
Landscape of Bone Marrow Metastasis in Human Neuroblastoma Unraveled by Transcriptomics and Deep Multiplex Imaging

 , , , , ,
, , , , ,  , ,
, ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Subject Details
2.1.1. Neuroblastoma Cell Lines
2.1.2. Bone Marrow Aspirates
2.1.3. Peripheral Blood-Derived MNCs
2.2. Biomarker Identification by Data Mining
2.3. Biomarker Validation
2.4. Multi Epitope Ligand Cartography
2.5. Sample Preparation, RNA-Isolation and RNA-Sequencing
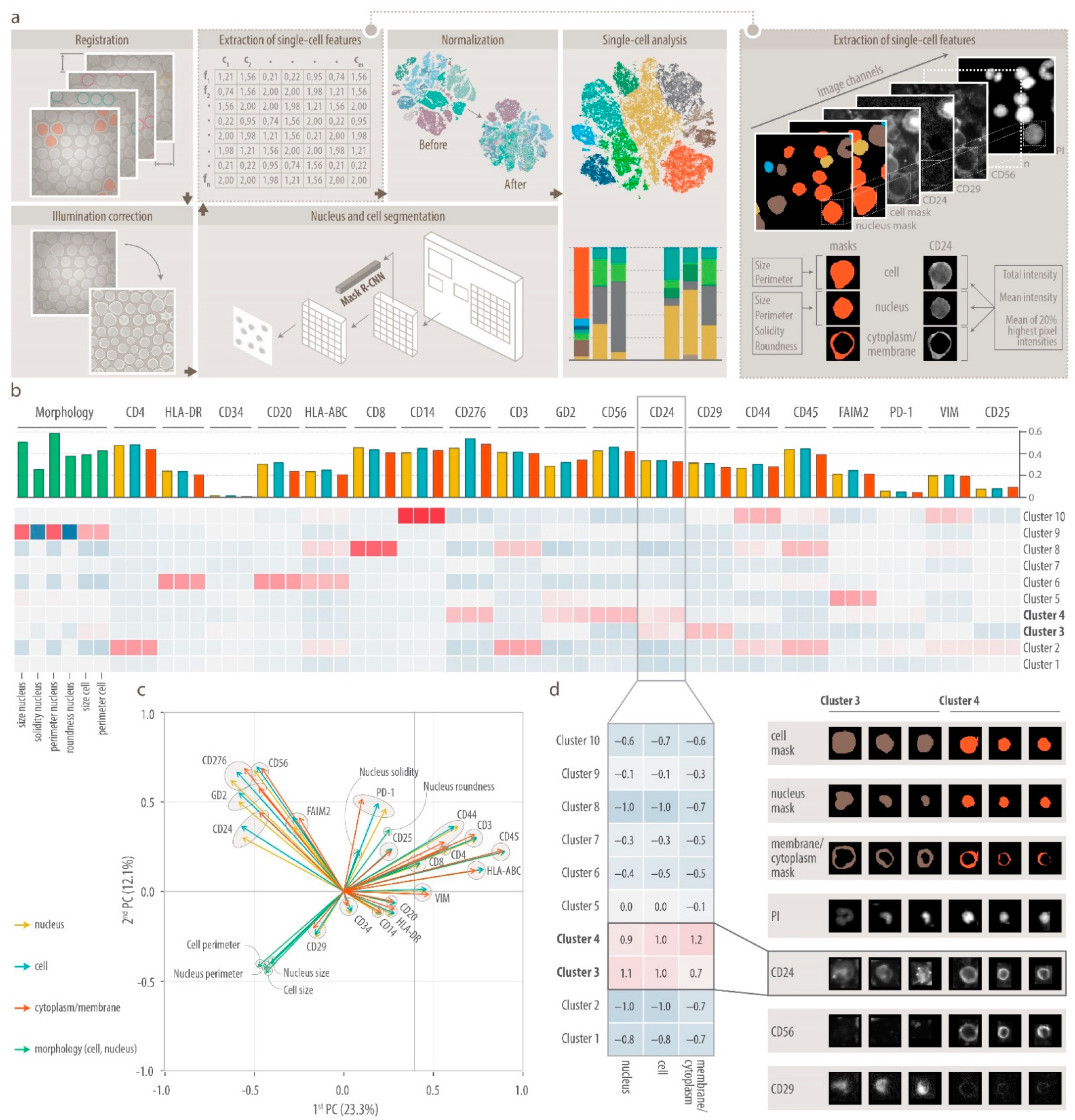
2.6. DeepFLEX
2.6.1. Image Processing
2.6.2. Segmentation
2.6.3. Feature Extraction
2.6.4. Normalization
2.6.5. Feature Validation
2.6.6. Single-Cell Analysis
2.7. RNA-Sequencing Analysis
3. Results
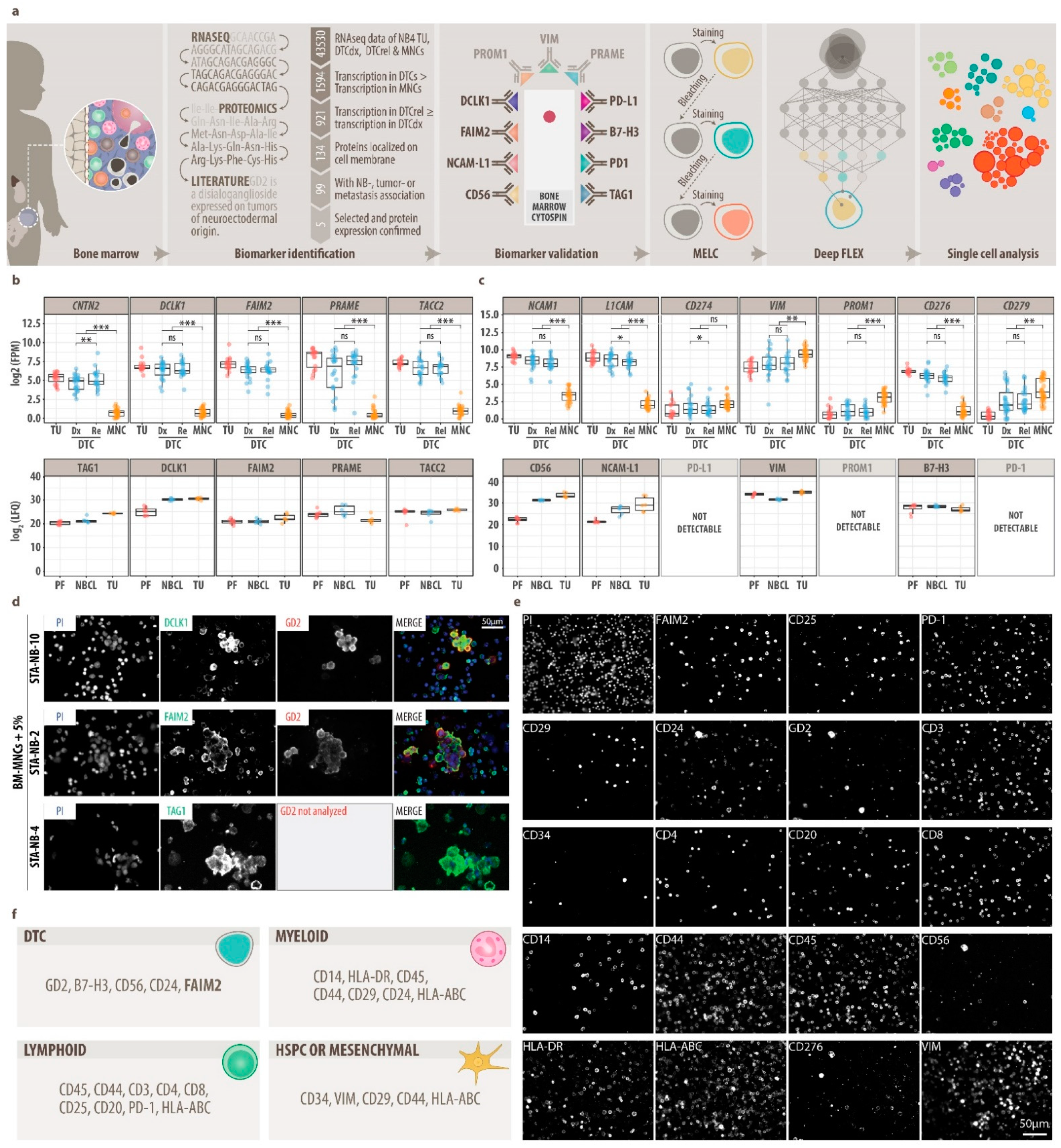
3.1. Comprehensive Single-Cell Multiplex Immunofluorescence Imaging Panel
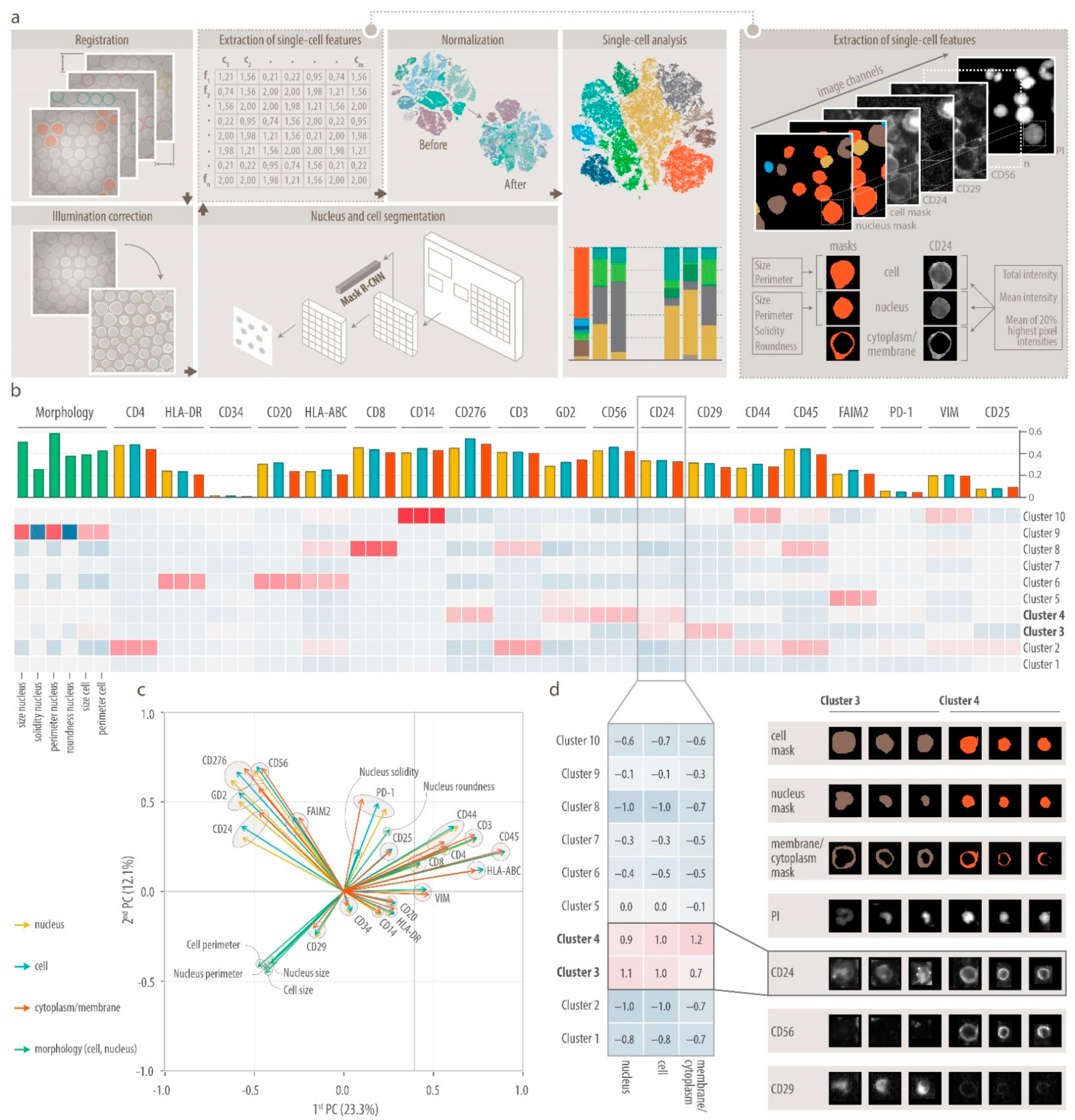
3.2. DeepFLEX Extracts Morphological Features and Protein Localization That Contribute to Cell Classification
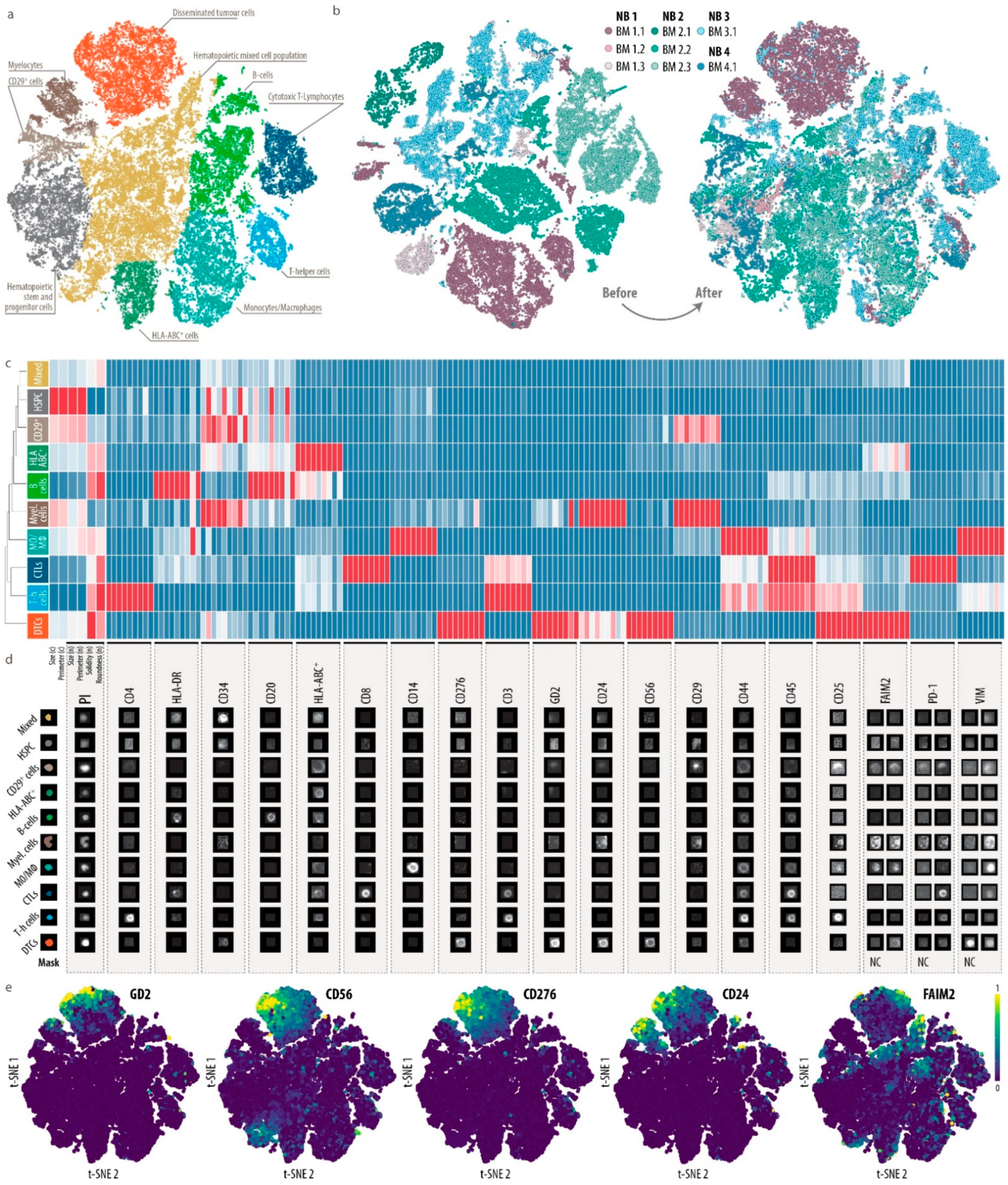
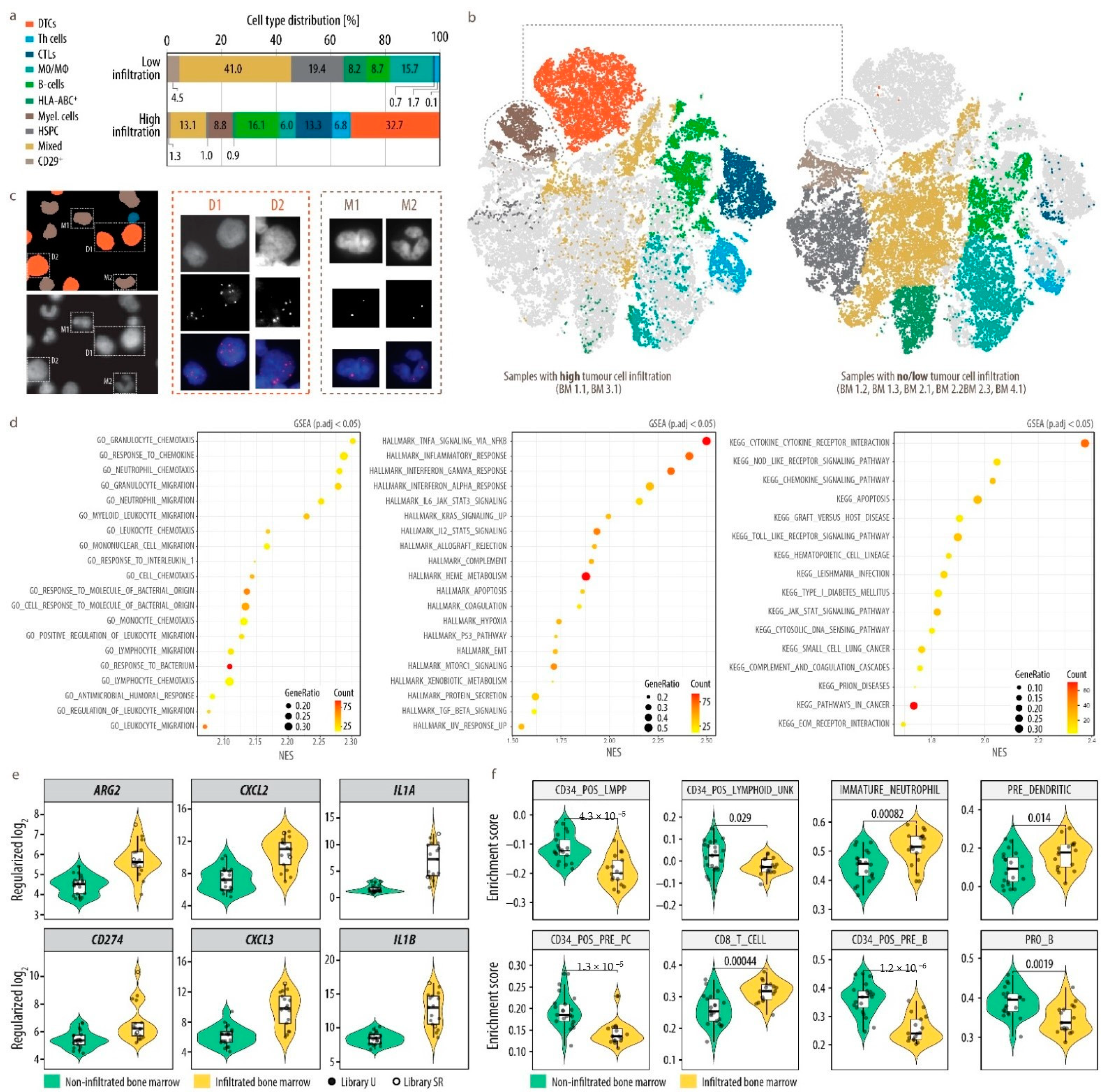
3.3. Single-Cell Map of Tumor Cells and the Microenvironment in Neuroblastoma Bone Marrow Metastases
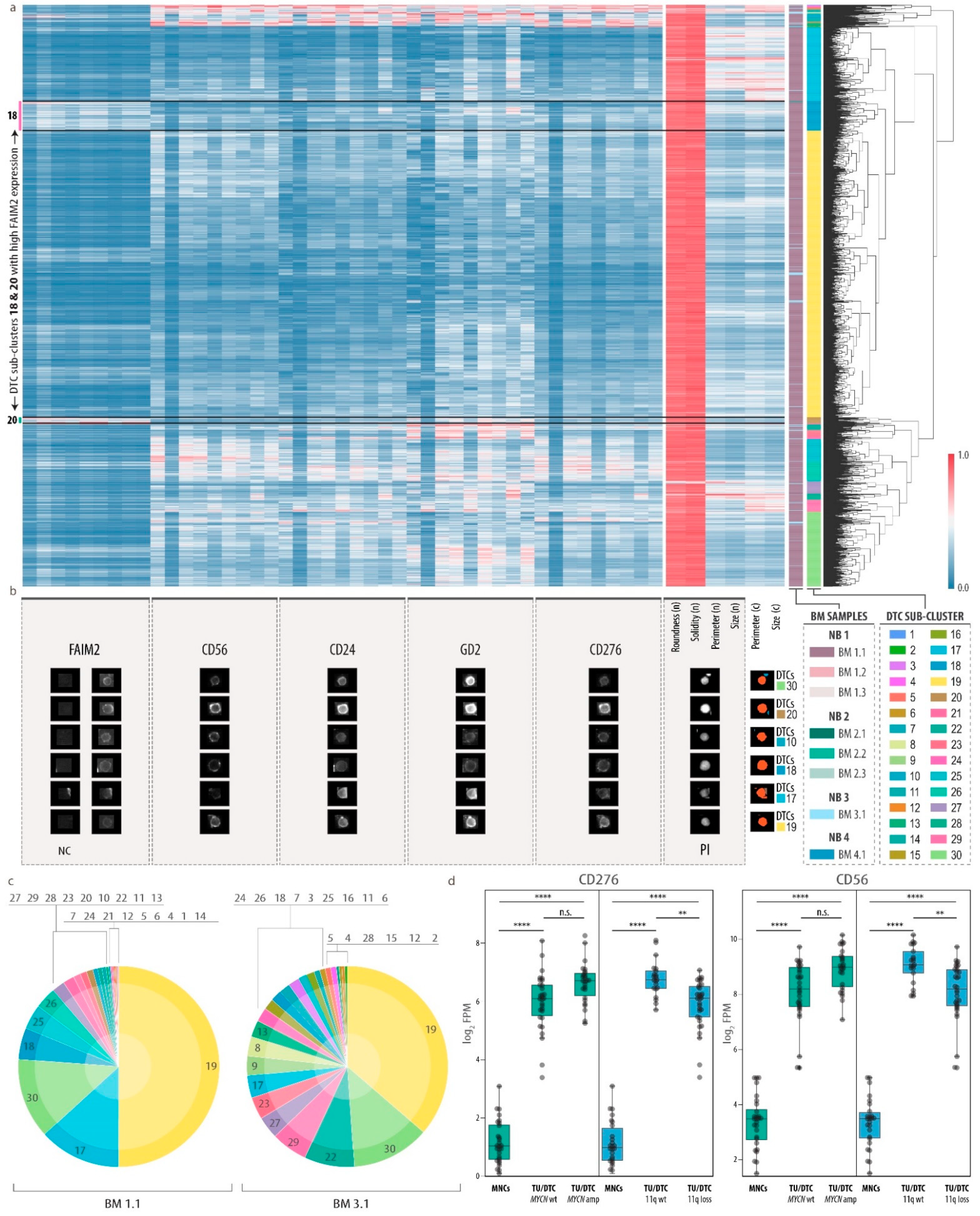
3.4. Heterogeneity of Disseminated Tumor Cells and FAIM2 as a Novel Complementary Marker
3.5. Analysis of Bone Marrow Microenvironmental Changes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W.; Amin, R.; Deasy, S.; Ha, N.H.; Wakefield, L. Genetic insights into the morass of metastatic heterogeneity. Nat. Rev. Cancer 2018, 18, 211–223. [Google Scholar] [CrossRef]
- Abbasi, M.R.; Rifatbegovic, F.; Brunner, C.; Mann, G.; Ziegler, A.; Pötschger, U.; Crazzolara, R.; Ussowicz, M.; Benesch, M.; Ebetsberger-Dachs, G.; et al. Impact of disseminated neuroblastoma cells on the identification of the relapse-seeding clone. Clin. Cancer Res. 2017, 23, 4224–4232. [Google Scholar] [CrossRef] [PubMed]
- Bogen, D.; Brunner, C.; Walder, D.; Ziegler, A.; Abbasi, R.; Ladenstein, R.L.; Noguera, R.; Martinsson, T.; Amann, G.; Schilling, F.H.; et al. The genetic tumor background is an important determinant for heterogeneous MYCN-amplified neuroblastoma. Int. J. Cancer 2016, 139, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Price, T.T.; Burness, M.L.; Ayelet, S.; Warner, M.J. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra73. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Bone marrow as a reservoir for disseminated tumor cells: A special source for liquid biopsy in cancer patients. Bonekey Rep. 2014, 3, 584. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Eber, M.R.; Berry, J.E.; Taichman, R.S. Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep. 2015, 4, 689. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 16078. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M.; Bagatell, R. Mechanisms of neuroblastoma regression. Nat. Rev. Clin. Oncol. 2014, 11, 704–713. [Google Scholar] [CrossRef]
- Hubertus Schulte, J.; Eggert, A. Neuroblastoma. Crit. Rev. Oncog. 2015, 20, 245–270. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O. Recent insights into the biology of neuroblastoma. Int. J. Cancer 2014, 135, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Merugu, S.; Chen, L.; Gavens, E.; Gabra, H.; Brougham, M.; Makin, G.; Ng, A.; Murphy, D.; Gabriel, A.S.; Robinson, M.L.; et al. Detection of Circulating and Disseminated Neuroblastoma Cells Using the ImageStream Flow Cytometer for Use as Predictive and Pharmacodynamic Biomarkers. Clin. Cancer Res. 2020, 26, 122–134. [Google Scholar] [CrossRef]
- Rifatbegovic, F.; Frech, C.; Abbasi, M.R.; Taschner-Mandl, S.; Weiss, T.; Schmidt, W.M.; Schmidt, I.; Ladenstein, R.; Ambros, I.M.; Ambros, P.F. Neuroblastoma cells undergo transcriptomic alterations upon dissemination into the bone marrow and subsequent tumor progression. Int. J. Cancer 2018, 142, 297–307. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- Van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Mazutis, L.; Rudensky, A.Y.; Pe, D.; Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment Resource. Cell 2018, 174, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell pathology landscape of breast cancer. Nature 2020, 578, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Keren, L.; Bosse, M.; Marquez, D.; Angoshtari, R.; Jain, S.; Varma, S.; Yang, S.R.; Kurian, A.; Van Valen, D.; West, R.; et al. A Structured Tumor-Immune Microenvironment in Triple Negative Breast Cancer Revealed by Multiplexed Ion Beam Imaging. Cell 2018, 174, 1373–1387. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.D.; Ye, C.J.; Yao, X.Y.; Luo, W.Q.; Cheng, X.M.; Miao, J.J.; Wang, J.F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733. [Google Scholar] [CrossRef]
- Jansky, S.; Sharma, A.K.; Körber, V.; Quintero, A.; Toprak, U.H.; Wecht, E.M.; Gartlgruber, M.; Greco, A.; Chomsky, E.; Grünewald, T.G.P.; et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat. Genet. 2021, 53, 683–693. [Google Scholar] [CrossRef]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Sci. Neurodev. 2017, 357, eeal3753. [Google Scholar] [CrossRef]
- Kildisiute, G.; Kholosy, W.M.; Young, M.D.; Roberts, K.; Elmentaite, R.; van Hooff, S.R.; Khabirova, E.; Piapi, A.; Thevanesan, C.; Blanco, E.B.; et al. Tumor to normal single cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci. Adv. 2021, 7, eabd3311. [Google Scholar] [CrossRef]
- Soldatov, R.; Kaucka, M.; Kastriti, E.M. Spatiotemporal structure of cell fate decisions in murine neural crest. Sci. Neurodev. 2019, 364, eaas9536. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932. [Google Scholar] [CrossRef]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Bodenmiller, B. Multiplexed Epitope-Based Tissue Imaging for Discovery and Healthcare Applications. Cell Syst. 2016, 2, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Méhes, G.; Luegmayr, A.; Ambros, I.M.; Ladenstein, R.; Ambros, P.F. Combined automatic immunological and molecular cytogenetic analysis allows exact identification and quantification of tumor cells in the bone marrow. Clin. Cancer Res. 2001, 7, 1969–1975. [Google Scholar] [PubMed]
- Méhes, G.; Luegmayr, A.; Kornmüller, R.; Ambros, I.M.; Ladenstein, R.; Gadner, H.; Ambros, P.F. Detection of disseminated tumor cells in neuroblastoma: 3 log improvement in sensitivity by automatic immunofluorescence plus FISH (AIPF) analysis compared with classical bone marrow cytology. Am. J. Pathol. 2003, 163, 393–399. [Google Scholar] [CrossRef]
- Ostalecki, C.; Lee, J.H.; Dindorf, J.; Collenburg, L.; Schierer, S.; Simon, B.; Schliep, S.; Kremmer, E.; Schuler, G.; Baur, A.S. Multiepitope tissue analysis reveals SPPL3-mediated ADAM10 activation as a key step in the transformation of melanocytes. Sci. Signal. 2017, 10, eaai8288. [Google Scholar] [CrossRef]
- Schubert, W.; Bonnekoh, B.; Pommer, A.J.; Philipsen, L.; Böckelmann, R.; Malykh, Y.; Gollnick, H.; Friedenberger, M.; Bode, M.; Dress, A.W.M. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006, 24, 1270–1278. [Google Scholar] [CrossRef]
- Ambros, I.M.; Rumpler, S.; Luegmayr, A.; Hattinger, C.M.; Strehl, S.; Kovar, H.; Gadner, H.; Ambros, P.F. Neuroblastoma cells can actively eliminate supernumerary MYCN gene copies by micronucleus formation - Sign of tumour cell revertance? Eur. J. Cancer 1997, 33, 2043–2049. [Google Scholar] [CrossRef]
- Abbasi, M.R.; Rifatbegovic, F.; Brunner, C.; Ladenstein, R.; Ambros, I.M.; Ambros, P.F. Bone marrows from neuroblastoma patients: An excellent source for tumor genome analyses. Mol. Oncol. 2015, 9, 545–554. [Google Scholar] [CrossRef]
- Guizar-Sicairos, M.; Thurman, S.T.; Fienup, J.R. Efficient subpixel image registration algorithms. Opt. Lett. 2008, 33, 156–158. [Google Scholar] [CrossRef]
- Smith, K.; Li, Y.; Piccinini, F.; Csucs, G.; Balazs, C.; Bevilacqua, A.; Horvath, P. CIDRE: An illumination-correction method for optical microscopy. Nat. Methods 2015, 12, 404–406. [Google Scholar] [CrossRef]
- Kromp, F.; Bozsaky, E.; Rifatbegovic, F.; Fischer, L.; Ambros, M.; Berneder, M.; Weiss, T.; Lazic, D.; Dörr, W.; Hanbury, A.; et al. An annotated fluorescence image dataset for training nuclear segmentation methods. Nat. Sci. Data 2020, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Kromp, F.; Fischer, L.; Bozsaky, E.; Ambros, I.M.; Dorr, W.; Beiske, K.; Ambros, P.F.; Hanbury, A.; Taschner-Mandl, S. Evaluation of Deep Learning architectures for complex immunofluorescence nuclear image segmentation. IEEE Trans. Med. Imaging 2021, 40, 1934–1949. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Chin, K.; Thibault, G.; Eng, J.; Burlingame, E.; Gray, J.W. RESTORE: Robust intEnSiTy nORmalization mEthod for multiplexed imaging. Commun. Biol. 2020, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Schlesner, M.; Hübschmann, D. cola: An R/Bioconductor package for consensus partitioning through a general framework. Nucleic Acids Res. 2021, 49, e15. [Google Scholar] [CrossRef] [PubMed]
- Josse, J.; Husson, F. {FactoMineR}: A Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Höllt, T.; Pezzotti, N.; van Unen, V.; Koning, F.; Eisemann, E.; Lelieveldt, B.; Vilanova, A. Cytosplore: Interactive Immune Cell Phenotyping for Large Single-Cell Datasets. Comput. Graph. Forum 2016, 35, 171–180. [Google Scholar] [CrossRef]
- Pezzotti, N.; Höllt, T.; Lelieveldt, B.; Eisemann, E.; Vilanova, A. Hierarchical Stochastic Neighbor Embedding. Comput. Graph. Forum 2016, 35, 21–30. [Google Scholar] [CrossRef]
- Comaniciu, D.; Meer, P. Mean shift: A robust approach toward feature space analysis. IEEE Trans. Pattern Anal. Mach. Intell. 2002, 24, 603–619. [Google Scholar] [CrossRef]
- Waskom, M.L. seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2016. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 21 July 2021).
- Krueger, F. Trim Galore: A Wrapper Tool around Gutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. Available online: https://www.bioinformatics.babraham.ac.uk/projects (accessed on 21 July 2021).
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020; Available online: https://github.com/igordot/msigdbr (accessed on 21 July 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the {tidyverse}. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Schadt, E.E. variancePartition: Interpreting drivers of variation in complex gene expression studies. BMC Bioinform. 2016, 17, 483. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omi. A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V. Fast gene set enrichment analysis. BioRxiv 2016, 060012. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Dolgalev, I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format 2020. Available online: https://CRAN.R-project.org/package=msigdbr (accessed on 21 July 2021).
- Hänzelmann, S.; Castelo, R.; Guinney, J. {GSVA}: Gene set variation analysis for microarray and {RNA-Seq} data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Hay, S.B.; Ferchen, K.; Chetal, K.; Grimes, L.H.; Salomonis, N. The human cell atlas bone marrow single-cell interactive web portal. Exp. Hematol. 2018, 68, 51–61. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. {limma} powers differential expression analyses for {RNA}-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Castriconi, R.; Dondero, A.; Augugliaro, R.; Cantoni, C.; Carnemolla, B.; Sementa, A.R.; Negri, F.; Conte, R.; Corrias, M.V.; Moretta, L.; et al. Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc. Natl. Acad. Sci. USA 2004, 101, 12640–12645. [Google Scholar] [CrossRef]
- Künkele, A.; Taraseviciute, A.; Finn, L.S.; Johnson, A.J.; Berger, C.; Finney, O.; Chang, C.A.; Rolczynski, L.S.; Brown, C.; Mgebroff, S.; et al. Preclinical assessment of CD171-directed CAR T-cell adoptive therapy for childhood neuroblastoma: CE7 epitope target safety and product manufacturing feasibility. Clin. Cancer Res. 2017, 23, 466–477. [Google Scholar] [CrossRef]
- Siebert, N.; Zumpe, M.; Jüttner, M.; Troschke-Meurer, S.; Lode, H.N. PD-1 blockade augments anti-neuroblastoma immune response induced by anti-GD2 antibody ch14.18/CHO. Oncoimmunology 2017, 6, e1343775. [Google Scholar] [CrossRef]
- Beiske, K.; Ambros, P.F.; Burchill, S.A.; Cheung, I.Y.; Swerts, K. Detecting minimal residual disease in neuroblastoma patients-the present state of the art. Cancer Lett. 2005, 228, 229–240. [Google Scholar] [CrossRef]
- Oetjen, K.A.; Lindblad, K.E.; Goswami, M.; Gui, G.; Dagur, P.K.; Lai, C.; Dillon, L.W.; McCoy, J.P.; Hourigan, C.S. Human bone marrow assessment by single-cell RNA sequencing, mass cytometry, and flow cytometry. JCI Insight 2018, 3, e124928. [Google Scholar] [CrossRef]
- Zhao, E.; Xu, H.; Wang, L.; Kryczek, I.; Wu, K.; Hu, Y.; Wang, G.; Zou, W. Bone marrow and the control of immunity. Cell. Mol. Immunol. 2012, 9, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Berthold, M.; Cebron, N.; Dill, F.; Gabriel, T.; Kötter, T.; Meinl, T. KNIME: The Konstanz Information Miner. Data Analysis, Machine Learning and Applications. Studies in Classification, Data Analysis, and Knowledge Organization; Springer: Berlin/Heidelberg, Germany, 2007; pp. 319–326. [Google Scholar]
- Carpenter, A.; Jones, T.; Lamprecht, M.; Clarke, C.; Kang, I.; Friman, O. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed]
- Czech, E.; Aksoy, B.A.; Aksoy, P.; Hammerbacher, J. Cytokit: A single-cell analysis toolkit for high dimensional fluorescent microscopy imaging. BMC Bioinform. 2019, 20, 448. [Google Scholar] [CrossRef] [PubMed]
- Holzwarth, K.; Köhler, R.; Philipsen, L.; Tokoyoda, K. Multiplexed fluorescence microscopy reveals heterogeneity among stromal cells in mouse bone marrow sections. Cytom. Part A 2018, 93, 876–888. [Google Scholar] [CrossRef]
- Rueden, C.; Schindelin, J.; Hiner, M.; DeZonia, B.; Walter, A.; Arena, E. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Schapiro, D.; Jackson, H.W.; Raghuraman, S.; Fischer, J.R.; Zanotelli, V.R.T.; Schulz, D.; Giesen, C.; Catena, R.; Varga, Z.; Bodenmiller, B. HistoCAT: Analysis of cell phenotypes and interactions in multiplex image cytometry data. Nat. Methods 2017, 14, 873–876. [Google Scholar] [CrossRef]
- Elghetany, M.T.; Patel, J. Assessment of CD24 expression on bone marrow neutrophilic granulocytes: CD24 is a marker for the myelocytic stage of development. Am. J. Hematol. 2002, 71, 348–349. [Google Scholar] [CrossRef]
- Fernández, M.; Segura, M.F.; Solé, C.; Colino, A.; Comella, J.X.; Ceña, V. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J. Neurochem. 2007, 103, 190–203. [Google Scholar] [CrossRef]
- Somia, N.V.; Schmitt, M.J.; Vetter, D.E.; Van Antwerp, D.; Heinemann, S.F.; Verma, I.M. LFG: An anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc. Natl. Acad. Sci. USA 2002, 96, 12667–12672. [Google Scholar] [CrossRef]
- Kang, H.C.; Kim, J.I.; Chang, H.K.; Woodard, G.; Choi, Y.S.; Ku, J.L.; Jablons, D.M.; Kim, I.J. FAIM2, as a novel diagnostic maker and a potential therapeutic target for small-cell lung cancer and atypical carcinoid. Sci. Rep. 2016, 6, 34022. [Google Scholar] [CrossRef]
- Bucan, V.; Reimers, K.; Choi, C.Y.; Eddy, M.T.; Vogt, P.M. The anti-apoptotic protein lifeguard is expressed in breast cancer cells and tissues. Cell. Mol. Biol. Lett. 2010, 15, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, B.; Taylor, V.; Welcher, A.; McClelland, M.; Suter, U. Neural membrane protein 35 (NMP35): A novel member of a gene family which is highly expressed in the adult nervous system. Mol Cell Neurosci. 1998, 11, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Planells-Ferrer, L.; Urresti, J.; Soriano, A.; Reix, S.; Murphy, D.M.; Ferreres, J.C.; Borràs, F.; Gallego, S.; Stallings, R.L.; Moubarak, R.S.; et al. MYCN repression of Lifeguard/FAIM2 enhances neuroblastoma aggressiveness. Cell Death Dis. 2014, 5, e1401. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Havens, A.M.; Pienta, K.J.; Taichman, R.S. The bone marrow niche: Habitat to hematopoietic and mesenchymal stem cells, and unwitting host to molecular parasites. Leukemia 2008, 22, 941–950. [Google Scholar] [CrossRef]
- Brück, O.; Blom, S.; Dufva, O.; Turkki, R.; Chheda, H.; Ribeiro, A.; Kovanen, P.; Aittokallio, T.; Koskenvesa, P.; Kallioniemi, O.; et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia 2018, 32, 1643–1656. [Google Scholar] [CrossRef]
- Van Galen, P.; Hovestadt, V.; Wadsworth, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281. [Google Scholar] [CrossRef]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef]
- Dhawan, A.; Friedrichs, J.; Bray, L.; Hofbauer, L.C.; Wobus, M.; Bornhäuser, M. Interaction of Tumor Cells with the Hematopoietic Stem and Progenitor Cell Niche. Blood 2014, 124, 5139. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Zhao, Y.; Rahmy, S.; Liu, Z.; Zhang, C.; Lu, X. Rational targeting of immunosuppressive neutrophils in cancer. Pharmacol. Ther. 2020, 212, 107556. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.B.G.; Coffelt, S.B.; Carlin, L.M. Neutrophil maturity in cancer. Front. Immunol. 2019, 10, 1912. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345. [Google Scholar] [CrossRef] [PubMed]
- Berbegall, A.P.; Villamón, E.; Piqueras, M.; Tadeo, I.; Djos, A.; Ambros, P.F.; Martinsson, T.; Ambros, I.M.; Cañete, A.; Castel, V.; et al. Comparative genetic study of intratumoral heterogenous MYCN amplified neuroblastoma versus aggressive genetic profile neuroblastic tumors. Oncogene 2016, 35, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Taschner-Mandl, S.; Janker, L.; Bileck, A.; Rifatbegovic, F.; Kromp, F.; Sorger, H.; Kauer, M.O.; Frech, C.; Windhager, R.; et al. Schwann cell plasticity regulates neuroblastic tumor cell differentiation via epidermal growth factor-like protein 8. Nat. Commun. 2021, 12, 1624. [Google Scholar] [CrossRef]
- Taschner-Mandl, S.; Schwarz, M.; Blaha, J.; Kauer, M.; Kromp, F.; Frank, N.; Rifatbegovic, F.; Weiss, T.; Ladenstein, R.; Hohenegger, M.; et al. Metronomic topotecan impedes tumor growth of MYCNamplified neuroblastoma cells in vitro and in vivo by therapy induced senescence. Oncotarget 2016, 7, 3571–3586. [Google Scholar] [CrossRef]
- Sidell, N.; Altman, A.; Haussler, M.R.; Seeger, R.C. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Exp. Cell Res. 1983, 148, 21–30. [Google Scholar] [CrossRef]
- Nakagawara, A.; Brodeur, G.M. Role of neurotrophins and their receptors in human neuroblastomas: A primary culture study. Eur. J. Cancer 1997, 33, 2050–2053. [Google Scholar] [CrossRef]
- Salzer, B.; Schueller, C.M.; Zajc, C.U.; Peters, T.; Schoeber, M.A.; Kovacic, B.; Buri, M.C.; Lobner, E.; Dushek, O.; Huppa, J.B.; et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Narath, R.; Ambros, I.; Kowalska, A.; Bozsaky, E.; Boukamp, P.; Ambros, P. Induction of senescence in MYCN amplified neuroblastoma cell lines by hydroxyurea. Genes Chromosom. Cancer 2007, 46, 130–142. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Antibody | Conjugate | Class|Host|Isotype | Clone | Supplier | Catalogue-Number | Optimal Dilution |
|---|---|---|---|---|---|---|---|
| Sw α Rb | FITC | polyclonal swine IgG | polyclonal | Dako | F0205 | 1:50 | |
| 1 | FAIM2 | unconj. | polyclonal rabbit IgG | polyclonal | ThermoFisher | PA5-20311 | 1:50 |
| Sw α Rb | FITC | polyclonal swine IgG | polyclonal | Dako | F0205 | 1:50 | |
| 2 | CD25 | PE | monoclonal mouse IgG | HI25a | ImmunoTools | 21810254 | 1:20 |
| Ms α Biot. | Cy3 | monoclonal mouse IgG | 3D6.6 | Jackson ImmunoResearch | 200-162-211 | 1:800 | |
| 3 | PD-1 | biotinylated | monoclonal mouse IgG1 | NAT105 | BioLegend | 367418 | 1:50 |
| Ms α Biot. | Cy3 | monoclonal mouse IgG | 3D6.6 | Jackson ImmunoResearch | 200-162-211 | 1:800 | |
| 4 | CD29 | FITC | monoclonal mouse IgG1 | HI29a | ImmunoTools | 21810293 | 1:20 |
| 5 | CD24 | FITC | monoclonal mouse IgG1 | SN3 | ImmunoTools | 21270243 | 1:20 |
| 6 | GD2 | FITC | monoclonal chinese hamster/humanized | ch14.18/deltaCHO | Tübingen | n.r. | 1:100 |
| 7 | CD3 | PE | monoclonal mouse IgG1 | UCHT1 | ImmunoTools | 21620034 | 1:20 |
| 8 | CD34 | PE | monoclonal mouse IgG1 | 4H11[APG] | ImmunoTools | 21270344 | 1:20 |
| 9 | CD4 | PE | monoclonal mouse IgG2a,k | VIT4 | Miltenyi Biotec | 130-113-214 | 1:20 |
| 10 | CD20 | PE | recombinant human IgG1 | REA780 | Miltenyi Biotec | 130-111-338 | 1:20 |
| 11 | CD8 | PE | monoclonal mouse IgG1 | HIT8a | ImmunoTools | 21810084 | 1:20 |
| 12 | CD14 | PE | monoclonal mouse IgG1 | 18D11 | ImmunoTools | 21620144 | 1:20 |
| 13 | CD44 | PE | monoclonal rat IgG2b | IM7 | ImmunoTools | 21850444 | 1:20 |
| 14 | CD45 | PE | monoclonal mouse IgG1 | HI30 | ImmunoTools | 21810454 | 1:20 |
| 15 | CD56 | PE | monoclonal mouse IgG1 | B-A19 | ImmunoTools | 21810564S | 1:20 |
| 16 | HLA-DR | PE | monoclonal mouse IgG1 | HI43 | ImmunoTools | 21819984 | 1:20 |
| 17 | HLA-ABC | PE | monoclonal mouse IgG2a | W6/32 | ImmunoTools | 21159034 | 1:20 |
| 18 | B7-H3 | PE | human IgG1 | REA1094 | Miltenyi Biotec | 130-118-570 | 1:40 |
| Gt α Ch | FITC | polyclonal goat IgG | polyclonal | ThermoFisher | A16055 | 1:500 | |
| 19 | Vimentin | unconj. | recombinant chicken IgY | polyclonal | Milipore/Chemicon | AB5733 | 1:100 |
| Gt α Ch | FITC | polyclonal goat IgG | polyclonal | ThermoFisher | A16055 | 1:500 | |
| 20 | Propidium Iodide | PI | n.r. | n.r. | Genaxxon bioscience | M3181.0010 | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazic, D.; Kromp, F.; Rifatbegovic, F.; Repiscak, P.; Kirr, M.; Mivalt, F.; Halbritter, F.; Bernkopf, M.; Bileck, A.; Ussowicz, M.; et al. Landscape of Bone Marrow Metastasis in Human Neuroblastoma Unraveled by Transcriptomics and Deep Multiplex Imaging. Cancers 2021, 13, 4311. https://doi.org/10.3390/cancers13174311
Lazic D, Kromp F, Rifatbegovic F, Repiscak P, Kirr M, Mivalt F, Halbritter F, Bernkopf M, Bileck A, Ussowicz M, et al. Landscape of Bone Marrow Metastasis in Human Neuroblastoma Unraveled by Transcriptomics and Deep Multiplex Imaging. Cancers. 2021; 13(17):4311. https://doi.org/10.3390/cancers13174311
Chicago/Turabian StyleLazic, Daria, Florian Kromp, Fikret Rifatbegovic, Peter Repiscak, Michael Kirr, Filip Mivalt, Florian Halbritter, Marie Bernkopf, Andrea Bileck, Marek Ussowicz, and et al. 2021. "Landscape of Bone Marrow Metastasis in Human Neuroblastoma Unraveled by Transcriptomics and Deep Multiplex Imaging" Cancers 13, no. 17: 4311. https://doi.org/10.3390/cancers13174311
APA StyleLazic, D., Kromp, F., Rifatbegovic, F., Repiscak, P., Kirr, M., Mivalt, F., Halbritter, F., Bernkopf, M., Bileck, A., Ussowicz, M., Ambros, I. M., Ambros, P. F., Gerner, C., Ladenstein, R., Ostalecki, C., & Taschner-Mandl, S. (2021). Landscape of Bone Marrow Metastasis in Human Neuroblastoma Unraveled by Transcriptomics and Deep Multiplex Imaging. Cancers, 13(17), 4311. https://doi.org/10.3390/cancers13174311