Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway


 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuroinflammation
2.1. Biological Background: The Role of Microglia in the CNS
2.2. Early Infections as Triggers for Immune Deregulation
2.3. Autoimmunity and Genetics
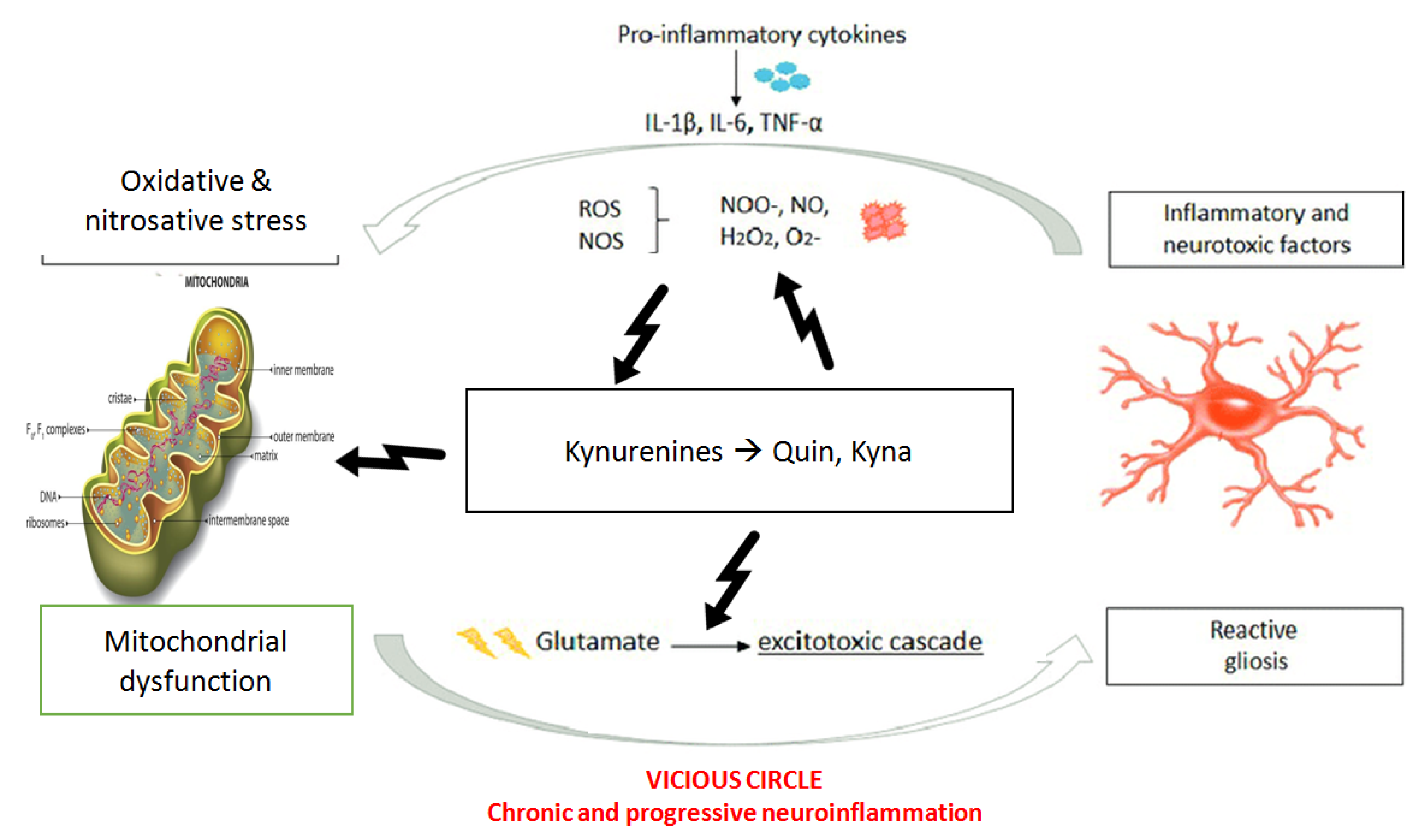
2.4. Inflammation, Mitochondria and Oxidative Stress
3. The Kynurenines Pathway
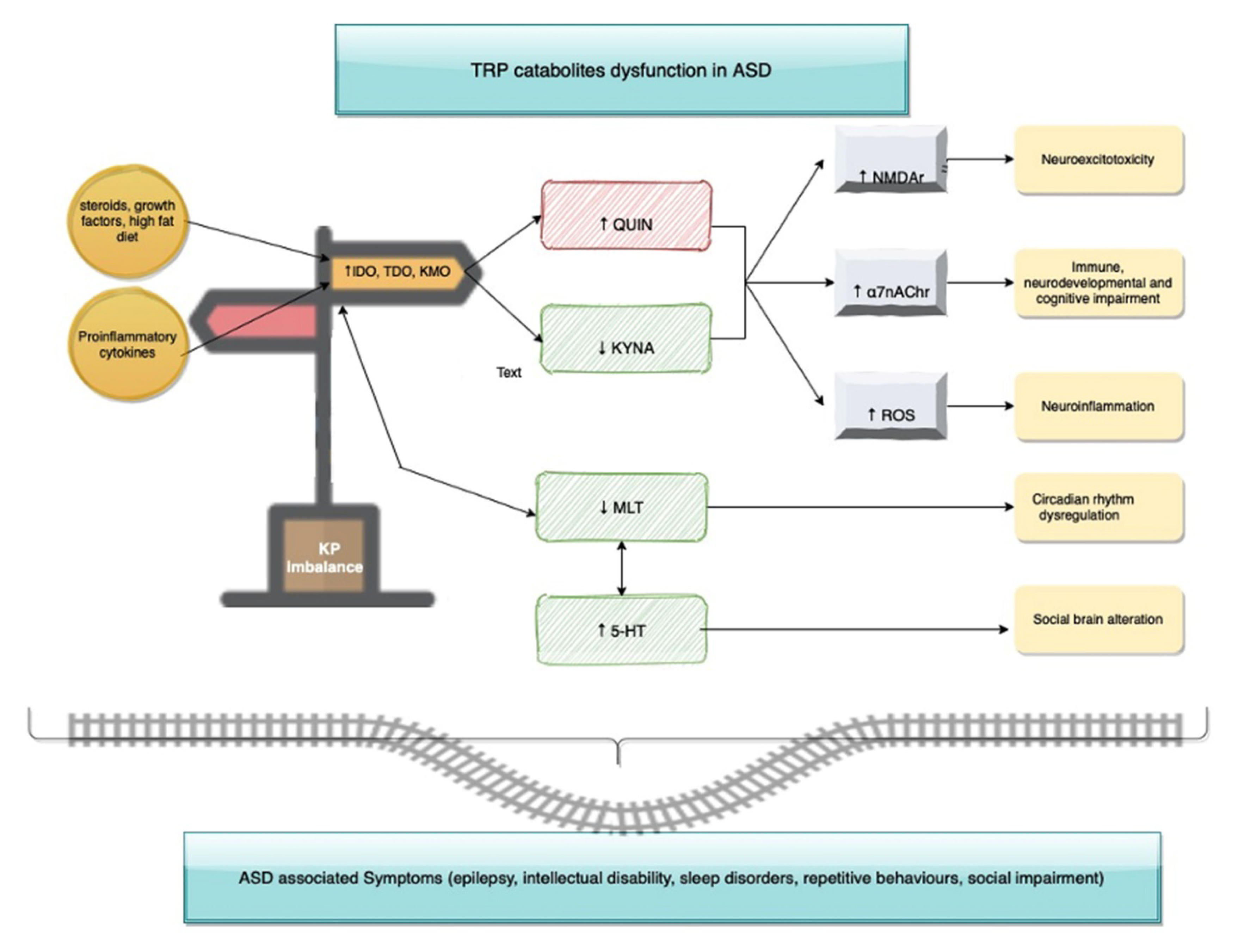
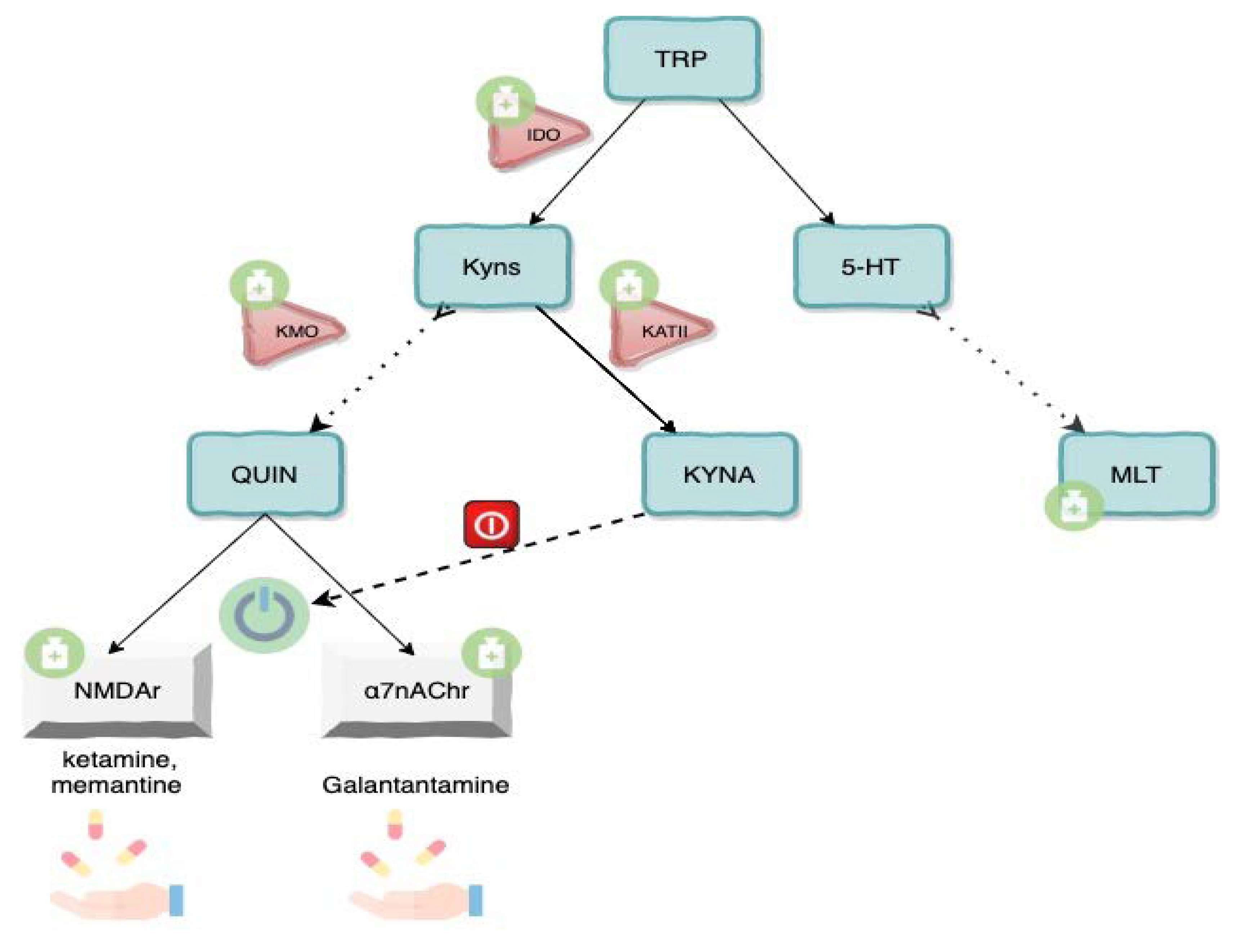
3.1. Tryptophan-Kynurenines Metabolism
3.2. Neuroactivity of Kynurenines
3.3. Kynurenine Pathway Enzymes and Inflammation
3.4. Tryptophan, Serotonin and Melatonin
3.5. Kynurenine Profile in Autism Spectrum Disorders
3.6. Future Perspectives and Drugs
4. Conclusions
- (1)
- a cascade that “sensitizes” the immune system with subsequent changes in cellular proliferation, activation of microglia and further increases in pro-inflammatory cytokines downstream;
- (2)
- increase of ROS and RNS, which leads to oxidative stress and further tissue damage;
- (3)
- mitochondrial dysfunction responsible for bioenergetic impairment and consequently neural suffering;
- (4)
- activation of Kynurenine pathways with an increase in neurotoxic metabolites and excitotoxicity causing long-term changes in glutamatergic function, trophic support and synaptic function.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013; ISBN 978-0-89042-555-8. [Google Scholar]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019, 51, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Gilman, S.R.; Chiang, A.H.; Sanders, S.J.; Vitkup, D. Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci. 2015, 18, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Li, X.; Zhong, Y. Inflammatory Cytokines: Potential Biomarkers of Immunologic Dysfunction in Autism Spectrum Disorders. Mediat. Inflamm. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, S.; Nacher, J.C.; Song, J.; Akutsu, T. An Overview of Bioinformatics Methods for Analyzing Autism Spectrum Disorders. CPD 2020, 25, 4552–4559. [Google Scholar] [CrossRef]
- Chin, E.W.M.; Goh, E.L.K. Behavioral Characterization of MeCP2 Dysfunction-Associated Rett Syndrome and Neuropsychiatric Disorders. In Psychiatric Disorders; Kobeissy, F.H., Ed.; Springer: New York, NY, USA, 2019; Volume 2011, pp. 593–605. ISBN 978-1-4939-9553-0. [Google Scholar]
- Wang, Z.; Hong, Y.; Zou, L.; Zhong, R.; Zhu, B.; Shen, N.; Chen, W.; Lou, J.; Ke, J.; Zhang, T.; et al. Reelin gene variants and risk of autism spectrum disorders: An integrated meta-analysis. Am. J. Med. Genet. 2014, 165, 192–200. [Google Scholar] [CrossRef]
- Fernandes, D.; Santos, S.D.; Coutinho, E.; Whitt, J.L.; Beltrão, N.; Rondão, T.; Leite, M.I.; Buckley, C.; Lee, H.-K.; Carvalho, A.L. Disrupted AMPA Receptor Function upon Genetic- or Antibody-Mediated Loss of Autism-Associated CASPR2. Cereb. Cortex 2019, 29, 4919–4931. [Google Scholar] [CrossRef]
- Canali, G.; Garcia, M.; Hivert, B.; Pinatel, D.; Goullancourt, A.; Oguievetskaia, K.; Saint-Martin, M.; Girault, J.-A.; Faivre-Sarrailh, C.; Goutebroze, L. Genetic variants in autism-related CNTNAP2 impair axonal growth of cortical neurons. Human Mol. Genet. 2018, 27, 1941–1954. [Google Scholar] [CrossRef] [Green Version]
- Taurines, R.; Schwenck, C.; Westerwald, E.; Sachse, M.; Siniatchkin, M.; Freitag, C. ADHD and autism: Differential diagnosis or overlapping traits? A selective review. ADHD Atten. Deficit Hyperact. Disord. 2012, 4, 115–139. [Google Scholar] [CrossRef]
- Wang, C.; Geng, H.; Liu, W.; Zhang, G. Prenatal, perinatal, and postnatal factors associated with autism: A meta-analysis. Medicine 2017, 96, e6696. [Google Scholar] [CrossRef]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef]
- Rice, D.; Barone, S. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 Suppl 3, 511–533. [Google Scholar] [CrossRef]
- Hahamy, A.; Behrmann, M.; Malach, R. The idiosyncratic brain: Distortion of spontaneous connectivity patterns in autism spectrum disorder. Nat. Neurosci. 2015, 18, 302–309. [Google Scholar] [CrossRef]
- Lacivita, E.; Perrone, R.; Margari, L.; Leopoldo, M. Targets for Drug Therapy for Autism Spectrum Disorder: Challenges and Future Directions. J. Med. Chem. 2017, 60, 9114–9141. [Google Scholar] [CrossRef]
- Hong, H.; Kim, B.S.; Im, H.-I. Pathophysiological Role of Neuroinflammation in Neurodegenerative Diseases and Psychiatric Disorders. Int. Neurourol. J. 2016, 20, S2–7. [Google Scholar] [CrossRef]
- Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Saugstad, O.D.; Ormstad, H. Kynurenine Pathway in Autism Spectrum Disorders in Children. Neuropsychobiology 2017, 76, 82–88. [Google Scholar] [CrossRef]
- Wang, Q.M.; Luo, A.Z.; Kong, X. Microglial Activation in Autism. NAJ Med. Sci. 2014, 7, 118–122. [Google Scholar] [CrossRef]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef]
- Bergink, V.; Gibney, S.M.; Drexhage, H.A. Autoimmunity, inflammation, and psychosis: A search for peripheral markers. Biol. Psychiatry 2014, 75, 324–331. [Google Scholar] [CrossRef]
- Albenzio, M.; Santillo, A.; Ciliberti, M.G.; Figliola, L.; Caroprese, M.; Polito, A.N.; Messina, G. Milk nutrition and childhood epilepsy: An ex vivo study on cytokines and oxidative stress in response to milk protein fractions. J. Dairy Sci. 2018, 101, 4842–4852. [Google Scholar] [CrossRef] [PubMed]
- Monji, A.; Kato, T.A.; Mizoguchi, Y.; Horikawa, H.; Seki, Y.; Kasai, M.; Yamauchi, Y.; Yamada, S.; Kanba, S. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2013, 42, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Takano, T. Role of Microglia in Autism: Recent Advances. Dev. Neurosci. 2015, 37, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.T.; Chana, G.; Pardo, C.A.; Achim, C.; Semendeferi, K.; Buckwalter, J.; Courchesne, E.; Everall, I.P. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 2010, 68, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef]
- Suzuki, K.; Sugihara, G.; Ouchi, Y.; Nakamura, K.; Futatsubashi, M.; Takebayashi, K.; Yoshihara, Y.; Omata, K.; Matsumoto, K.; Tsuchiya, K.J.; et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 2013, 70, 49–58. [Google Scholar] [CrossRef]
- Rodriguez, J.I.; Kern, J.K. Evidence of microglial activation in autism and its possible role in brain underconnectivity. Neuron Glia Biol. 2011, 7, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef]
- Morgan, J.T.; Barger, N.; Amaral, D.G.; Schumann, C.M. Stereological Study of Amygdala Glial Populations in Adolescents and Adults with Autism Spectrum Disorder. PLoS ONE 2014, 9, e110356. [Google Scholar] [CrossRef] [Green Version]
- Shigemori, T.; Sakai, A.; Takumi, T.; Itoh, Y.; Suzuki, H. Altered Microglia in the Amygdala Are Involved in Anxiety-related Behaviors of a Copy Number Variation Mouse Model of Autism. J. Nippon Med. Sch. 2015, 82, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Benros, M.E.; Nielsen, P.R.; Nordentoft, M.; Eaton, W.W.; Dalton, S.O.; Mortensen, P.B. Autoimmune Diseases and Severe Infections as Risk Factors for Schizophrenia: A 30-Year Population-Based Register Study. AJP 2011, 168, 1303–1310. [Google Scholar] [CrossRef]
- Gentile, I.; Zappulo, E.; Militerni, R.; Pascotto, A.; Borgia, G.; Bravaccio, C. Etiopathogenesis of autism spectrum disorders: Fitting the pieces of the puzzle together. Med. Hypotheses 2013, 81, 26–35. [Google Scholar] [CrossRef]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, M.W.; Larsen, N.; Grove, J.; Bonefeld-Jørgensen, E.C.; Nørgaard-Pedersen, B.; Hougaard, D.M.; Mortensen, E.L. Neonatal chemokine levels and risk of autism spectrum disorders: Findings from a Danish historic birth cohort follow-up study. Cytokine 2013, 61, 370–376. [Google Scholar] [CrossRef]
- Müller, N.; Weidinger, E.; Leitner, B.; Schwarz, M.J. The role of inflammation in schizophrenia. Front. Neurosci. 2015, 9, 372. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites with Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef]
- Comi, A.M.; Zimmerman, A.W.; Frye, V.H.; Law, P.A.; Peeden, J.N. Familial Clustering of Autoimmune Disorders and Evaluation of Medical Risk Factors in Autism. J. Child. Neurol. 1999, 14, 388–394. [Google Scholar] [CrossRef]
- Hughes, H.K.; Mills Ko, E.; Rose, D.; Ashwood, P. Immune Dysfunction and Autoimmunity as Pathological Mechanisms in Autism Spectrum Disorders. Front. Cell. Neurosci. 2018, 12, 405. [Google Scholar] [CrossRef] [Green Version]
- Mouridsen, S.E.; Rich, B.; Isager, T.; Nedergaard, N.J. Autoimmune diseases in parents of children with infantile autism: A case-control study. Dev. Med. Child. Neurol. 2007, 49, 429–432. [Google Scholar] [CrossRef]
- Sweeten, T.L.; Bowyer, S.L.; Posey, D.J.; Halberstadt, G.M.; McDougle, C.J. Increased Prevalence of Familial Autoimmunity in Probands with Pervasive Developmental Disorders. PEDIATRICS 2003, 112, e420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.S.; Surcel, H.-M.; Hinkka-Yli-Salomäki, S.; Cheslack-Postava, K.; Bao, Y.; Sourander, A. Maternal thyroid autoantibody and elevated risk of autism in a national birth cohort. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2015, 57, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-H.; Su, T.-P.; Chen, Y.-S.; Hsu, J.-W.; Huang, K.-L.; Chang, W.-H.; Chen, T.-J.; Bai, Y.-M. Comorbidity of allergic and autoimmune diseases in patients with autism spectrum disorder: A nationwide population-based study. Res. Autism Spectr. Disord. 2013, 7, 205–212. [Google Scholar] [CrossRef]
- Enstrom, A.M.; Van de Water, J.A.; Ashwood, P. Autoimmunity in autism. Curr. Opin. Investig. Drugs 2009, 10, 463–473. [Google Scholar] [PubMed]
- Enstrom, A.; Krakowiak, P.; Onore, C.; Pessah, I.N.; Hertz-Picciotto, I.; Hansen, R.L.; Van de Water, J.A.; Ashwood, P. Increased IgG4 levels in children with autism disorder. Brain Behav. Immun. 2009, 23, 389–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Aggarwal, S.; Rashanravan, B.; Lee, T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J. Neuroimmunol. 1998, 85, 106–109. [Google Scholar] [CrossRef]
- Enstrom, A.M.; Onore, C.E.; Van de Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Al-Hakbany, M.; Awadallah, S.; AL-Ayadhi, L. The Relationship of HLA Class I and II Alleles and Haplotypes with Autism: A Case Control Study. Autism Res. Treat. 2014, 2014, 1–6. [Google Scholar] [CrossRef]
- Elmer, B.M.; McAllister, A.K. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci. 2012, 35, 660–670. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.-L.; Wu, Y.-Y.; Chen, C.-H.; Gau, S.S.-F.; Huang, Y.-S.; Chien, W.-H.; Hu, F.-C.; Chao, Y.-L. Association of HLA-DRB1 alleles and neuropsychological function in autism. Psychiatr. Genet. 2012, 22, 46–49. [Google Scholar] [CrossRef]
- Johnson, W.G.; Buyske, S.; Mars, A.E.; Sreenath, M.; Stenroos, E.S.; Williams, T.A.; Stein, R.; Lambert, G.H. HLA-DR4 as a Risk Allele for Autism Acting in Mothers of Probands Possibly During Pregnancy. Arch. Pediatr. Adolesc. Med. 2009, 163, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennabi, M.; Gaman, A.; Delorme, R.; Boukouaci, W.; Manier, C.; Scheid, I.; Mohammed, N.S.; Bengoufa, D.; Charron, D.; Krishnamoorthy, R.; et al. HLA-class II haplotypes and Autism Spectrum Disorders. Sci. Rep. 2018, 8, 7639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harville, T.; Rhodes-Clark, B.; Bennuri, S.C.; Delhey, L.; Slattery, J.; Tippett, M.; Wynne, R.; Rose, S.; Kahler, S.; Frye, R.E. Inheritance of HLA-Cw7 Associated With Autism Spectrum Disorder (ASD). Front. Psychiatry 2019, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.R.; Sweeten, T.L.; Johnson, R.C.; Odell, D.; Westover, J.B.; Bray-Ward, P.; Ward, D.C.; Davies, C.J.; Thomas, A.J.; Croen, L.A.; et al. Common Genetic Variants Found in HLA and KIR Immune Genes in Autism Spectrum Disorder. Front. Neurosci. 2016, 10, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, A.R.; Westover, J.B.; Gibbons, C.; Johnson, R.C.; Ward, D.C. Activating killer-cell immunoglobulin-like receptors (KIR) and their cognate HLA ligands are significantly increased in autism. Brain Behav. Immun. 2012, 26, 1122–1127. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.S.; Sajuthi, S.; Langefeld, C.D.; Walker, S.J. Immune function genes CD99L2, JARID2 and TPO show association with autism spectrum disorder. Mol. Autism 2012, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Curatolo, P.; Moavero, R.; de Vries, P.J. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015, 14, 733–745. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Nat. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Dupont, J.; Yakar, S.; Karas, M.; LeRoith, D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene 2004, 23, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Porokhovnik, L. Individual Copy Number of Ribosomal Genes as a Factor of Mental Retardation and Autism Risk and Severity. Cells 2019, 8, 1151. [Google Scholar] [CrossRef] [Green Version]
- Velinov, M. Genomic Copy Number Variations in the Autism Clinic—Work in Progress. Front. Cell. Neurosci. 2019, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.M.; Walsh, C.A. Recent Advances in Understanding the Genetic Architecture of Autism. Annu. Rev. Genom. Hum. Genet. 2020, 21, 289–304. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Eapen, V. Converging Pathways in Autism Spectrum Disorders: Interplay between Synaptic Dysfunction and Immune Responses. Front. Hum. Neurosci. 2013, 7, 738. [Google Scholar] [CrossRef]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef]
- Young, A.M.H.; Campbell, E.; Lynch, S.; Suckling, J.; Powis, S.J. Aberrant NF-KappaB Expression in Autism Spectrum Condition: A Mechanism for Neuroinflammation. Front. Psychiatry 2011, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Young, A.M.H.; Chakrabarti, B.; Roberts, D.; Lai, M.-C.; Suckling, J.; Baron-Cohen, S. From molecules to neural morphology: Understanding neuroinflammation in autism spectrum condition. Mol. Autism 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.; Anderson, G.; Dean, O.; Berk, M.; Galecki, P.; Martin-Subero, M.; Maes, M. The glutathione system: A new drug target in neuroimmune disorders. Mol. Neurobiol. 2014, 50, 1059–1084. [Google Scholar] [CrossRef]
- Scaglia, F. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 2010, 16, 136–143. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Watkins, C.C.; Andrews, S.R. Clinical studies of neuroinflammatory mechanisms in schizophrenia. Schizophr. Res. 2016, 176, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, G.; Maes, M. Redox Regulation and the Autistic Spectrum: Role of Tryptophan Catabolites, Immuno-inflammation, Autoimmunity and the Amygdala. Curr. Neuropharmacol. 2014, 12, 148–167. [Google Scholar] [CrossRef]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Goldani, A.A.S.; Downs, S.R.; Widjaja, F.; Lawton, B.; Hendren, R.L. Biomarkers in Autism. Front. Psychiatry 2014, 5, 100. [Google Scholar] [CrossRef]
- Parellada, M.; Moreno, C.; Mac-Dowell, K.; Leza, J.C.; Giraldez, M.; Bailón, C.; Castro, C.; Miranda-Azpiazu, P.; Fraguas, D.; Arango, C. Plasma antioxidant capacity is reduced in Asperger syndrome. J. Psychiatr. Res. 2012, 46, 394–401. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. 2011, 69, 41R–47R. [Google Scholar] [CrossRef]
- Ghanizadeh, A. Increased Glutamate and Homocysteine and Decreased Glutamine Levels in Autism: A Review and Strategies for Future Studies of Amino Acids in Autism. Dis. Markers 2013, 35, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Cavaliere, G.; Viggiano, E.; Trinchese, G.; De Filippo, C.; Messina, A.; Monda, V.; Valenzano, A.; Cincione, R.I.; Zammit, C.; Cimmino, F.; et al. Long feeding high-fat diet induces hypothalamic oxidative stress and inflammation, and prolonged hypothalamic AMPK activation in rat animal model. Front. Physiol. 2018, 9, 818. [Google Scholar] [CrossRef]
- Palmieri, L.; Persico, A.M. Mitochondrial dysfunction in autism spectrum disorders: Cause or effect? Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1797, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Markham, A.; Bains, R.; Franklin, P.; Spedding, M. Changes in mitochondrial function are pivotal in neurodegenerative and psychiatric disorders: How important is BDNF? Br. J. Pharmacol. 2014, 171, 2206–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkhalter, J.; Fiumelli, H.; Allaman, I.; Chatton, J.-Y.; Martin, J.-L. Brain-Derived Neurotrophic Factor Stimulates Energy Metabolism in Developing Cortical Neurons. J. Neurosci. 2003, 23, 8212–8220. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Gleichmann, M.; Mattson, M.P. Neuronal Calcium Homeostasis and Dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.T.; Strömland, K.; Ventura, L.; Johansson, M.; Bandim, J.M.; Gillberg, C. Autism associated with conditions characterized by developmental errors in early embryogenesis: A mini review. Int. J. Dev. Neurosci. 2005, 23, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism. Open. Access 2016, 6, 1000190. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.P.; Franco, N.F.; Varney, B.; Sundaram, G.; Brown, D.A.; de Bie, J.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Expression of the Kynurenine Pathway in Human Peripheral Blood Mononuclear Cells: Implications for Inflammatory and Neurodegenerative Disease. PLoS ONE 2015, 10, e0131389. [Google Scholar] [CrossRef] [Green Version]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2017, 112, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.-M.; Kim, Y.-K. Network beyond IDO in psychiatric disorders: Revisiting neurodegeneration hypothesis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, D.G.; Ramirez-Ortega, D.; Pineda, B.; Castro, N.; Rios, C.; de la Cruz, V.P. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Iannone, L.F.; Preda, A.; Blottière, H.M.; Clarke, G.; Albani, D.; Belcastro, V.; Carotenuto, M.; Cattaneo, A.; Citraro, R.; Ferraris, C.; et al. Microbiota-gut brain axis involvement in neuropsychiatric disorders. Expert Rev. Neurother. 2019, 19, 1037–1050. [Google Scholar] [CrossRef]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Möller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.; Zhang, Z.; Nance, E.; Drewes, J.L.; Lesniak, W.G.; Singh, S.; Chugani, D.C.; Rangaramanujam, K.; Graham, D.R.; Kannan, S. Maternal Inflammation Results in Altered Tryptophan Metabolism in Rabbit Placenta and Fetal Brain. Dev. Neurosci. 2017, 39, 399–412. [Google Scholar] [CrossRef]
- Notarangelo, F.M.; Pocivavsek, A. Elevated kynurenine pathway metabolism during neurodevelopment: Implications for brain and behavior. Neuropharmacology 2017, 112, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxidative Med. Cell. Longev. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.K.; Essa, M.M.; de Paula Martins, R.; Lovejoy, D.B.; Bilgin, A.A.; Waly, M.I.; Al-Farsi, Y.M.; Al-Sharbati, M.; Al-Shaffae, M.A.; Guillemin, G.J. Altered kynurenine pathway metabolism in autism: Implication for immune-induced glutamatergic activity: Altered kynurenine pathway metabolism in ASD. Autism Res. 2016, 9, 621–631. [Google Scholar] [CrossRef]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Brown, M.S.; Singel, D.; Hepburn, S.; Rojas, D.C. Increased Glutamate Concentration in the Auditory Cortex of Persons with Autism and First-Degree Relatives: A 1 H-MRS Study: Increased glutamate concentration in autism. Autism Res. 2013, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ghaleiha, A.; Asadabadi, M.; Mohammadi, M.-R.; Shahei, M.; Tabrizi, M.; Hajiaghaee, R.; Hassanzadeh, E.; Akhondzadeh, S. Memantine as adjunctive treatment to risperidone in children with autistic disorder: A randomized, double-blind, placebo-controlled trial. Int. J. Neuropsychopharmacol. 2013, 16, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Rios, C.; Santamaria, A. Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem. Res. 1991, 16, 1139–1143. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef] [PubMed]
- Yakel, J.L. Nicotinic ACh receptors in the hippocampal circuit; functional expression and role in synaptic plasticity. J. Physiol 2014, 592, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Luchicchi, A.; Bloem, B.; Viaña, J.N.M.; Mansvelder, H.D.; Role, L.W. Illuminating the role of cholinergic signaling in circuits of attention and emotionally salient behaviors. Front. Synaptic Neurosci. 2014, 6, 24. [Google Scholar] [CrossRef]
- Funakoshi, H.; Kanai, M.; Nakamura, T. Modulation of tryptophan metabolism, promotion of neurogenesis and alteration of anxiety-related behavior in tryptophan 2,3-dioxygenase-deficient mice. Int. J. Tryptophan Res. 2011, 4, 7–18. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Imamura, Y.; Saito, K.; Sakai, D.; Motoyama, J. Altered kynurenine pathway metabolites in a mouse model of human attention-deficit hyperactivity/autism spectrum disorders: A potential new biological diagnostic marker. Sci. Rep. 2019, 9, 13182. [Google Scholar] [CrossRef]
- Tordjman, S.; Anderson, G.M.; Kermarrec, S.; Bonnot, O.; Geoffray, M.-M.; Brailly-Tabard, S.; Chaouch, A.; Colliot, I.; Trabado, S.; Bronsard, G.; et al. Altered circadian patterns of salivary cortisol in low-functioning children and adolescents with autism. Psychoneuroendocrinology 2014, 50, 227–245. [Google Scholar] [CrossRef]
- White, S.W.; Roberson-Nay, R. Anxiety, Social Deficits, and Loneliness in Youth with Autism Spectrum Disorders. J. Autism Dev. Disord. 2009, 39, 1006–1013. [Google Scholar] [CrossRef]
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320, 595–597. [Google Scholar] [CrossRef] [Green Version]
- Mosienko, V.; Bert, B.; Beis, D.; Matthes, S.; Fink, H.; Bader, M.; Alenina, N. Exaggerated aggression and decreased anxiety in mice deficient in brain serotonin. Transl. Psychiatry 2012, 2, e122. [Google Scholar] [CrossRef] [Green Version]
- McDougle, C.J.; Naylor, S.T.; Cohen, D.J.; Aghajanian, G.K.; Heninger, G.R.; Price, L.H. Effects of tryptophan depletion in drug-free adults with autistic disorder. Arch. Gen. Psychiatry 1996, 53, 993–1000. [Google Scholar] [CrossRef]
- Vitalis, T.; Parnavelas, J.G. The Role of Serotonin in Early Cortical Development. Dev. Neurosci. 2003, 25, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Azmitia, E.C. Modern views on an ancient chemical: Serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res. Bull. 2001, 56, 413–424. [Google Scholar] [CrossRef]
- Takeuchi, Y. Serotonergic Neurotransmission in Autism Spectrum Disorders. In Autism—A Neurodevelopmental Journey from Genes to Behaviour; Eapen, V., Ed.; InTech: Rijeka, Croatia, 2011; ISBN 978-953-307-493-1. [Google Scholar]
- Jenkins, T.; Nguyen, J.; Polglaze, K.; Bertrand, P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef]
- Nishizawa, S.; Benkelfat, C.; Young, S.N.; Leyton, M.; Mzengeza, S.; de Montigny, C.; Blier, P.; Diksic, M. Differences between males and females in rates of serotonin synthesis in human brain. Proc. Nat. Acad. Sci. USA 1997, 94, 5308–5313. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, D.; Riedel, W.J.; Sambeth, A. Effects of acute tryptophan depletion on memory, attention and executive functions: A systematic review. Neurosci. Biobehav. Rev. 2009, 33, 926–952. [Google Scholar] [CrossRef]
- Monda, V.; Salerno, M.; Sessa, F.; Bernardini, R.; Valenzano, A.; Marsala, G.; Zammit, C.; Avola, R.; Carotenuto, M.; Messina, G.; et al. Functional Changes of Orexinergic Reaction to Psychoactive Substances. Mol. Neurobiol. 2018, 55, 6362–6368. [Google Scholar] [CrossRef] [PubMed]
- Ruddick, J.P.; Evans, A.K.; Nutt, D.J.; Lightman, S.L.; Rook, G.A.W.; Lowry, C.A. Tryptophan metabolism in the central nervous system: Medical implications. Expert Rev. Mol. Med. 2006, 8, 1–27. [Google Scholar] [CrossRef]
- Dunn, A.J.; Welch, J. Stress-and Endotoxin-Induced Increases in Brain Tryptophan and Serotonin Metabolism Depend on Sympathetic Nervous System Activity. J. Neurochem. 1991, 57, 1615–1622. [Google Scholar] [CrossRef]
- Kennett, G.A.; Curzon, G.; Hunt, A.; Patel, A.J. Immobilization Decreases Amino Acid Concentrations in Plasma but Maintains or Increases Them in Brain. J. Neurochem. 1986, 46, 208–212. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Hawkins, R.A. Na+ -dependent transport of large neutral amino acids occurs at the abluminal membrane of the blood-brain barrier. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1167–E1173. [Google Scholar] [CrossRef] [Green Version]
- Ormstad, H.; Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Halvorsen, B.; Saugstad, O.D.; Isaksen, J.; Maes, M. Serum tryptophan, tryptophan catabolites and brain-derived neurotrophic factor in subgroups of youngsters with autism spectrum disorders. CNSNDDT 2018, 17, 626–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.M.; Malow, B.A. Sleep and autism spectrum disorders. Pediatr. Clin. N. Am. 2011, 58, 685–698. [Google Scholar] [CrossRef]
- Messina, A.; Monda, V.; Sessa, F.; Valenzano, A.; Salerno, M.; Bitetti, I.; Precenzano, F.; Marotta, R.; Lavano, F.; Lavano, S.M.; et al. Sympathetic, Metabolic Adaptations, and Oxidative Stress in Autism Spectrum Disorders: How Far From Physiology? Front. Physiol. 2018, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Shokouhi, G.; Tubbs, R.S.; Shoja, M.M.; Hadidchi, S.; Ghorbanihaghjo, A.; Roshangar, L.; Farahani, R.M.; Mesgari, M.; Oakes, W.J. Neuroprotective effects of high-dose vs low-dose melatonin after blunt sciatic nerve injury. Childs Nerv. Syst. 2007, 24, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Hüther, G.; et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Li, Y.; Hu, N.; Yang, D.; Oxenkrug, G.; Yang, Q. Regulating the balance between the kynurenine and serotonin pathways of tryptophan metabolism. FEBS J. 2017, 284, 948–966. [Google Scholar] [CrossRef] [Green Version]
- Míguez, J.M.; Simonneaux, V.; Pévet, P. Evidence for a Regulatory Role of Melatonin on Serotonin Release and Uptake in the Pineal Gland. J. Neuroendocrinol. 1995, 7, 949–956. [Google Scholar] [CrossRef]
- El-Ansary, A.; Al-Ayadhi, L. Neuroinflammation in autism spectrum disorders. J. Neuroinflamm. 2012, 9, 768. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Matsuzaki, H.; Iwata, K.; Kameno, Y.; Shimmura, C.; Kawai, S.; Yoshihara, Y.; Wakuda, T.; Takebayashi, K.; Takagai, S.; et al. Plasma Cytokine Profiles in Subjects with High-Functioning Autism Spectrum Disorders. PLoS ONE 2011, 6, e20470. [Google Scholar] [CrossRef]
- El-Ansary, A.; Al-Ayadhi, L. GABAergic/glutamatergic imbalance relative to excessive neuroinflammation in autism spectrum disorders. J. Neuroinflamm. 2014, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.R.; Namboodiri, M.A. Tryptophan and the immune response. Immunol. Cell Biol. 2003, 81, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin: Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Essa, M.M.; Braidy, N.; Vijayan, K.R.; Subash, S.; Guillemin, G.J. Excitotoxicity in the Pathogenesis of Autism. Neurotox. Res. 2013, 23, 393–400. [Google Scholar] [CrossRef]
- Havelund, J.F.; Andersen, A.D.; Binzer, M.; Blaabjerg, M.; Heegaard, N.H.H.; Stenager, E.; Færgeman, N.J.; Gramsbergen, J.B. Changes in kynurenine pathway metabolism in Parkinson patients with L-DOPA-induced dyskinesia. J. Neurochem. 2017, 142, 756–766. [Google Scholar] [CrossRef]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; Darlington, L.G. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders: Kynurenines and CNS disorders. Br. J. Pharmacol. 2013, 169, 1211–1227. [Google Scholar] [CrossRef]
- Nabi, R.; Serajee, F.J.; Chugani, D.C.; Zhong, H.; Huq, A.H.M.M. Association of tryptophan 2,3 dioxygenase gene polymorphism with autism. Am. J. Med. Genet. 2004, 125B, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Rind, H.B.; Russo, A.F.; Whittemore, S.R. Developmental regulation of tryptophan hydroxylase messenger RNA expression and enzyme activity in the raphe and its target fields. Neuroscience 2000, 101, 665–677. [Google Scholar] [CrossRef]
- Cascio, L.; Chen, C.; Pauly, R.; Srikanth, S.; Jones, K.; Skinner, C.D.; Stevenson, R.E.; Schwartz, C.E.; Boccuto, L. Abnormalities in the genes that encode Large Amino Acid Transporters increase the risk of Autism Spectrum Disorder. Mol. Genet. Genom. Med. 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Haslinger, D.; Waltes, R.; Yousaf, A.; Lindlar, S.; Schneider, I.; Lim, C.K.; Tsai, M.-M.; Garvalov, B.K.; Acker-Palmer, A.; Krezdorn, N.; et al. Loss of the Chr16p11.2 ASD candidate gene QPRT leads to aberrant neuronal differentiation in the SH-SY5Y neuronal cell model. Mol. Autism 2018, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-J.; Choi, S.Y.; Kim, E. NMDA receptor dysfunction in autism spectrum disorders. Curr. Opin. Pharmacol. 2015, 20, 8–13. [Google Scholar] [CrossRef]
- Bortz, D.M.; Wu, H.-Q.; Schwarcz, R.; Bruno, J.P. Oral administration of a specific kynurenic acid synthesis (KAT II) inhibitor attenuates evoked glutamate release in rat prefrontal cortex. Neuropharmacology 2017, 121, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisholm, K.; Lin, A.; Abu-Akel, A.; Wood, S.J. The association between autism and schizophrenia spectrum disorders: A review of eight alternate models of co-occurrence. Neurosci. Biobehav. Rev. 2015, 55, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, K.R.; Castellano-Gonzalez, G.; Guillemin, G.J.; Lovejoy, D.B. Major Developments in the Design of Inhibitors along the Kynurenine Pathway. CMC 2017, 24, 2471–2495. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, D.A.; Frye, R.E. Melatonin in autism spectrum disorders: A systematic review and meta-analysis: Review. Dev. Med. Child Neurol. 2011, 53, 783–792. [Google Scholar] [CrossRef]
- Tordjman, S.; Najjar, I.; Bellissant, E.; Anderson, G.; Barburoth, M.; Cohen, D.; Jaafari, N.; Schischmanoff, O.; Fagard, R.; Lagdas, E.; et al. Advances in the Research of Melatonin in Autism Spectrum Disorders: Literature Review and New Perspectives. IJMS 2013, 14, 20508–20542. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.P.H.; Werner, M.U.; Rosenkilde, M.M.; Fenger, A.Q.; Petersen, M.C.; Rosenberg, J.; Gögenur, I. Pharmacokinetics of high-dose intravenous melatonin in humans. J. Clin. Pharmacol. 2016, 56, 324–329. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savino, R.; Carotenuto, M.; Polito, A.N.; Di Noia, S.; Albenzio, M.; Scarinci, A.; Ambrosi, A.; Sessa, F.; Tartaglia, N.; Messina, G. Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway. Brain Sci. 2020, 10, 631. https://doi.org/10.3390/brainsci10090631
Savino R, Carotenuto M, Polito AN, Di Noia S, Albenzio M, Scarinci A, Ambrosi A, Sessa F, Tartaglia N, Messina G. Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway. Brain Sciences. 2020; 10(9):631. https://doi.org/10.3390/brainsci10090631
Chicago/Turabian StyleSavino, Rosa, Marco Carotenuto, Anna Nunzia Polito, Sofia Di Noia, Marzia Albenzio, Alessia Scarinci, Antonio Ambrosi, Francesco Sessa, Nicola Tartaglia, and Giovanni Messina. 2020. "Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway" Brain Sciences 10, no. 9: 631. https://doi.org/10.3390/brainsci10090631
APA StyleSavino, R., Carotenuto, M., Polito, A. N., Di Noia, S., Albenzio, M., Scarinci, A., Ambrosi, A., Sessa, F., Tartaglia, N., & Messina, G. (2020). Analyzing the Potential Biological Determinants of Autism Spectrum Disorder: From Neuroinflammation to the Kynurenine Pathway. Brain Sciences, 10(9), 631. https://doi.org/10.3390/brainsci10090631