, Which Encodes Adiponutrin, Is Associated with Liver Fat Content in Humans. Diabetologia 2009, 52, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Day, C. The Genetics of Nonalcoholic Fatty Liver Disease: Spotlight on PNPLA3 and TM6SF2. Semin. Liver Dis. 2015, 35, 270–290. [Google Scholar]
- Niriella, M.A.; Kasturiratne, A.; Pathmeswaran, A.; De Silva, S.T.; Perera, K.R.; Subasinghe, S.K.C.E.; Kodisinghe, S.K.; Piyaratna, T.A.C.L.; Vithiya, K.; Dassanayaka, A.S.; et al. Lean Non-Alcoholic Fatty Liver Disease (Lean NAFLD): Characteristics, Metabolic Outcomes and Risk Factors from a 7-Year Prospective, Community Cohort Study from Sri Lanka. Hepatol. Int. 2018, 13, 314–322. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.S.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic Variants Regulating Insulin Receptor Signalling Are Associated with the Severity of Liver Damage in Patients with Non-Alcoholic Fatty Liver Disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Anstee, Q.; Valenti, L. Genetic Predisposition in NAFLD and NASH: Impact on Severity of Liver Disease and Response to Treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, H.J.; Lee, K.E.; Kim, D.J.; Kim, S.K.; Ahn, C.W.; Lim, S.-K.; Kim, K.R.; Lee, H.C.; Huh, K.B.; et al. Metabolic Significance of Nonalcoholic Fatty Liver Disease in Nonobese, Nondiabetic Adults. Arch. Intern. Med. 2004, 164, 2169. [Google Scholar] [CrossRef] [Green Version]
- Gaiani, S.; Avogaro, A.; Bombonato, G.C.; Bolognesi, M.; Amor, F.; Vigili de Kreutzenberg, S.; Guarneri, G.; Sacerdoti, D. Nonalcoholic Fatty Liver Disease (NAFLD) in Nonobese Patients with Diabetes: Prevalence and Relationships with Hemodynamic Alterations Detected with Doppler Sonography. J. Ultrasound 2009, 12, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, A.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R. Drug-Induced Steatohepatitis. Expert Opin. Drug Metab. Toxicol. 2017, 13, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, S.; Shapiro, H.; Rozin, S.; Shibolet, O.; Elinav, E. Non-Alcoholic Fatty Liver and the Gut Microbiota. Mol. Metab. 2016, 5, 782–794. [Google Scholar] [CrossRef]
- O’Hara, A.M.; Shanahan, F. The Gut Flora as a Forgotten Organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [Green Version]
- Wiest, R.; Albillos, A.; Trauner, M.; Bajaj, J.; Jalan, R. Targeting the Gut-Liver Axis in Liver Disease. J. Hepatol. 2017, 67, 1084–1103. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased Intestinal Permeability and Tight Junction Alterations in Nonalcoholic Fatty Liver Disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Wong, V.W.S.; Wong, G.L.H.; Chan, H.Y.; Yeung, D.K.W.; Chan, R.S.M.; Chim, A.M.L.; Chan, C.K.M.; Tse, Y.K.; Woo, J.; Chu, W.C.W.; et al. Bacterial Endotoxin and Non-Alcoholic Fatty Liver Disease in the General Population: A Prospective Cohort Study. Aliment. Pharmacol. Ther. 2015, 42, 731–740. [Google Scholar] [CrossRef]
- Pang, J.; Xu, W.; Zhang, X.; Wong, G.L.H.; Chan, A.W.H.; Chan, H.Y.; Tse, C.H.; Shu, S.S.T.; Choi, P.C.L.; Chan, H.L.Y.; et al. Significant Positive Association of Endotoxemia with Histological Severity in 237 Patients with Non-Alcoholic Fatty Liver Disease. Aliment. Pharmacol. Ther. 2017, 46, 175–182. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, R.D.; Baker, S.S. Gut Microbiome and Nonalcoholic Fatty Liver Diseases. Pediatr. Res. 2015, 77, 245–251. [Google Scholar] [CrossRef]
- Abu-Shanab, A.; Quigley, E.M.M. The Role of the Gut Microbiota in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Mehal, W.Z. The Inflammasome in Liver Injury and Non-Alcoholic Fatty Liver Disease. Dig. Dis. 2014, 32, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Zheng, D.; Shibolet, O.; Elinav, E. The Role of the Microbiome in NAFLD and NASH. EMBO Mol. Med. 2019, 11, e9302. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.; Li, A.; John, N.; Sallam, S.; Shah, N.; Kwong, W.; Cholankeril, G.; Kim, D.; Ahmed, A. The Therapeutic Implications of the Gut Microbiome and Probiotics in Patients with NAFLD. Diseases 2019, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.-S.; Chitturi, S.; Wong, G.L.-H.; Yu, J.; Chan, H.L.-Y.; Farrell, G.C. Pathogenesis and Novel Treatment Options for Non-Alcoholic Steatohepatitis. Lancet Gastroenterol. Hepatol. 2016, 1, 56–67. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F.W. Steatohepatitis: A Tale of Two"Hits"? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Woodcock, J.; Griffin, J.P.; Behrman, R.E. Development of Novel Combination Therapies. N. Engl. J. Med. 2011, 364, 985–987. [Google Scholar] [CrossRef] [Green Version]
- GENFIT Launches a Combination Therapy Clinical Program in NASH. Available online: https://www.Genfit.Com/Press-Release/Genfit-Launches-a-Combination-Therapy-Clinical-Program-in-NASH/ (accessed on 24 November 2019).
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular Mechanism of PPARα Action and Its Impact on Lipid Metabolism, Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | PPAR Isotype | Primary Outcome | Duration | Status | Trial Number/Reference |
|---|---|---|---|---|---|
| Phase 4 | |||||
| Lobeglitazone | α/γ | Changes from baseline in controlled attenuation parameters | 24 weeks | Completed (no results posted) | NCT02285205 [51] |
| Rosiglitazone Alpha-lipoic acid Rosiglitazone + alpha-lipoic acid | γ | Improvement in NASH histological scoring system | 24 weeks | Terminated (because of withdrawal of Avandia sale due to its risks outweigh its benefits) | NCT01406704 |
| Phase 3 | |||||
| Elafibranor | α/δ | NASH resolution without worsening of fibrosis | 72 weeks 4 years | Recruiting | NCT02704403 |
| Pioglitazone Vitamin E | γ | Change in standardized scoring of liver biopsies | 96 weeks | Completed (has results) | NCT00063622 [52] |
| Phase 2 | |||||
| Lanifibranor | α/δ/γ | Decrease of at least two points in SAF (Steatosis, Activity, and Fibrosis) activity score combining hepatocellular inflammation and ballooning without the worsening of fibrosis | 24 weeks | Active | NCT03008070 |
| Lifestyle-intervention Pioglitazone Berberine | γ | Improved metabolic parameters (glucose, lipid, liver enzymes, etc.,). | 16 weeks | Completed (results submitted) | NCT00633282 [53] |
| Pioglitazone | γ | Reduction in the NASH activity index by three points or more with improvements of at least one point each in steatosis, parenchymal inflammation, and hepatocellular injury | 48 weeks | Completed (has results) | NCT00062764 |
| Pioglitazone | γ | Improvement in hepatic histology as determined by the reduction of at least three points in the NASH activity score | 48 weeks | Completed (no results posted) | NCT00013598 |
| Saroglitazar | α/γ | Percentage change from baseline in serum ALT levels | 16 weeks | Active | NCT03061721 |
| Saroglitazar magnesium | α/γ | Safety measured by adverse events, vital signs, physical exams, body weight, electrocardiograms, and lab results | 24 weeks | Recruiting | NCT03639623 |
| Seladelpar | δ | Evaluation of the hepatic fat fraction, safety, and tolerability in NASH patients | 12 weeks | Suspended (unexpected histological findings) | NCT03551522 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2020, 9, 37. https://doi.org/10.3390/cells9010037
Boeckmans J, Natale A, Rombaut M, Buyl K, Rogiers V, De Kock J, Vanhaecke T, Rodrigues RM. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells. 2020; 9(1):37. https://doi.org/10.3390/cells9010037
Chicago/Turabian StyleBoeckmans, Joost, Alessandra Natale, Matthias Rombaut, Karolien Buyl, Vera Rogiers, Joery De Kock, Tamara Vanhaecke, and Robim M. Rodrigues. 2020. "Anti-NASH Drug Development Hitches a Lift on PPAR Agonism" Cells 9, no. 1: 37. https://doi.org/10.3390/cells9010037