
The Role of DNA Damage Response in Dysbiosis-Induced Colorectal Cancer

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Human Microbiota
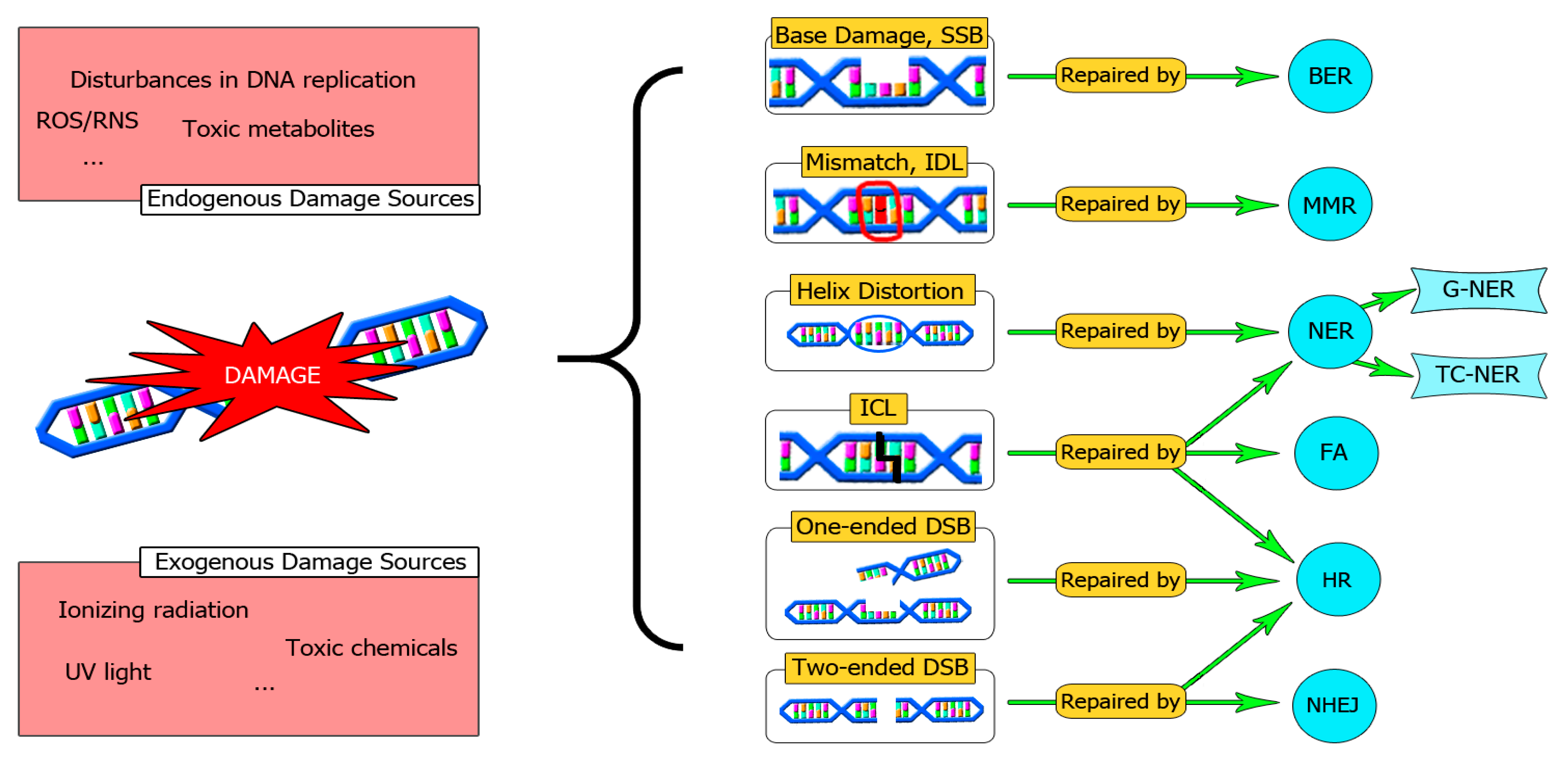
2. DNA Damage and Cancer, Old Friends
3. Colorectal Cancer: The Origin
3.1. The Poisoned Inheritance
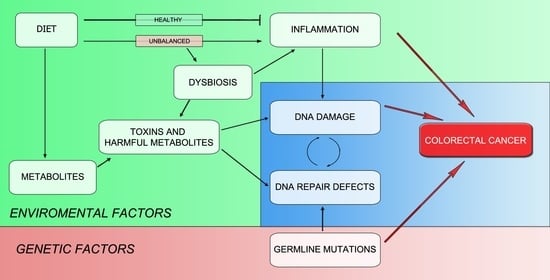
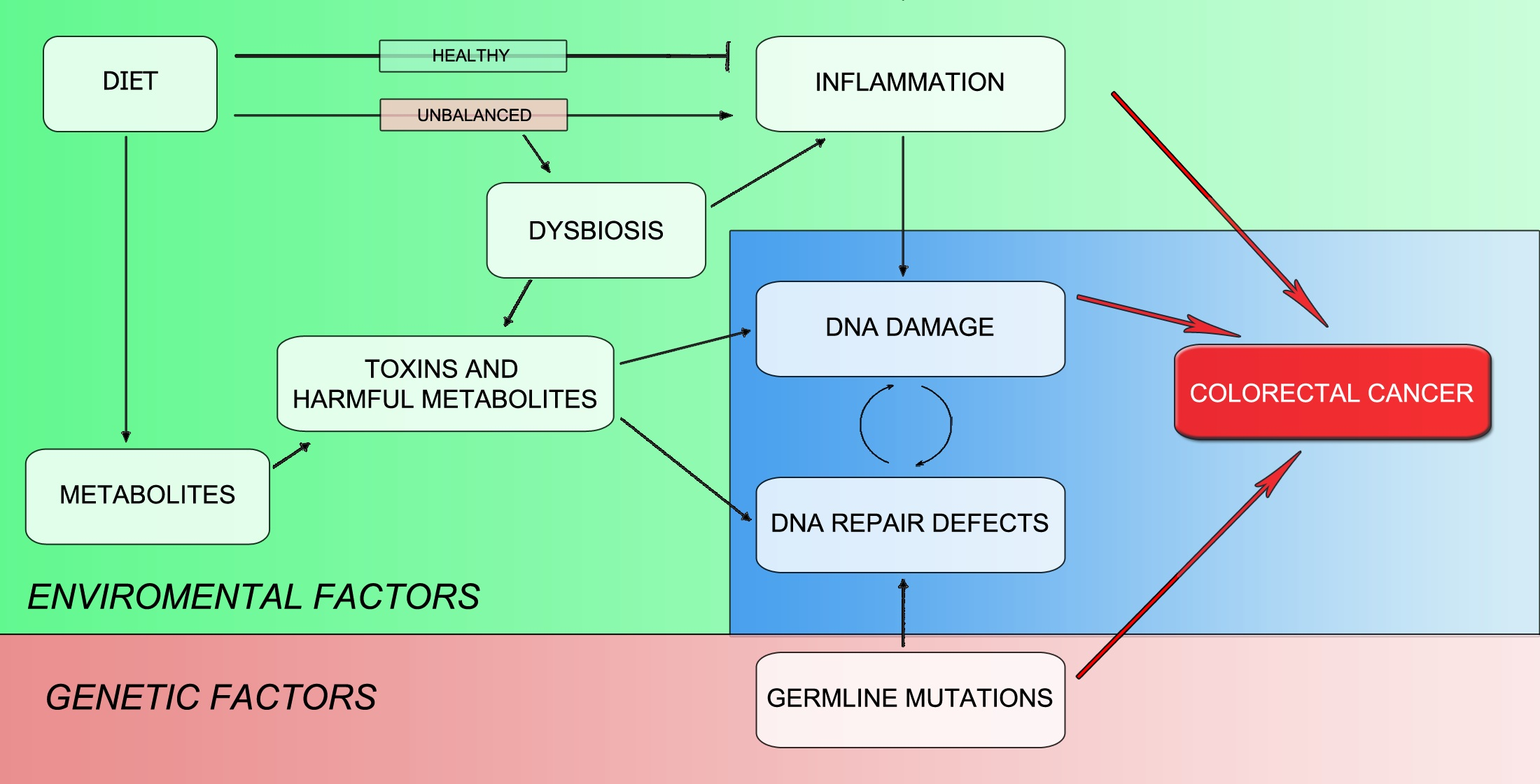
3.2. Environment Is the Key
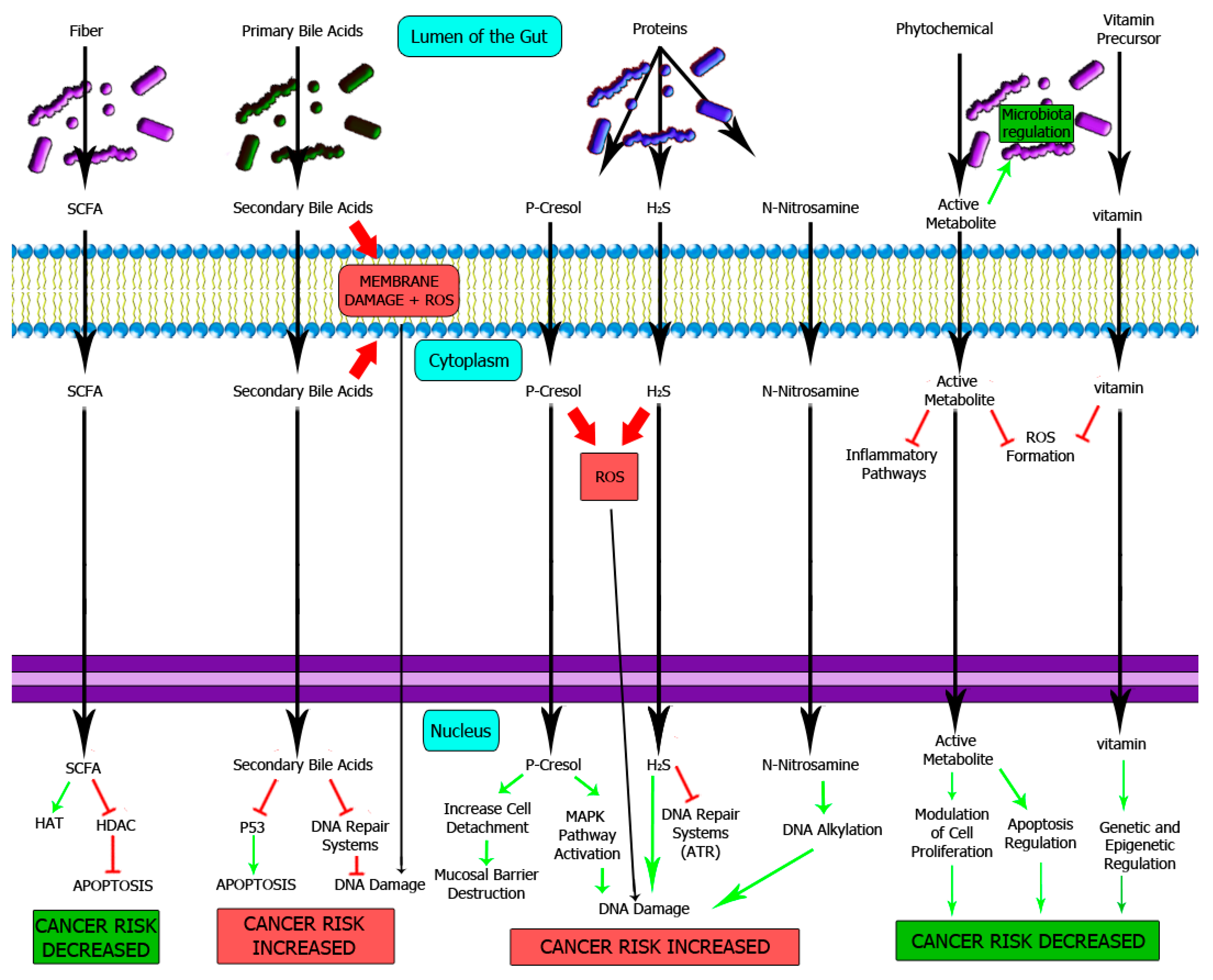
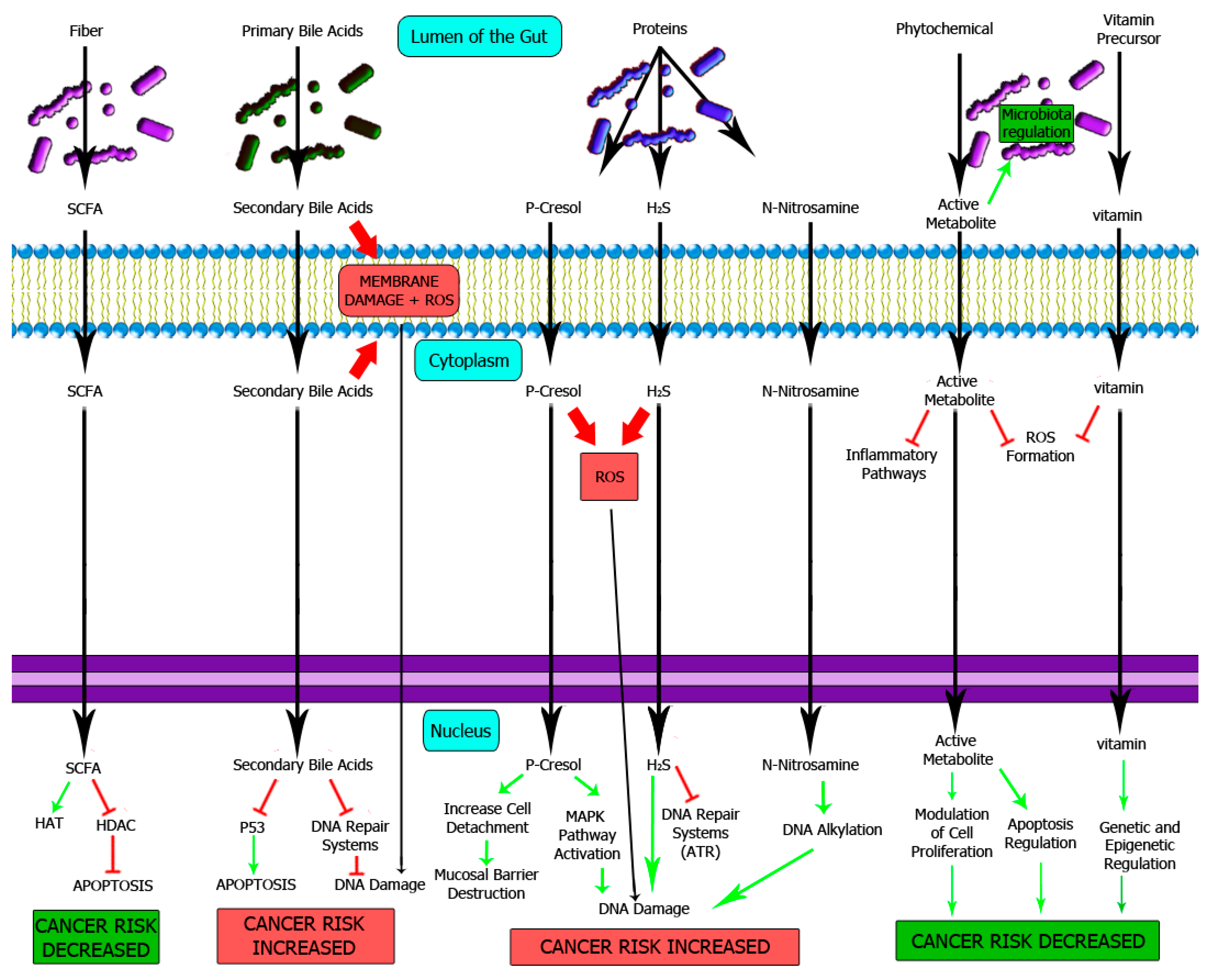
4. DNA Damage Induced by Metabolites from Diet
4.1. Metabolites from Carbohydrates
4.2. Metabolites from Fat and Bile Acids
4.3. Metabolites from Proteins
4.3.1. p-Cresol
4.3.2. H2S
4.3.3. N-Nitrosamines
4.3.4. Ammonia
4.4. Phytochemicals and Vitamins
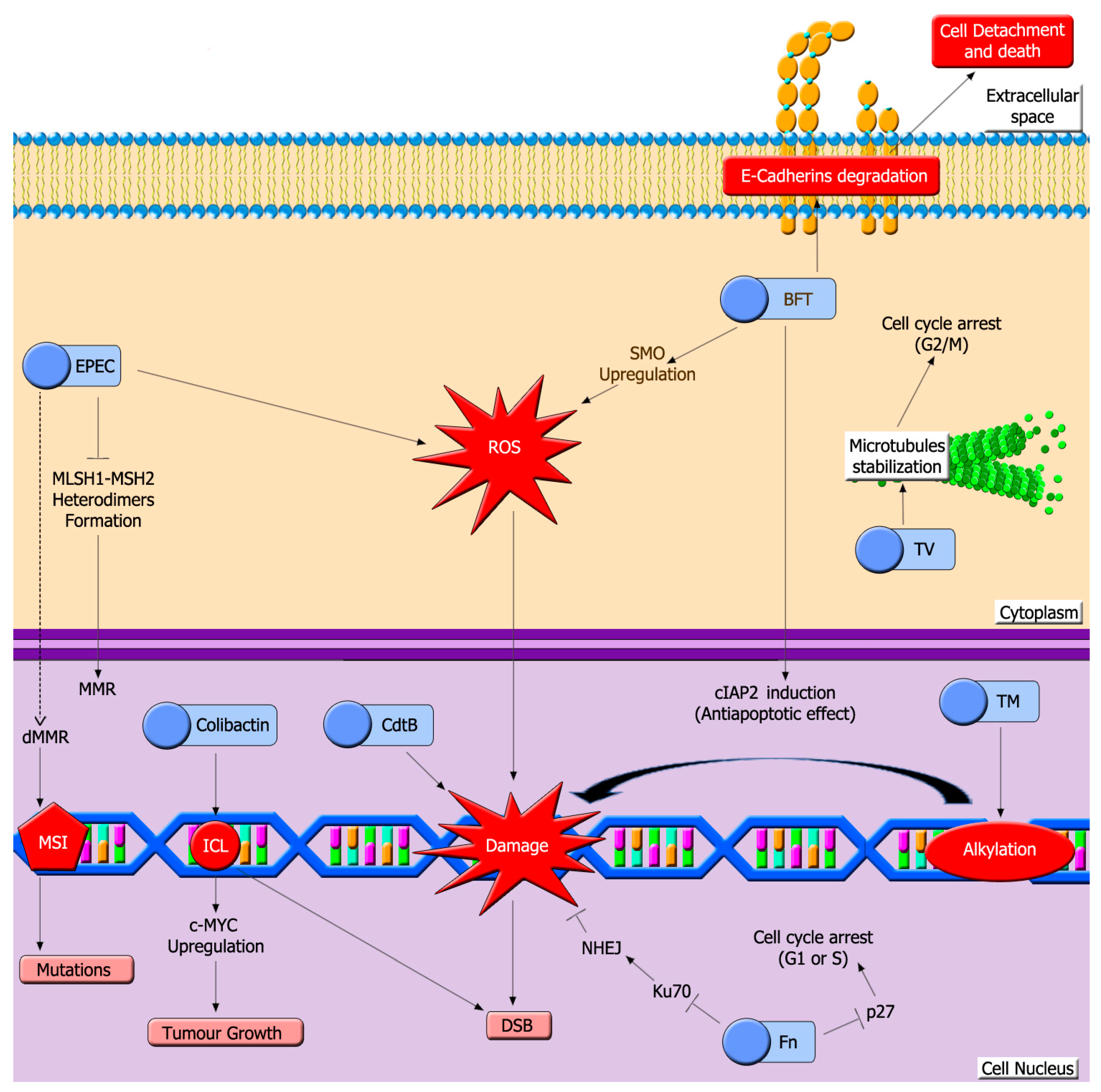
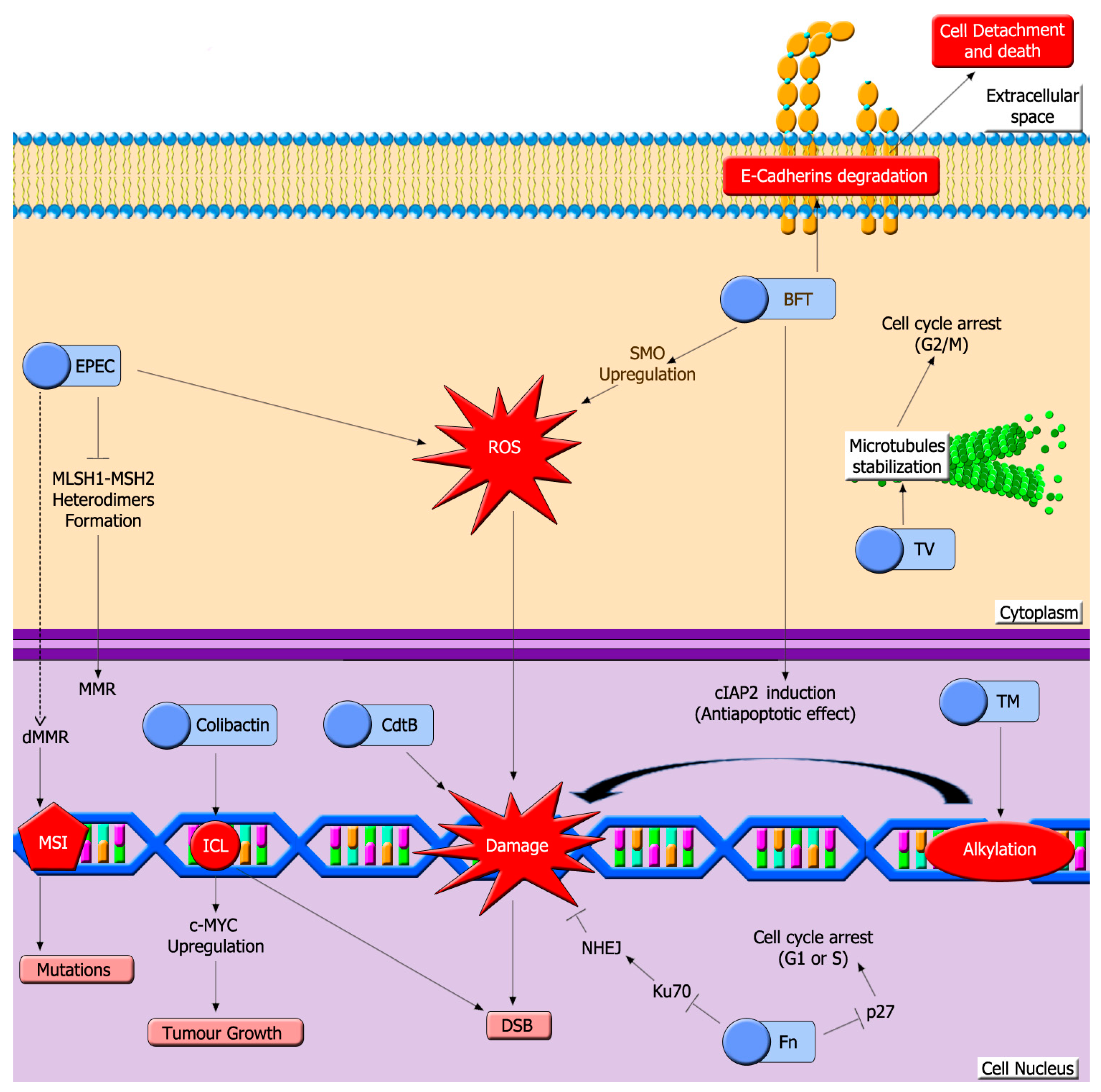
5. Microbiota Genotoxins
5.1. Colibactin
5.2. Toxins Generated by EPEC
5.3. Cytolethal Distending Toxins (Cdt)
5.4. Bacteroides Fragilis Toxin (BFT)
5.5. Fusobacterium Nucleatum (Fn) Toxins
5.6. Klebsiella Oxytoca Enterotoxins
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dominguez-Bello, M.G.; Godoy-Vitorino, F.; Knight, R.; Blaser, M.J. Role of the microbiome in human development. Gut 2019, 68, 1108–1114. [Google Scholar] [CrossRef]
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Skolnick, S.D.; Greig, N.H. Microbes and Monoamines: Potential Neuropsychiatric Consequences of Dysbiosis. Trends Neurosci. 2019, 42, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, A.M.; Sarvetnick, N.E. Current understanding of the role of gut dysbiosis in type 1 diabetes. J. Diabetes 2019, 11, 632–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.M.; Jobin, C. The Microbiome and Cancer: Is the “Oncobiome” Mirage Real? Trends Cancer 2015, 1, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.H.; Diven, M.A.; Huff, L.W.; Paulos, C.M. Harnessing the Microbiome to Enhance Cancer Immunotherapy. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The Role of IL-1β in the Early Tumour Cell–Induced Angiogenic Response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef] [Green Version]
- Kipanyula, M.J.; Seke Etet, P.F.; Vecchio, L.; Farahna, M.; Nukenine, E.N.; Nwabo Kamdje, A.H. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell. Signal. 2013, 25, 403–416. [Google Scholar] [CrossRef]
- Ono, M. Molecular links between tumour angiogenesis and inflammation: Inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008, 99, 1501–1506. [Google Scholar] [CrossRef]
- Helleday, T. DNA repair as treatment target. Eur. J. Cancer 2011, 47, S333–S335. [Google Scholar] [CrossRef]
- Puigvert, J.C.; Sanjiv, K.; Helleday, T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016, 283, 232–245. [Google Scholar] [CrossRef]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Mammalian DNA base excision repair: Dancing in the moonlight. DNA Repair 2020, 93, 102921. [Google Scholar] [CrossRef] [PubMed]
- Orta, M.L.; Höglund, A.; Calderón-Montaño, J.M.; Domínguez, I.; Burgos-Morón, E.; Visnes, T.; Pastor, N.; Ström, C.; López-lázaro, M.; Helleday, T. The PARP inhibitor Olaparib disrupts base excision repair of 5-aza-2′-deoxycytidine lesions. Nucleic Acids Res. 2014, 42, 9108–9120. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P.L. DNA Mismatch Repair: Functions and Mechanisms. Chem. Rev. 2006. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [Green Version]
- Reilly, N.M.; Novara, L.; Di Nicolantonio, F.; Bardelli, A. Exploiting DNA repair defects in colorectal cancer. Mol. Oncol. 2019, 13, 681–700. [Google Scholar] [CrossRef] [Green Version]
- Gelsomino, F.; Barbolini, M.; Spallanzani, A.; Pugliese, G.; Cascinu, S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat. Rev. 2016, 51, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Ruiz-Bañobre, J.; Goel, A. DNA Mismatch Repair Deficiency and Immune Checkpoint Inhibitors in Gastrointestinal Cancers. Gastroenterology 2019, 156, 890–903. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, R.; Ozluk, E.; Islam, M.Z.; Hopper, B.; Meram, A.; Ghali, G.; Kevil, C.G. Hydrogen sulfide and DNA repair. Redox Biol. 2021, 38, 101675. [Google Scholar] [CrossRef]
- Bradbury, A.; Hall, S.; Curtin, N.; Drew, Y. Targeting ATR as Cancer Therapy: A new era for synthetic lethality and synergistic combinations? Pharmacol. Ther. 2020, 207, 107450. [Google Scholar] [CrossRef]
- Chen, J.; Shen, X.; Pardue, S.; Meram, A.T.; Rajendran, S.; Ghali, G.E.; Kevil, C.G.; Shackelford, R.E. The Ataxia telangiectasia-mutated and Rad3-related protein kinase regulates cellular hydrogen sulfide concentrations. DNA Repair 2019, 73, 55–63. [Google Scholar] [CrossRef]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.K. DNA-PKcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 532, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiffin, N.; Hosking, F.J.; Farrington, S.M.; Palles, C.; Dobbins, S.E.; Zgaga, L.; Lloyd, A.; Kinnersley, B.; Gorman, M.; Tenesa, A.; et al. Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum. Mol. Genet. 2014, 23, 4729–4737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantor, M.; Sobrado, J.; Patel, S.; Eiseler, S.; Ochner, C. Hereditary Colorectal Tumours: A Literature Review on MUTYH-Associated Polyposis. Gastroenterol. Res. Pract. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Puccini, A.; Berger, M.D.; Naseem, M.; Tokunaga, R.; Battaglin, F.; Cao, S.; Hanna, D.L.; McSkane, M.; Soni, S.; Zhang, W.; et al. Colorectal cancer: Epigenetic alterations and their clinical implications. Biochim. Biophys. Acta - Rev. Cancer 2017, 1868, 439–448. [Google Scholar] [CrossRef]
- Hale, V.L.; Jeraldo, P.; Chen, J.; Mundy, M.; Yao, J.; Priya, S.; Keeney, G.; Lyke, K.; Ridlon, J.; White, B.A.; et al. Distinct microbes, metabolites, and ecologies define the microbiome in deficient and proficient mismatch repair colorectal cancers. Genome Med. 2018, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Mármol, I.; Sánchez-de-Diego, C.; Dieste, A.P.; Cerrada, E.; Yoldi, M.J.R. Colorectal carcinoma: A general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumourigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- O’Keefe, S.J.D. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.; Xie, M.; Yu, H.; Chen, H. Intestinal dysbacteriosis activates tumour-associated macrophages to promote epithelial-mesenchymal transition of colorectal cancer. Innate Immun. 2018, 24, 480–489. [Google Scholar] [CrossRef] [Green Version]
- Irwin, C.R.; Myrillas, T.T.; Traynor, P.; Leadbetter, N.; Cawston, T.E. The Role of Soluble Interleukin (IL)-6 Receptor in Mediating the effects of IL-6 on Matrix Metalloproteinase-1 and Tissue Inhibitor of Metalloproteinase-1 Expression by Gingival Fibroblasts. J. Periodontol. 2002, 73, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, H.; Song, J.; Sugimoto, M.; Hagihara, K.; Kishimoto, T.; Yoshizaki, K.; Nishimoto, N. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 1521–1529. [Google Scholar] [CrossRef]
- Cai, Q.; Huang, H.; Bai, B.; Lin, S.; Gao, Y.; Xia, Y.; Wang, X.; Lu, J. IL-6 Promotes Cell Proliferation and Antiapoptosis through Activation of the JAK/STAT3 Pathway in Patients with NK/T—Cell Lymphoma and Correlates with Poor Treatmemt Outcome. Blood 2013, 122, 1758. [Google Scholar] [CrossRef]
- McDermott, E.P.; O’Neill, L.A.J. Ras participates in the activation of p38 MAPK by interleukin-1 by associating with IRAK, IRAK2, TRAF6, and TAK-1. J. Biol. Chem. 2002, 277, 7808–7815. [Google Scholar] [CrossRef] [Green Version]
- Palsson, E.M.; Popoff, M.; Thelestam, M.; O’Neill, L.A.J. Divergent roles for Ras and Rap in the activation of p38 mitogen- activated protein kinase by interleukin-1. J. Biol. Chem. 2000, 275, 7818–7825. [Google Scholar] [CrossRef] [Green Version]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumour invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human Intestinal Lumen and Mucosa-Associated Microbiota in Patients with Colorectal Cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.M.; Liu, H.Q.; Liu, S.R.; Tang, S.P.; Yang, L.; Feng, G.S. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1β in vivo and in vitro. Breast Cancer Res. Treat. 2005, 89, 5–14. [Google Scholar] [CrossRef]
- Pannone, G.; Santoro, A.; Feola, A.; Bufo, P.; Papagerakis, P.; Muzio, L.; Staibano, S.; Ionna, F.; Longo, F.; Franco, R.; et al. The Role of E-Cadherin Down-Regulation in Oral Cancer: CDH1 Gene Expression and Epigenetic Blockage. Curr. Cancer Drug Targets 2014, 14, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Lin, S.; Li, Y.; Zamyatnin, A.A.; Werner, J.; Bazhin, A.V. Reactive oxygen species and colorectal cancer. J. Cell. Physiol. 2018, 233, 5119–5132. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumour progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrazabal, T.; Thakur, B.K.; Kang, M.; Malaise, Y.; Streutker, C.; Wong, E.O.Y.; Copeland, J.; Gryfe, R.; Guttman, D.S.; Navarre, W.W.; et al. Limiting oxidative DNA damage reduces microbe-induced colitis-associated colorectal cancer. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, H.; Bartsch, H. Chronic infections and inflammatory processes as cancer risk factors: Possible role of nitric oxide in carcinogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1994, 305, 253–264. [Google Scholar] [CrossRef]
- Lancaster, J.R.; Xie, K. Tumours face NO problems? Cancer Res. 2006, 66, 6459–6462. [Google Scholar] [CrossRef] [Green Version]
- Gaston, B.; Drazen, J.M.; Loscalzo, J.; Stamler, J.S. The biology of nitrogen oxides in the airways. Am. J. Respir. Crit. Care Med. 1994, 149, 538–551. [Google Scholar] [CrossRef]
- Edwards, R.A.; Witherspoon, M.; Wang, K.; Afrasiabi, K.; Pham, T.; Birnbaumer, L.; Lipkin, S.M. Epigenetic repression of DNA mismatch repair by inflammation and hypoxia in inflammatory bowel disease-associated colorectal cancer. Cancer Res. 2009, 69, 6423–6429. [Google Scholar] [CrossRef] [Green Version]
- Sárközy, M.; Kovács, Z.Z.A.; Kovács, M.G.; Gáspár, R.; Szűcs, G.; Dux, L. Mechanisms and Modulation of Oxidative/Nitrative Stress in Type 4 Cardio-Renal Syndrome and Renal Sarcopenia. Front. Physiol. 2018, 9, 1648. [Google Scholar] [CrossRef]
- Vermeer, I.T.M.; Van Maanen, J.M.S. Nitrate exposure and the endogenous formation of carcinogenic nitrosamines in humans. Rev. Environ. Health 2001, 16, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, I.T.M.; Henderson, L.Y.; Moonen, E.J.C.; Engels, L.G.J.B.; Dallinga, J.W.; Van Maanen, J.M.S.; Kleinjans, C.S. Neutrophil-mediated formation of carcinogenic N-nitroso compounds in an in vitro model for intestinal inflammation. Toxicol. Lett. 2004, 154, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Zenser, T.V.; Lakshmi, V.M.; Schut, H.A.J.; Zhou, H.j.; Josephy, P.D. Activation of aminoimidazole carcinogens by nitrosation: Mutagenicity and nucleotide adducts. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2009, 673, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Alhinai, E.A.; Walton, G.E.; Commane, D.M. The role of the gut microbiota in colorectal cancer causation. Int. J. Mol. Sci. 2019, 20, 5295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Abu-Ghazaleh, N.; Chua, W.J.; Gopalan, V. Intestinal microbiota and its association with colon cancer and red/processed meat consumption. J. Gastroenterol. Hepatol. 2021, 36, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keefe, S.J.D.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldati, L.; Di Renzo, L.; Jirillo, E.; Ascierto, P.A.; Marincola, F.M.; De Lorenzo, A. The influence of diet on anti-cancer immune responsiveness. J. Transl. Med. 2018, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 1500902. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhu, M.J. Butyrate Inhibits Indices of Colorectal Carcinogenesis via Enhancing α-Ketoglutarate-Dependent DNA Demethylation of Mismatch Repair Genes. Mol. Nutr. Food Res. 2018, 62, 1700932. [Google Scholar] [CrossRef]
- Lucas, C.; Barnich, N.; Nguyen, H.T.T. Microbiota, Inflammation and Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 1310. [Google Scholar] [CrossRef] [Green Version]
- Abdulla Al Hinai, E.; Kullamethee, P.; Rowland, I.R.; Swann, J.; Walton, G.E.; Commane, D.M. Gut Microbes Modelling the role of microbial p-cresol in colorectal genotoxicity Modelling the role of microbial p-cresol in colorectal genotoxicity. Gut Microbes 2018. [Google Scholar] [CrossRef] [Green Version]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of human gut microbiota in control subjects and patients with colorectal carcinoma in adenoma: Terminal restriction fragment length polymorphism and next-generation sequencing analyses. Oncol. Rep. 2016, 35, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Jahani-Sherafat, S.; Alebouyeh, M.; Moghim, S.; Amoli, H.A.; Ghasemian-Safaei, H. Role of gut microbiota in the pathogenesis of colorectal cancer; A review article. Gastroenterol. Hepatol. Bed Bench 2018, 11, 101–109. [Google Scholar] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Salek Farrokhi, A.; Mohammadlou, M.; Abdollahi, M.; Eslami, M.; Yousefi, B. Histone Deacetylase Modifications by Probiotics in Colorectal Cancer. J. Gastrointest. Cancer 2020, 51, 754–764. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Payne, C. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin. Exp. Gastroenterol. 2008, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Hadden, T.; Yu, Y.; Majumdar, A.P.N. Bile acid: A potential inducer of colon cancer stem cells. Stem Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Wolf, P.G.; Gaskins, H.R. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016, 7, 201–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosignoli, P.; Fabiani, R.; De Bartolomeo, A.; Fuccelli, R.; Pelli, M.A.; Morozzi, G. Genotoxic effect of bile acids on human normal and tumour colon cells and protection by dietary antioxidants and butyrate. Eur. J. Nutr. 2008, 47, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.K.; Muir, J.G.; Gibson, P.R. Review article: Insights into colonic protein fermentation, its modulation and potential health implications. Aliment. Pharmacol. Ther. 2016, 43, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andriamihaja, M.; Lan, A.; Beaumont, M.; Audebert, M.; Wong, X.; Yamada, K.; Yin, Y.; Tomé, D.; Carrasco-Pozo, C.; Gotteland, M.; et al. The deleterious metabolic and genotoxic effects of the bacterial metabolite p-cresol on colonic epithelial cells. Free Radic. Biol. Med. 2015, 85, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Lecerf, J.M.; Dépeint, F.; Clerc, E.; Dugenet, Y.; Niamba, C.N.; Rhazi, L.; Cayzeele, A.; Abdelnour, G.; Jaruga, A.; Younes, H.; et al. Xylo-oligosaccharide (XOS) in combination with inulin modulates both the intestinal environment and immune status in healthy subjects, while XOS alone only shows prebiotic properties. Br. J. Nutr. 2012, 108, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Boutwell, R.K.; Bosch, D.K. The tumour-promoting action of phenol and related compounds for mouse skin. Cancer Res. 1959, 19, 413–424. [Google Scholar]
- Attene-Ramos, M.S.; Nava, G.M.; Muellner, M.G.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ. Mol. Mutagen. 2010, 51, 304–314. [Google Scholar] [CrossRef]
- Aroca, A.; Gotor, C.; Bassham, D.C.; Romero, L.C. Hydrogen Sulfide: From a Toxic Molecule to a Key Molecule of Cell Life. Antioxidants 2020, 9, 621. [Google Scholar] [CrossRef]
- Wu, D.; Si, W.; Wang, M.; Lv, S.; Ji, A.; Li, Y. Hydrogen sulfide in cancer: Friend or foe? Nitric Oxide - Biol. Chem. 2015, 50, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wang, M.; Ju, L.; Wang, C.; Zhu, Y. Hydrogen sulfide induces human colon cancer cell proliferation: Role of Akt, ERK and p21. Cell Biol. Int. 2010, 34, 565–572. [Google Scholar] [CrossRef]
- Wu, Y.C.; Wang, X.J.; Yu, L.; Chan, F.K.L.; Cheng, A.S.L.; Yu, J.; Sung, J.J.Y.; Wu, W.K.K.; Cho, C.H. Hydrogen sulfide lowers proliferation and induces protective autophagy in colon epithelial cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, H.; Teng, T.; Duan, S.; Ji, A.; Li, Y. Hydrogen sulfide and autophagy: A double edged sword. Pharmacol. Res. 2018, 131, 120–127. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 2006, 4, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Attene-Ramos, M.S.; Wagner, E.D.; Gaskins, H.R.; Plewa, M.J. Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 2007, 5, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Güerci, A.; Liviac, D.; Marcos, R. Detection of excision repaired DNA damage in the comet assay by using Ara-C and hydroxyurea in three different cell types. Cell Biol. Toxicol. 2009, 25, 73–80. [Google Scholar] [CrossRef]
- Deplancke, B.; Gaskins, H.R. Hydrogen sulfide induces serum-independent cell cycle entry in nontransformed rat intestinal epithelial cells. FASEB J. 2003, 17, 1310–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.L. The toxicity of hydrogen sulphide and other sulphides. Q. J. Exp. Physiol. Cogn. Med. Sci. 1967, 52, 231–248. [Google Scholar] [CrossRef]
- Shattuck-Brandt, R.L.; Varilek, G.W.; Radhika, A.; Yang, F.; Washington, M.K.; DuBois, R.N. Cyclooxygenase 2 expression is increased in the stroma of colon carcinomas from IL-10(-/-) mice. Gastroenterology 2000, 118, 337–345. [Google Scholar] [CrossRef]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Zhi, L.; Ang, A.D.; Zhang, H.; Moore, P.K.; Bhatia, M. Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-κB pathway. J. Leukoc. Biol. 2007, 81, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Horrell, E.M.W.; Christian, P.A.; Vanover, J.C.; Boulanger, M.C.; Zou, Y.; D’Orazio, J.A. PKA-Mediated Phosphorylation of ATR Promotes Recruitment of XPA to UV-Induced DNA Damage. Mol. Cell 2014, 54, 999–1011. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.E.; Haza, A.I.; Arranz, N.; García, A.; Morales, P.; Delgado, M.E.; Haza, A.A.I.; Arranz, A.N.; García, A.; Morales, A.P. Dietary polyphenols protect against N-nitrosamines and benzo(a)pyrene-induced DNA damage (strand breaks and oxidized purines/pyrimidines) in HepG2 human hepatoma cells. Eur. J. Nutr. 2008, 47, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Guttenplan, J.B. N-Nitrosamines: Bacterial mutagenesis and in vitro metabolism. Mutat. Res. 1987, 186, 81–134. [Google Scholar] [CrossRef]
- Zink, C.N.; Soissons, N.; Fishbein, J.C. Products of the direct reaction of the diazonium ion of a metabolite of the carcinogen N -nitrosomorpholine with purines of nucleosides and dna. Chem. Res. Toxicol. 2010, 23, 1223–1233. [Google Scholar] [CrossRef]
- Arranz, N.; Haza, A.I.; García, A.; Rafter, J.; Morales, P. Protective effect of vitamin C towards N-nitrosamine-induced DNA damage in the single-cell gel electrophoresis (SCGE)/HepG2 assay. Toxicol. Vitr. 2007, 21, 1311–1317. [Google Scholar] [CrossRef]
- Arranz, N.; Haza, A.I.; García, A.; Möller, L.; Rafter, J.; Morales, P. Protective effects of organosulfur compounds towards N-nitrosamine-induced DNA damage in the single-cell gel electrophoresis (SCGE)/HepG2 assay. Food Chem. Toxicol. 2007, 45, 1662–1669. [Google Scholar] [CrossRef]
- Bobermin, L.D.; Souza, D.O.; Gonçalves, C.A.; Quincozes-Santos, A. Resveratrol prevents ammonia-induced mitochondrial dysfunction and cellular redox imbalance in C6 astroglial cells. Nutr. Neurosci. 2018, 21, 276–285. [Google Scholar] [CrossRef]
- Görg, B.; Karababa, A.; Häussinger, D. Hepatic Encephalopathy and Astrocyte Senescence. J. Clin. Exp. Hepatol. 2018, 8, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, S.; Jiang, Y.; Zhao, Y.; Sun, L.; Zheng, B.; Chen, L.; Liu, Z.; Zheng, X.; Yi, K.; et al. Effects of ammonia on apoptosis and oxidative stress in bovine mammary epithelial cells. Mutagenesis 2018, 33. [Google Scholar] [CrossRef]
- Wilson, A.S.; Koller, K.R.; Ramaboli, M.C.; Nesengani, L.T.; Ocvirk, S.; Chen, C.; Flanagan, C.A.; Sapp, F.R.; Merritt, Z.T.; Bhatti, F.; et al. Diet and the Human Gut Microbiome: An International Review. Dig. Dis. Sci. 2020, 65, 723–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espín, J.C.; González-Sarrías, A.; Tomás-Barberán, F.A. The gut microbiota: A key factor in the therapeutic effects of (poly)phenols. Biochem. Pharmacol. 2017, 139, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.P.; Zhao, Y.; Huang, F.; Chen, J.; Yao, Y.H.; Li, J.; Wu, X.N. Equol inhibits proliferation of human gastric carcinoma cells via modulating Akt pathway. World J. Gastroenterol. 2015, 21, 10385–10399. [Google Scholar] [CrossRef]
- Herman, C.; Adlercreãoetz, T.; Goldin, B.R.; Gorbach, S.L.; Hã-Ckerstedt, K.A.V.; Watanabe, S.; Hämäläinen, E.K.; Helene Markkanen, M.; Mäkelä, T.H.; Wähälä, K.T.; et al. Soybean Phytoestrogen Intake and Cancer Risk12. J. Nutr. 1995, 125, 757S–770S. [Google Scholar]
- Arora, A.; Nair, M.G.; Strasburg, G.M. Antioxidant activities of isoflavones and their biological metabolites in a liposomal system. Arch. Biochem. Biophys. 1998, 356, 133–141. [Google Scholar] [CrossRef]
- Casto, B.C.; Knobloch, T.J.; Galioto, R.L.; Yu, Z.; Accurso, B.T.; Warner, B.M. Chemoprevention of oral cancer by lyophilized strawberries. Anticancer Res. 2013, 33, 4757–4766. [Google Scholar]
- Balansky, R.; Ganchev, G.; Iltcheva, M.; Kratchanova, M.; Denev, P.; Kratchanov, C.; Polasa, K.; D’Agostini, F.; Steele, V.E.; De Flora, S. Inhibition of lung tumour development by berry extracts in mice exposed to cigarette smoke. Int. J. Cancer 2012, 131, 1991–1997. [Google Scholar] [CrossRef]
- Somasagara, R.R.; Hegde, M.; Chiruvella, K.K.; Musini, A.; Choudhary, B.; Raghavan, S.C. Extracts of Strawberry Fruits Induce Intrinsic Pathway of Apoptosis in Breast Cancer Cells and Inhibits Tumour Progression in Mice. PLoS ONE 2012, 7, e47021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: A review. Eur. J. Nutr. 2015, 54, 325–341. [Google Scholar] [CrossRef] [Green Version]
- Shi, N.; Clinton, S.K.; Liu, Z.; Wang, Y.; Riedl, K.M.; Schwartz, S.J.; Zhang, X.; Pan, Z.; Chen, T. Strawberry phytochemicals inhibit azoxymethane/dextran sodium sulfate-induced colorectal carcinogenesis in Crj: CD-1 mice. Nutrients 2015, 7, 1696–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrera-Quintanar, L.; Roa, R.I.L.; Quintero-Fabián, S.; Sánchez-Sánchez, M.A.; Vizmanos, B.; Ortuño-Sahagún, D. Phytochemicals that influence gut microbiota as prophylactics and for the treatment of obesity and inflammatory diseases. Mediators Inflamm. 2018, 2018. [Google Scholar] [CrossRef]
- Yuan, X.; Long, Y.; Ji, Z.; Gao, J.; Fu, T.; Yan, M.; Zhang, L.; Su, H.; Zhang, W.; Wen, X.; et al. Green Tea Liquid Consumption Alters the Human Intestinal and Oral Microbiome. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Lázaro, M.; Calderón-Montaño, J.M.; Burgos-Morón, E.; Austin, C.A. Green tea constituents (-)-epigallocatechin-3-gallate (EGCG) and gallic acid induce topoisomerase I- and topoisomerase II-DNA complexes in cells mediated by pyrogallol-induced hydrogen peroxide. Mutagenesis 2011, 26, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Crascì, L.; Lauro, M.R.; Puglisi, G.; Panico, A. Natural antioxidant polyphenols on inflammation management: Anti-glycation activity vs metalloproteinases inhibition. Crit. Rev. Food Sci. Nutr. 2018, 58, 893–904. [Google Scholar] [CrossRef]
- Neukam, K.; Pastor, N.; Cortés, F. Tea flavanols inhibit cell growth and DNA topoisomerase II activity and induce endoreduplication in cultured Chinese hamster cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2008, 654, 8–12. [Google Scholar] [CrossRef]
- Duthie, S.J. Epigenetic modifications and human pathologies: Cancer and CVD. In Proceedings of the Nutrition Society; Cambridge University Press: Cambridge, UK, 2011; Volume 70, pp. 47–56. [Google Scholar]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Catala, G.N.; Bestwick, C.S.; Russell, W.R.; Tortora, K.; Giovannelli, L.; Moyer, M.P.; Lendoiro, E.; Duthie, S.J. Folate, genomic stability and colon cancer: The use of single cell gel electrophoresis in assessing the impact of folate in vitro, in vivo and in human biomonitoring. Mutat. Res. - Genet. Toxicol. Environ. Mutagen. 2019, 843, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. In Advances in Food and Nutrition Research; Academic Press Inc.: Amsterdan, The Netherlands, 2018; Volume 83, pp. 83–149. ISBN 9780128118030. [Google Scholar]
- Sheflin, A.M.; Whitney, A.K.; Weir, T.L. Cancer-Promoting Effects of Microbial Dysbiosis. Curr. Oncol. Rep. 2014, 16, 406. [Google Scholar] [CrossRef] [Green Version]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More than a new bacterial toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.R.; Li, J.; Cai, W.; Lai, J.Y.H.; McKinnie, S.M.K.; Zhang, W.P.; Moore, B.S.; Zhang, W.; Qian, P.Y. Macrocyclic colibactin induces DNA double-strand breaks via copper-mediated oxidative cleavage. Nat. Chem. 2019, 11, 880–889. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.P. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Nougayrède, J.-P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Xue, M.; Wernke, K.M.; Herzon, S.B. Depurination of colibactin-derived interstrand cross-links. Biochemistry 2020, 59, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Vizcaino, M.I.; Crawford, J.M. The colibactin warhead crosslinks DNA. Nat. Chem. 2015, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Sun, Q.; Li, D. Sumoylation in Cellular Senescence and Aging. Curr. Mol. Med. 2017, 16, 871–876. [Google Scholar] [CrossRef]
- Leibiger, K.; Schweers, J.M.; Schütz, M. Biogenesis and function of the autotransporter adhesins YadA, intimin and invasin. Int. J. Med. Microbiol. 2019, 309, 331–337. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Short, A.J.; Donnenberg, M.S.; Bader, S.; Harrison, D.J. Attaching and Effacing Escherichia coli Downregulate DNA Mismatch Repair Protein In Vitro and Are Associated with Colorectal Adenocarcinomas in Humans. PLoS ONE 2009, 4, e5517. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, O.D.K.; Scanlon, K.M.; Donnenberg, M.S. An escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. MBio 2013, 4, e5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.L.; Marra, G.; Chauhan, D.P.; Ha, H.T.; Chang, D.K.; Ricciardiello, L.; Randolph, A.; Carethers, J.M.; Richard Boland, C. Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 2002, 283, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Feng, Y.; Ge, L.; Parry, N.; Muthupalani, S.; Fox, J.G. Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell. Microbiol. 2017, 19, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumourigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumour progression? FEBS J. 2011, 278, 4577–4588. [Google Scholar] [CrossRef] [Green Version]
- Frisan, T.; Cortes-Bratti, X.; Chaves-Olarte, E.; Stenerlow, B.; Thelestam, M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double-strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlöw, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumourigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, A.C.; Destefano Shields, C.E.; Wu, S.; Huso, D.L.; Wu, X.Q.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumourigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef] [Green Version]
- Sears, C.L.; Geis, A.L.; Housseau, F. Bacteroides fragilis subverts mucosal biology: From symbiont to colon carcinogenesis. J. Clin. Invest. 2014, 124, 4166–4172. [Google Scholar] [CrossRef] [Green Version]
- Koi, M.; Okita, Y.M.; Carethers, J. Fusobacterium nucleatum Infection in Colorectal Cancer: Linking Inflammation, DNA Mismatch Repair and Genetic and Epigenetic Alterations. J. Anus Rectum Colon 2018, 2, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, F.; Zhang, Y.; Lu, Z.; Zhang, S.; Pan, Y. Fusobacterium nucleatum Caused DNA Damage and Promoted Cell Proliferation by the Ku70/p53 Pathway in Oral Cancer Cells. DNA Cell Biol. 2020, 39, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Beaugerie, L.; Metz, M.; Barbut, F.; Bellaiche, G.; Bouhnik, Y.; Raskine, L.; Nicolas, J.C.; Chatelet, F.P.; Lehn, N.; Petit, J.C. Klebsiella oxytoca as an agent of antibiotic-associated hemorrhagic colitis. Clin. Gastroenterol. Hepatol. 2003, 1, 370–376. [Google Scholar] [CrossRef]
- Abbas, A.F.; Al-Saadi, A.G.M.; Alkhudhairy, M.K. Biofilm Formation and Virulence Determinants of Klebsiella oxytoca Clinical Isolates from Patients with Colorectal Cancer. J. Gastrointest. Cancer 2020, 51, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Unterhauser, K.; Pöltl, L.; Schneditz, G.; Kienesberger, S.; Glabonjat, R.A.; Kitsera, M.; Pletz, J.; Josa-Prado, F.; Dornisch, E.; Lembacher-Fadum, C.; et al. Klebsiella oxytoca enterotoxins tilimycin and tilivalline have distinct host DNA-damaging and microtubule-stabilizing activities. Proc. Natl. Acad. Sci. USA 2019, 116, 3774–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hering, N.A.; Fromm, A.; Bücker, R.; Gorkiewicz, G.; Zechner, E.; Högenauer, C.; Fromm, M.; Schulzke, J.D.; Troeger, H. Tilivalline-and tilimycin-independent effects of klebsiella oxytoca on tight junction-mediated intestinal barrier impairment. Int. J. Mol. Sci. 2019, 20, 5595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivas-Domínguez, A.; Pastor, N.; Martínez-López, L.; Colón-Pérez, J.; Bermúdez, B.; Orta, M.L. The Role of DNA Damage Response in Dysbiosis-Induced Colorectal Cancer. Cells 2021, 10, 1934. https://doi.org/10.3390/cells10081934
Rivas-Domínguez A, Pastor N, Martínez-López L, Colón-Pérez J, Bermúdez B, Orta ML. The Role of DNA Damage Response in Dysbiosis-Induced Colorectal Cancer. Cells. 2021; 10(8):1934. https://doi.org/10.3390/cells10081934
Chicago/Turabian StyleRivas-Domínguez, Antonio, Nuria Pastor, Laura Martínez-López, Julia Colón-Pérez, Beatriz Bermúdez, and Manuel Luis Orta. 2021. "The Role of DNA Damage Response in Dysbiosis-Induced Colorectal Cancer" Cells 10, no. 8: 1934. https://doi.org/10.3390/cells10081934
APA StyleRivas-Domínguez, A., Pastor, N., Martínez-López, L., Colón-Pérez, J., Bermúdez, B., & Orta, M. L. (2021). The Role of DNA Damage Response in Dysbiosis-Induced Colorectal Cancer. Cells, 10(8), 1934. https://doi.org/10.3390/cells10081934