Assessment of Tumor Mutational Burden in Pediatric Tumors by Real-Life Whole-Exome Sequencing and In Silico Simulation of Targeted Gene Panels: How the Choice of Method Could Affect the Clinical Decision?

 ,
,
Abstract
:1. Introduction
2. Results
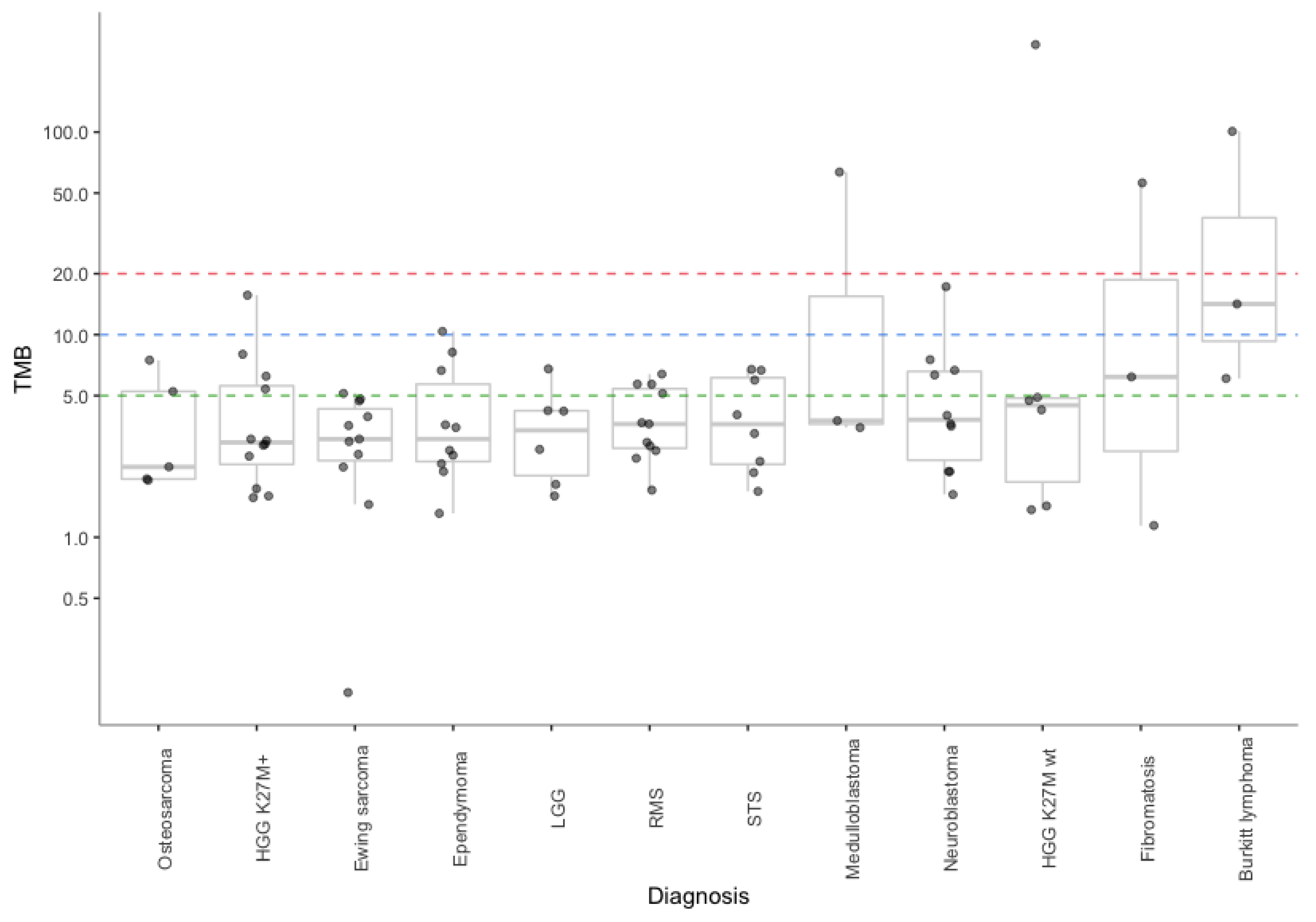
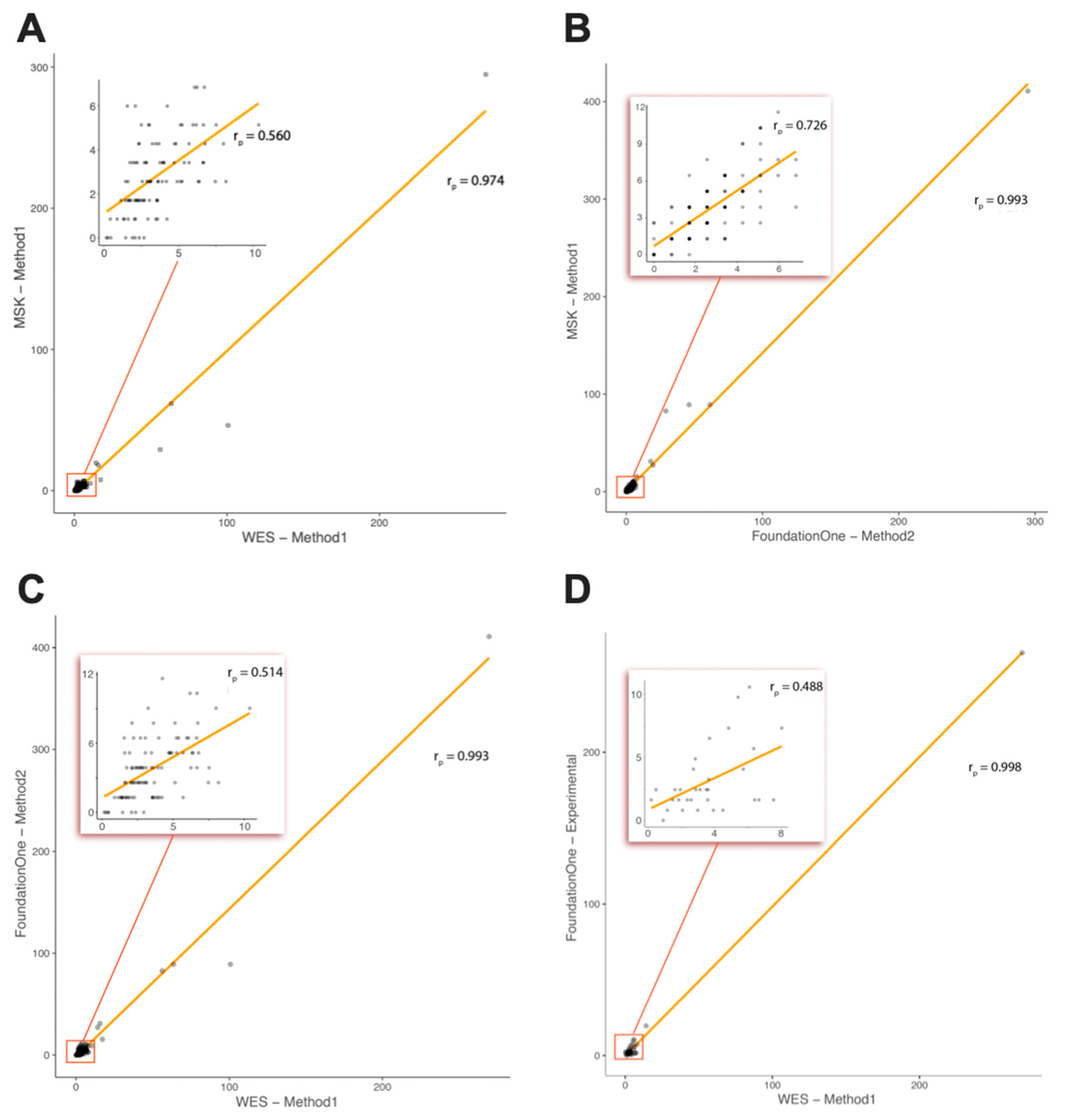
2.1. Comparison of TMB between Real-Life WES and In Silico Targeted Gene Panels
2.2. Comparison of TMB between Real-Life WES and the Foundation Medicine Inc. (FMI) Testing Service (Subcohort of Patients)
2.3. WES-Based TMB Values during Tumor Progression
2.4. Consequence of TMB Assessment Method for ICI Indication
3. Discussion
4. Materials and Methods
4.1. Patients and Biological Specimens
4.2. DNA Isolation
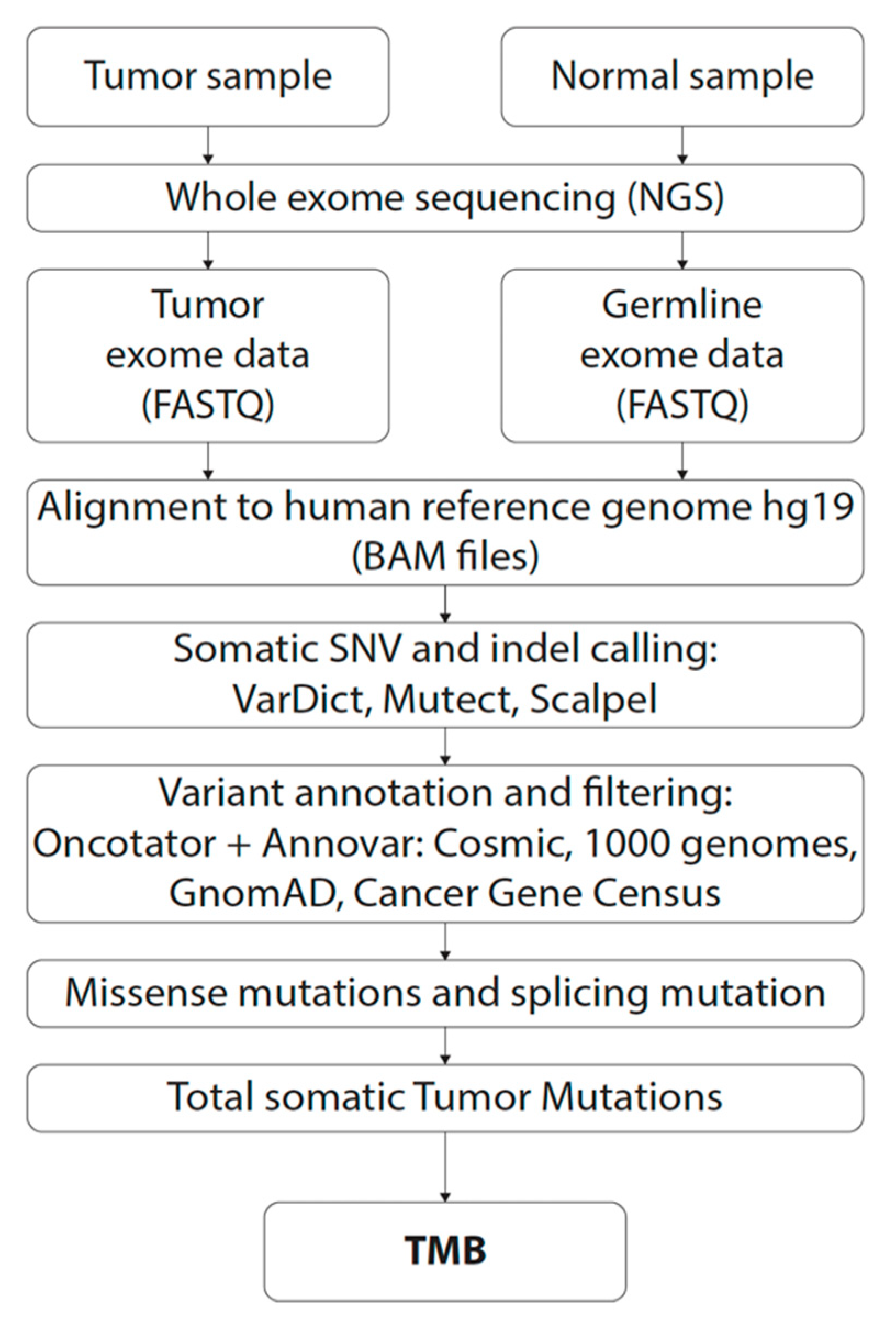
4.3. Whole Exome Sequencing
4.4. Bioinformatic Analysis
4.5. Tumor Mutational Burden Estimation
- All coding sequences (whole exome; 35 Mb; using M1 for TMB calculation);
- The coding sequences of genes analyzed by the FMI (F1CDx panel; 324 cancer-related genes; 0,8 Mbl using M2 for TMB calculation);
- The coding sequences of genes analyzed by the Memorial Sloan Kettering Cancer Center (MSK-IMPACT; 468 cancer-related genes; 1.22 Mb; using M1 for TMB calculation)
4.6. Comparative Study with the Foundation Medicine Inc. (FMI) Sequencing Service
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Sasson, A.; Srinivasan, S.; Golhar, R.; Greenawalt, D.M.; Geese, W.J.; Green, G.; Zerba, K.; Kirov, S.; Szustakowski, J. Bioinformatic Methods and Bridging of Assay Results for Reliable Tumor Mutational Burden Assessment in Non-Small-Cell Lung Cancer. Mol. Diagn. Ther. 2019, 23, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.L.; et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef]
- Buchhalter, I.; Rempel, E.; Endris, V.; Allgauer, M.; Neumann, O.; Volckmar, A.L.; Kirchner, M.; Leichsenring, J.; Lier, A.; von Winterfeld, M.; et al. Size matters: Dissecting key parameters for panel-based tumor mutational burden analysis. Int. J. Cancer 2019, 144, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Warr, A.; Robert, C.; Hume, D.; Archibald, A.; Deeb, N.; Watson, M. Exome Sequencing: Current and Future Perspectives. G3 Genes Genomes Genet. 2015, 5, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.X.; Wang, Z.X.; Zhao, Q.; Wang, F.; Xu, R.H. Designing gene panels for tumor mutational burden estimation: The need to shift from ‘correlation’ to ‘accuracy’. J. Immunother. Cancer 2019, 7, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Pan, K.; Fakih, M.; Pal, S.; Salgia, R. Value-based genomics. Oncotarget 2018, 9, 15792–15815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Endris, V.; Buchhalter, I.; Allgauer, M.; Rempel, E.; Lier, A.; Volckmar, A.L.; Kirchner, M.; von Winterfeld, M.; Leichsenring, J.; Neumann, O.; et al. Measurement of tumor mutational. burden (TMB) in routine molecular diagnostics: In silico and real-life analysis of three larger gene panels. Int. J. Cancer 2019, 144, 2303–2312. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Allgauer, M.; Litchfield, K.; Rempel, E.; Christopoulos, P.; Kazdal, D.; Endris, V.; Thomas, M.; Frohling, S.; Peters, S.; et al. Optimizing panel-based tumor mutational burden (TMB) measurement. Ann. Oncol. 2019, 30, 1496–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 201178. [Google Scholar]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Ramos, A.H.; Lichtenstein, L.; Gupta, M.; Lawrence, M.S.; Pugh, T.J.; Saksena, G.; Meyerson, M.; Getz, G. Oncotator: Cancer variant annotation tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef]
- Fang, H.; Bergmann, E.A.; Arora, K.; Vacic, V.; Zody, M.C.; Iossifov, I.; O’Rawe, J.A.; Wu, Y.; Jimenez Barron, L.T.; Rosenbaum, J.; et al. Indel variant analysis of short-read sequencing data with Scalpel. Nat. Protoc. 2016, 11, 2529–2548. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Buttner, R.; Longshore, J.W.; Lopez-Rios, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deans, Z.C.; Costa, J.L.; Cree, I.; Dequeker, E.; Edsjo, A.; Henderson, S.; Hummel, M.; Ligtenberg, M.J.; Loddo, M.; Machado, J.C.; et al. Integration of next-generation sequencing in clinical diagnostic molecular pathology laboratories for analysis of solid tumours; an expert opinion on behalf of IQN Path ASBL. Virchows Arch. 2017, 470, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Krieken, H.; Deans, S.; Hall, J.A.; Normanno, N.; Ciardiello, F.; Douillard, J.Y. Quality to rely on: Meeting report of the 5th Meeting of External Quality Assessment,c Naples 2016. ESMO Open 2016, 1, e000114. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Diagnosis | TMB WES—M1 * Real-Life (Median/Value) | (Min–Max) | TMB MSK—M1 * In Silico (Median/Value) | (Min–Max) | TMB F1CDx—M2 ** In Silico (Median/Value) | (Min–Max) |
|---|---|---|---|---|---|---|
| HGG glioma H3K27M+ | 2.9 | (1.6–15.7) | 4.7 | (2.6–17.9) | 4.5 | (2.6–31) |
| Rhabdomyosarcoma | 3.6 | (1.7–6.4) | 2.6 | (1.7–4.3) | 2.6 | (0–5.2) |
| Ewing sarcoma | 3.1 | (0.2–5.1) | 2.6 | (0–5.1) | 2.6 | (0–7.8) |
| Ependymoma | 3.1 | (1.3–10.4) | 1.7 | (0–5.1) | 3.2 | (1.3–9) |
| Neuroblastoma | 3.8 | (1.6–17.2) | 3.0 | (0.9–7.7) | 4.5 | (1.3–15.5) |
| Soft tissue sarcoma | 3.6 | (1.7–6.7) | 3.4 | (0–6.8) | 3.2 | (0–9) |
| Low-grade glioma | 3.5 | (1.6–6.8) | 2.1 | (0.9–4.3) | 3.9 | (1.3–5.2) |
| High-grade glioma H3K27M wt | 4.5 | (1.4–269.8) | 3.4 | (0.9–294.7) | 5.2 | (1.3–410.9) |
| Osteosarcoma | 2.2 | (1.9–7.5) | 3.4 | (0–5.1) | 5.2 | (1.3–6.5) |
| Burkitt lymphoma | 14.2 | (6.1–100.7) | 19.6 | (6.8–46.1) | 27.1 | (6.5–89.2) |
| Medulloblastoma | 3.8 | (3.5–63.6) | 3.4 | (0.9–61.5) | 3.9 | (1.3–89.2) |
| Fibromatosis | 6.2 | (1.1–56.2) | 5.1 | (1.7–29) | 10.3 | (1.3–82.7) |
| Wilms tumor | 3.1 | (2.3–3.9) | 3.4 | (2.6–4.3) | 2.6 | (1.3–3.9) |
| Renal cell carcinoma | 1.8 | (1.5–2.1) | 4.3 | (2.6–6.0) | 4.5 | (1.3–7.8) |
| Adrenocortical carcinoma | 0.9 | - | 0.9 | - | 1.3 | - |
| Plexus choroideus carcinoma | 5.2 | - | 2.6 | - | 5.2 | - |
| Hepatocellular carcinoma | 3.6 | - | 0.9 | - | 3.9 | - |
| Disseminated adenocarcinoma | 2.3 | - | 4.3 | - | 6.5 | - |
| Familiar infantile myofibromatosis | 2.1 | - | 1.7 | - | 0.0 | - |
| Myeloid sarcoma | 0.3 | - | 0.0 | - | 0.0 | - |
| Undifferentiated embryonal tumor of spinal canal | 3.1 | - | 2.6 | - | 2.6 | - |
| Nongerminomatous Germ Cell tumor CNS | 2.3 | - | 1.7 | - | 1.3 | - |
| Epithelial hepatoblastoma | 0.5 | - | 0.0 | - | 0.0 | - |
| Spindle cell hemangioma | 2.1 | - | 0.9 | - | 2.6 | - |
| Fibrodysplasia ossificans progressiva | 3.1 | - | 2.6 | - | 2.6 | - |
| Hepatosplenic T-lymphoma | 0.4 | - | 0.9 | - | 0.0 | - |
| Multisystemic Langerhans cell histiocytosis | 3.1 | - | 2.6 | - | 3.9 | - |
| Gastrointestinal stromal tumor | 2.7 | - | 3.4 | - | 6.5 | - |
| Gender | Age at Diagnosis | Diagnosis | TMB F1Heme Real-Life (Mut/Mb) | TMB WES—M1 * Real-Life (Mut/Mb) | Same Sample (Yes/No) |
|---|---|---|---|---|---|
| F | 9 | Renal cell carcinoma | 1.63 | 1.45 | yes |
| F | 7 | Diffuse intrinsic pontine glioma H3K27M+ | 2.44 | 1.60 | yes |
| M | 13 | Desmoid fibromatosis | 0.81 | 1.14 | yes |
| M | 6 | Spindle cell hemangioma | 0.81 | 2.05 | yes |
| F | 14 | Gastrointestinal stromal tumor | 4.07 | 2.71 | yes |
| F | 14 | Osteosarcoma | 2.44 | 1.91 | yes |
| M | 2 | Langerhans cell histiocytosis | 2.44 | 3.11 | yes |
| M | 11 | Wilms tumor | 1.63 | 2.34 | yes |
| M | 11 | Ewing sarcoma | 1.63 | 2.57 | yes |
| F | 7 | Ependymoma | 2.44 | 3.48 | yes |
| M | 18 | Embryonal rhabdomyosarcoma | 4.89 | 2.82 | yes |
| F | 14 | Ewing sarcoma | 1.63 | 3.57 | yes |
| F | 6 | Wilms tumor | 0.81 | 3.91 | yes |
| F | 18 | Ewing sarcoma | 0.81 | 2.97 | yes |
| M | 9 | Alveolar rhabdomyosarcoma | 3.26 | 3.62 | yes |
| F | 5 | Diffuse intrinsic pontine glioma | 2.44 | 2.85 | yes |
| M | 10 | Ewing sarcoma | 1.63 | 0.17 | yes |
| F | 1 | Neuroblastoma | 1.63 | 7.53 | yes |
| F | 10 | Ewing sarcoma | 7.33 | 4.82 | yes |
| M | 20 | Glioblastoma H3G34R+ | 7.33 | 8.02 | yes |
| F | 2 | Neuroblastoma | 5.70 | 6.33 | yes |
| F | 1 | Embryonal rhabdomyosarcoma | 1.63 | 6.39 | yes |
| M | 3 | Burkitt lymphoma | 10.59 | 6.08 | yes |
| M | 7 | Burkitt lymphoma | 19.55 | 14.18 | yes |
| M | 18 | Glioblastoma | 265.56 | 269.75 | yes |
| F | 10 | Low-grade astroblastoma | 1.63 | 1.83 | no |
| M | 4 | Adrenocortical carcinoma | 0.00 | 0.88 | no |
| M | 15 | Hepatocellular carcinoma | 2.44 | 3.59 | no |
| M | 3 | Epithelial hepatoblastoma | 2.44 | 0.46 | no |
| M | 5 | Embryonal rhabdomyosarcoma | 6.52 | 3.68 | no |
| M | 3 | Embryonal rhabdomyosarcoma | 4.07 | 5.71 | no |
| F | 7 | Glioblastoma | 0.81 | 4.48 | no |
| M | 1 | Anaplastic ependymoma | 1.63 | 6.65 | no |
| F | 4 | Diffuse intrinsic pontine glioma H3K27M+ | 9.78 | 5.39 | no |
| Gender | Age at Diagnosis | Diagnosis | Diagnosis/Relapse | Year of Biopsy | TMB (WES M1 *) Real-Life |
|---|---|---|---|---|---|
| F | 9 | Supratentorial ependymoma | local relapse | 2016 | 2.31 |
| local relapse | 2018 | 3.88 | |||
| F | 1 | Neuroblastoma | metastatic relapse | 2017 | 7.53 |
| metastatic relapse | 2018 | 3.17 | |||
| M | 11 | Ewing sarcoma | primary tumor | 2017 | 2.57 |
| local relapse | 2018 | 4.19 | |||
| M | 5 | DIPG | primary tumor | 2015 | 2.51 |
| local relapse | 2018 | 6.68 | |||
| F | 10 | LG astroblastoma | primary tumor | 2017 | 1.83 |
| local relapse | 2018 | 3.05 | |||
| M | 3 | Epithelial hepatoblastoma | primary tumor | 2016 | 0.46 |
| local relapse | 2018 | 2.48 | |||
| F | 2 | Ependymoma | primary tumor | 2014 | 10.38 |
| metastatic relapse | 2018 | 10.53 | |||
| M | 18 | Osteosarcoma | metastatic relapse | 2018 | 7.47 |
| metastatic relapse | 2018 | 8.10 | |||
| M | 1 | Infantile myofibromatosis | metastatic relapse | 2015 | 2.08 |
| metastatic relapse | 2018 | 1.88 |
| TMB—M1 * In Silico (Number of Cases Above Cut-Off) | TMB—M2 ** In Silico (Number of Cases Above Cut-Off) | |||||
|---|---|---|---|---|---|---|
| Cut-off for ICIs Indication (mut/Mb) | ≥5 | ≥10 | ≥20 | ≥5 | ≥10 | ≥20 |
| WES | 30 | 8 | 4 | 75 | 25 | 7 |
| MSK-IMPACT | 23 | 6 | 4 | 61 | 12 | 6 |
| F1CDx | 24 | 7 | 5 | 42 | 11 | 6 |
| Diagnosis | Number of Patients | Gender Ratio (F/M) | Age Median | Age (Min–Max) | Type of Sample Ratio (Primary Tumor/Local or Metastatic Relapse) |
|---|---|---|---|---|---|
| High-grade glioma H3K27M+ | 12 | 8/2 | 9 | 4–20 | 12/0 |
| Rhabdomyosarcoma | 11 | 7/4 | 5 | 0–18 | 6/5 |
| Ewing sarcoma | 11 | 6/5 | 11 | 8–18 | 2/9 |
| Neuroblastoma | 10 | 6/4 | 2 | 1–8 | 1/9 |
| Ependymoma | 10 | 6/4 | 5.5 | 1–16 | 4/6 |
| Non-rhabdomyosarcoma soft-tissue sarcomas | 8 | 2/6 | 12 | 8–19 | 0/8 |
| High-grade glioma H3K27M wt | 6 | 0/6 | 16 | 8–23 | 5/1 |
| Low-grade glioma | 6 | 1/5 | 9.5 | 3–19 | 1/5 |
| Osteosarcoma | 5 | 4/1 | 18 | 14–28 | 0/5 |
| Burkitt lymphoma | 3 | 0/3 | 7 | 3–12 | 0/3 |
| Medulloblastoma | 3 | 0/3 | 4 | 2–5 | 1/2 |
| Fibromatosis | 3 | 1/2 | 17 | 13–20 | 1/2 |
| Wilms tumor | 2 | 1/1 | 8.5 | 6–11 | 1/1 |
| Renal cell carcinoma | 2 | 1/1 | 13.5 | 9–18 | 1/0 |
| Adrenocortical carcinoma | 1 | F | 4 | - | primary tumor |
| Choroid plexus carcinoma | 1 | M | 1 | - | primary tumor |
| Hepatocellular carcinoma | 1 | M | 15 | - | primary tumor |
| Lung adenocarcinoma | 1 | F | 15 | - | metastatic relapse |
| Familiar infantile myofibromatosis | 1 | M | 1 | - | primary tumor |
| Myeloid sarcoma | 1 | F | 5 | - | primary tumor |
| Undifferentiated embryonal tumor of spinal canal | 1 | M | 2 | - | primary tumor |
| CNS germ cell tumor | 1 | M | 11 | - | local relapse |
| Epithelial hepatoblastoma | 1 | M | 3 | - | primary tumor |
| Spindle cell hemangioendothelioma | 1 | M | 6 | - | primary vascular malformation |
| Fibrodysplasia ossificans progressiva | 1 | F | 1 | - | primary tumor |
| Hepatosplenic T-lymphoma | 1 | M | 17 | - | diagnostic aspiration/bone marrow |
| Multiple system Langerhans cell histiocytosis | 1 | M | 2 | - | metastasis |
| Gastrointestinal stromal tumor | 1 | F | 14 | - | metastatic relapse |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noskova, H.; Kyr, M.; Pal, K.; Merta, T.; Mudry, P.; Polaskova, K.; Ivkovic, T.C.; Adamcova, S.; Hornakova, T.; Jezova, M.; et al. Assessment of Tumor Mutational Burden in Pediatric Tumors by Real-Life Whole-Exome Sequencing and In Silico Simulation of Targeted Gene Panels: How the Choice of Method Could Affect the Clinical Decision? Cancers 2020, 12, 230. https://doi.org/10.3390/cancers12010230
Noskova H, Kyr M, Pal K, Merta T, Mudry P, Polaskova K, Ivkovic TC, Adamcova S, Hornakova T, Jezova M, et al. Assessment of Tumor Mutational Burden in Pediatric Tumors by Real-Life Whole-Exome Sequencing and In Silico Simulation of Targeted Gene Panels: How the Choice of Method Could Affect the Clinical Decision? Cancers. 2020; 12(1):230. https://doi.org/10.3390/cancers12010230
Chicago/Turabian StyleNoskova, Hana, Michal Kyr, Karol Pal, Tomas Merta, Peter Mudry, Kristyna Polaskova, Tina Catela Ivkovic, Sona Adamcova, Tekla Hornakova, Marta Jezova, and et al. 2020. "Assessment of Tumor Mutational Burden in Pediatric Tumors by Real-Life Whole-Exome Sequencing and In Silico Simulation of Targeted Gene Panels: How the Choice of Method Could Affect the Clinical Decision?" Cancers 12, no. 1: 230. https://doi.org/10.3390/cancers12010230