
The Microenvironment of Small Intestinal Neuroendocrine Tumours Contains Lymphocytes Capable of Recognition and Activation after Expansion

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
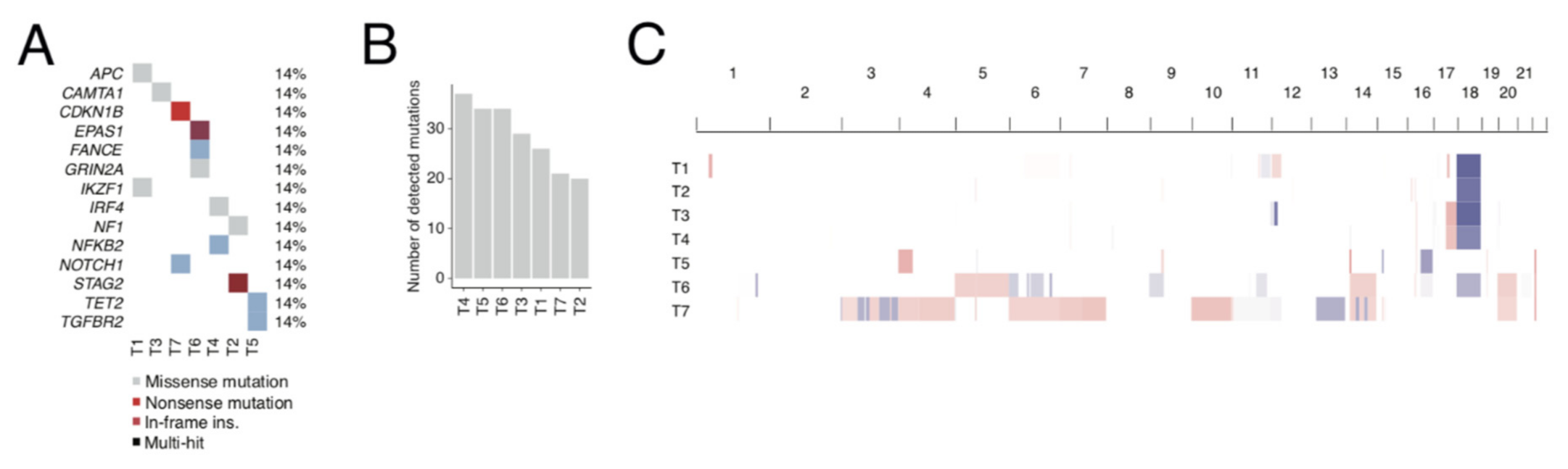
2.1. Exome and RNA Sequencing of Metastatic SINET Reveals Insight into Genetics and Immunology
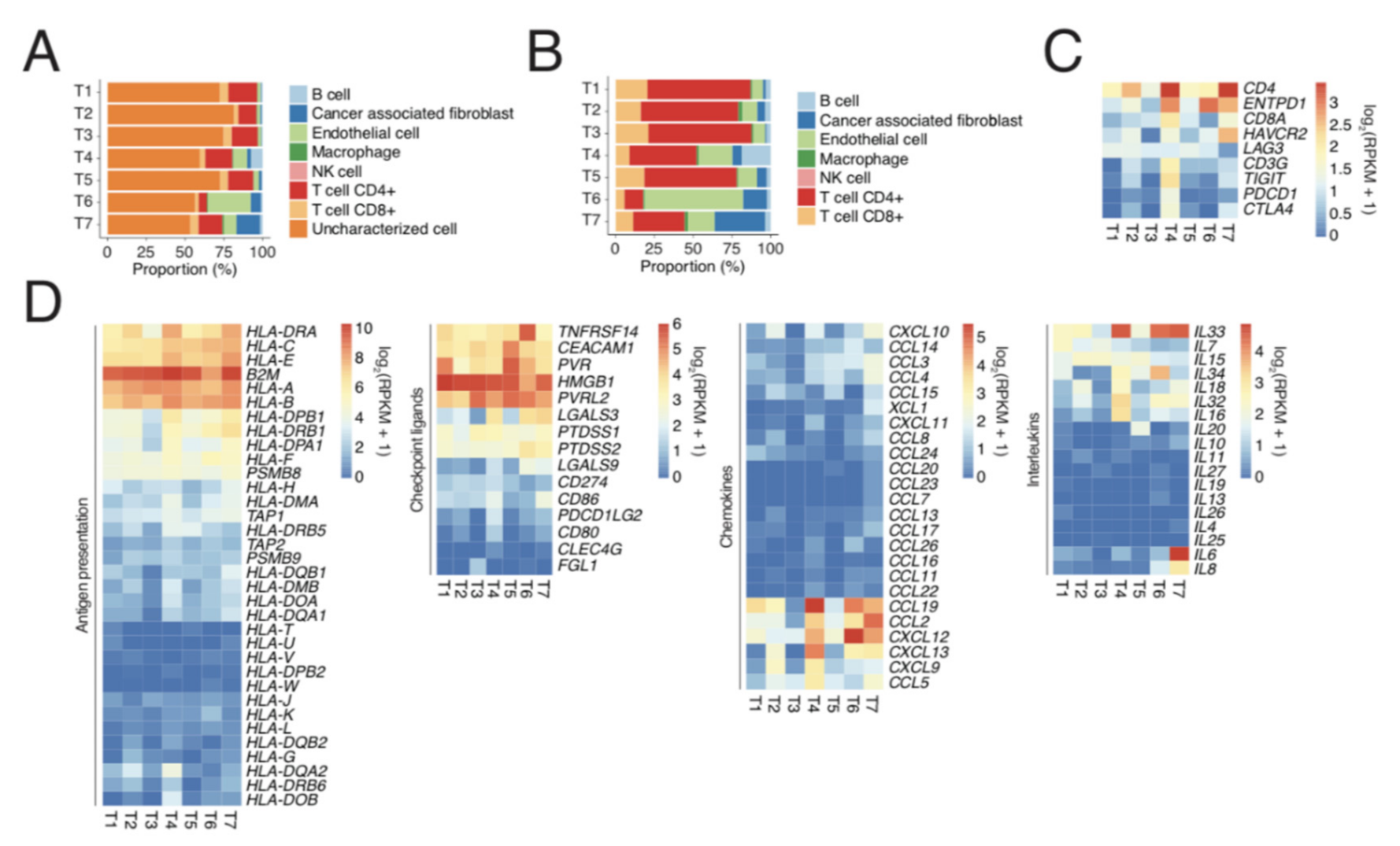
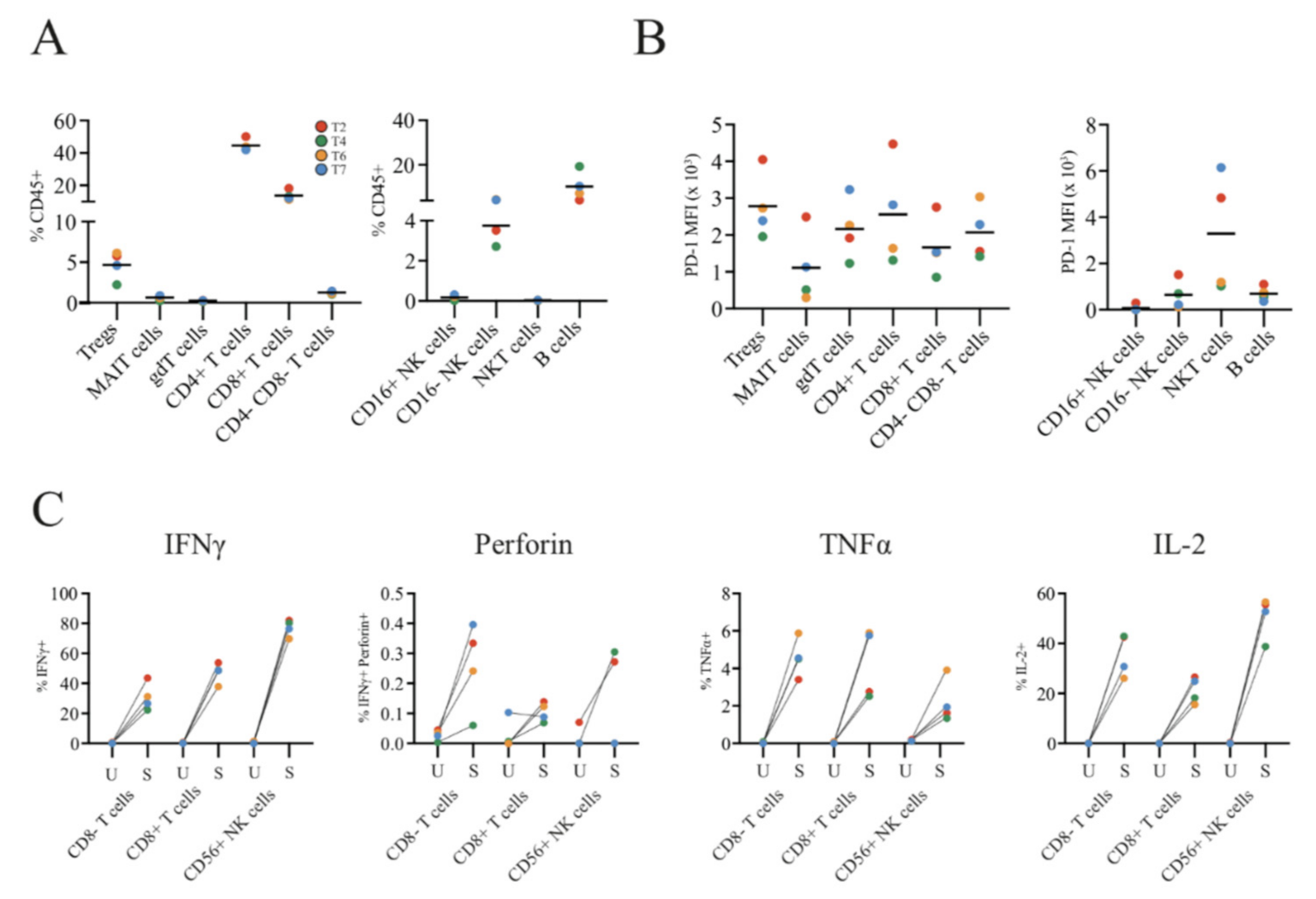
2.2. Single-Cell Analyses by IHC and Flow Cytometry of Tumours and TILs Show Immune Heterogeneity
2.3. SINET Cells Can Survive in Long-Term Tumour Grafts but Are Resistant to T Cell Killing In Vivo
3. Material and Methods
3.1. Whole-Exome Sequencing
3.2. Immunohistochemistry
3.3. Generation of Tumour-Infiltrating Immune Cells
3.4. Characterisation and Stimulation of Tumour-Infiltrating Immune Cells
3.5. Degranulation Assay
3.6. Patient-Derived Xenografts
3.7. Preprocessing of RNA-Seq Data
3.8. Immune Cell Deconvolution
3.9. Preprocessing of Exome Sequencing Data
3.10. Mutation Calling
3.11. Copy Number Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.M.; Kiezun, A.; Ramos, A.H.; Serra, S.; Pedamallu, C.S.; Qian, Z.R.; Banck, M.S.; Kanwar, R.; Kulkarni, A.A.; Karpathakis, A.; et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat. Genet. 2013, 45, 1483–1486. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214. [Google Scholar] [CrossRef]
- Busse, A.; Mochmann, L.H.; Spenke, C.; Arsenic, R.; Briest, F.; Jöhrens, K.; Lammert, H.; Sipos, B.; Kühl, A.A.; Wirtz, R.; et al. Immunoprofiling in Neuroendocrine Neoplasms Unveil Immunosuppressive Microenvironment. Cancers 2020, 12, 3448. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.; Caldas, C.; Clarke, R.; De Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [Green Version]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef]
- Williams, J.A. Using PDX for Preclinical Cancer Drug Discovery: The Evolving Field. J. Clin. Med. 2018, 7, 41. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.; Kim, K.; Kim, S.H.; Chung, Y.J.; Lee, C. Studying cancer immunotherapy using patient-derived xenografts (PDXs) in humanized mice. Exp. Mol. Med. 2018, 50, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wege, A.K.; Ernst, W.; Eckl, J.; Frankenberger, B.; Vollmann-Zwerenz, A.; Männel, D.N.; Ortmann, O.; Kroemer, A.; Brockhoff, G. Humanized tumor mice--a new model to study and manipulate the immune response in advanced cancer therapy. Int. J. Cancer 2011, 129, 2194–2206. [Google Scholar] [CrossRef]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Söderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8, 707. [Google Scholar] [CrossRef]
- Kolby, L.; Bernhardt, P.; Ahlman, H.; Wängberg, B.; Johanson, V.; Wigander, A.; Forssell-Aronsson, E.; Karlsson, S.; Ahrén, B.; Stenman, G. A transplantable human carcinoid as model for somatostatin receptor-mediated and amine transporter-mediated radionuclide uptake. Am. J. Pathol. 2001, 158, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Hofving, T.; Karlsson, J.; Nilsson, O.; Nilsson, J.A. H-STS, L-STS and KRJ-I are not authentic GEPNET cell lines. Nat. Genet. 2019, 51, 1426–1427. [Google Scholar] [CrossRef]
- Andersson, E.; Swärd, C.; Stenman, G.; Ahlman, H.; Nilsson, O. High-resolution genomic profiling reveals gain of chromosome 14 as a predictor of poor outcome in ileal carcinoids. Endocr. Relat. Cancer 2009, 16, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulke, M.H.; Freed, E.; Chiang, D.Y.; Philips, J.; Zahrieh, D.; Glickman, J.N.; Shivdasani, R.A. High-resolution analysis of genetic alterations in small bowel carcinoid tumors reveals areas of recurrent amplification and loss. Genes Chromosomes Cancer 2008, 47, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Karpathakis, A.; Dibra, H.; Pipinikas, C.; Feber, A.; Morris, T.; Francis, J.; Oukrif, D.; Mandair, D.; Pericleous, M.; Mohmaduvesh, M.; et al. Prognostic Impact of Novel Molecular Subtypes of Small Intestinal Neuroendocrine Tumor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Nonaka, D.; Breitwieser, W.; Ashton, G.; Barriuso, J.; McNamara, M.G.; Moghadam, S.; Rogan, J.; Mansoor, W.; Hubner, R.A.; et al. PD-L1 expression and presence of TILs in small intestinal neuroendocrine tumours. Oncotarget 2018, 9, 14922–14938. [Google Scholar] [CrossRef] [Green Version]
- da Silva, A.; Bowden, M.; Zhang, S.; Masugi, Y.; Thorner, A.R.; Herbert, Z.T.; Zhou, C.W.; Brais, L.; Chan, J.A.; Hodi, F.S.; et al. Characterization of the Neuroendocrine Tumor Immune Microenvironment. Pancreas 2018, 47, 1123–1129. [Google Scholar] [CrossRef]
- Cives, M.; Strosberg, J.; Al Diffalha, S.; Coppola, D. Analysis of the immune landscape of small bowel neuroendocrine tumors. Endocr. Relat. Cancer 2019, 26, 119–130. [Google Scholar] [CrossRef]
- Muul, L.M.; Spiess, P.J.; Director, E.P.; Rosenberg, S.A. Identification of specific cytolytic immune responses against autologous tumor in humans bearing malignant melanoma. J. Immunol. 1987, 138, 989–995. [Google Scholar]
- Carter, L.; Fouser, L.A.; Jussif, J.; Fitz, L.; Deng, B.; Wood, C.R.; Collins, M.; Honjo, T.; Freeman, G.J.; Carreno, B.M. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur. J. Immunol. 2002, 32, 634–643. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, L.; Serra, S.; Law, C.; Wei, A.; Stockley, T.; Ezzat, S.; Asa, S.L. Establishment and Characterization of a Human Neuroendocrine Tumor Xenograft. Endocr. Pathol. 2016, 27, 97–103. [Google Scholar] [CrossRef]
- Ny, L.; Rizzo, L.Y.; Belgrano, V.; Karlsson, J.; Jespersen, H.; Carstam, L.; Bagge, R.; Nilsson, L.; Nilsson, J. Supporting clinical decision making in advanced melanoma by preclinical testing in personalized immune-humanized xenograft mouse models. Ann. Oncol. 2020, 31, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.D.; Bagci, P.; Ohike, N.; Saka, B.; Seven, I.E.; Dursun, N.; Balci, S.; Gucer, H.; Jang, K.-T.; Tajiri, T.; et al. Calculation of the Ki67 index in pancreatic neuroendocrine tumors: A comparative analysis of four counting methodologies. Mod. Pathol. 2015, 28, 686–694. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Schallmach, E.; Kubi, A.; Shalmon, B.; Hardan, I.; Catane, R.; et al. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J. Immunother. 2009, 32, 415–423. [Google Scholar] [CrossRef]
- Tran, K.Q.; Zhou, J.; Durflinger, K.H.; Langhan, M.M.; Shelton, T.E.; Wunderlich, J.R.; Robbins, P.F.; Rosenberg, S.A.; Dudley, M.E. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J. Immunother. 2008, 31, 742–751. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.; Junker, N.; Ellebaek, E.; Andersen, M.H.; Straten, P.T.; Svane, I.M. Characterization and comparison of "standard" and "young" tumour-infiltrating lymphocytes for adoptive cell therapy at a Danish translational research institution. Scand. J. Immunol. 2012, 75, 157–167. [Google Scholar] [CrossRef]
- Liang, F.; Rezapour, A.; Falk, P.; Angenete, E.; Yrlid, U. Cryopreservation of Whole Tumor Biopsies from Rectal Cancer Patients Enable Phenotypic and In Vitro Functional Evaluation of Tumor-Infiltrating T Cells. Cancers 2021, 13, 2428. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Vikman, S.; Giandomenico, V.; Sommaggio, R.; Öberg, K.; Essand, M.; Tötterman, T.H. CD8+ T cells against multiple tumor-associated antigens in peripheral blood of midgut carcinoid patients. Cancer Immunol. Immunother. 2008, 57, 399–409. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.-C.S.; Rancan, C.; et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e1613. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender | Age at Surgery | Disease Stage | Dead/Alive | Tumour ID | Tumour Metastasis Site | Tumour Grade (% Ki67) |
|---|---|---|---|---|---|---|---|
| P1 | M | 79 | IIIB | AWD | T1 | LN | G1 (1.5) |
| P2 | F | 69 | IIIB | AWD | T2 | LN | G1 (0.5) |
| P3 | M | 76 | IV | AWD | T3 | Hepatic | G1 (2.6) |
| IV | AWD | T4 | LN | G2 (3.7) | |||
| P4 | M | 57 | IV | AWD | T5 | LN | G1 (0.9) |
| P5 | M | 84 | IV | AWD | T6 | LN | G1 (1.3) |
| P6 | M | 75 | IV | AWD | T7 | LN | G2 (3.3) |
| Transplantation Site | Subcutaneous | Orthotopic | |
|---|---|---|---|
| Patient Tumour Site | Lymph Node | Hepatic | Hepatic |
| From surgery | 0/15 | 1/4 | NA |
| From cryofrozen | 0/14 | 0/5 | 0/16 |
| Total | 0/29 | 1/8 | 0/16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hofving, T.; Liang, F.; Karlsson, J.; Yrlid, U.; Nilsson, J.A.; Nilsson, O.; Nilsson, L.M. The Microenvironment of Small Intestinal Neuroendocrine Tumours Contains Lymphocytes Capable of Recognition and Activation after Expansion. Cancers 2021, 13, 4305. https://doi.org/10.3390/cancers13174305
Hofving T, Liang F, Karlsson J, Yrlid U, Nilsson JA, Nilsson O, Nilsson LM. The Microenvironment of Small Intestinal Neuroendocrine Tumours Contains Lymphocytes Capable of Recognition and Activation after Expansion. Cancers. 2021; 13(17):4305. https://doi.org/10.3390/cancers13174305
Chicago/Turabian StyleHofving, Tobias, Frank Liang, Joakim Karlsson, Ulf Yrlid, Jonas A. Nilsson, Ola Nilsson, and Lisa M. Nilsson. 2021. "The Microenvironment of Small Intestinal Neuroendocrine Tumours Contains Lymphocytes Capable of Recognition and Activation after Expansion" Cancers 13, no. 17: 4305. https://doi.org/10.3390/cancers13174305
APA StyleHofving, T., Liang, F., Karlsson, J., Yrlid, U., Nilsson, J. A., Nilsson, O., & Nilsson, L. M. (2021). The Microenvironment of Small Intestinal Neuroendocrine Tumours Contains Lymphocytes Capable of Recognition and Activation after Expansion. Cancers, 13(17), 4305. https://doi.org/10.3390/cancers13174305