
Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension

 ,
,  , ,
, , 
Abstract
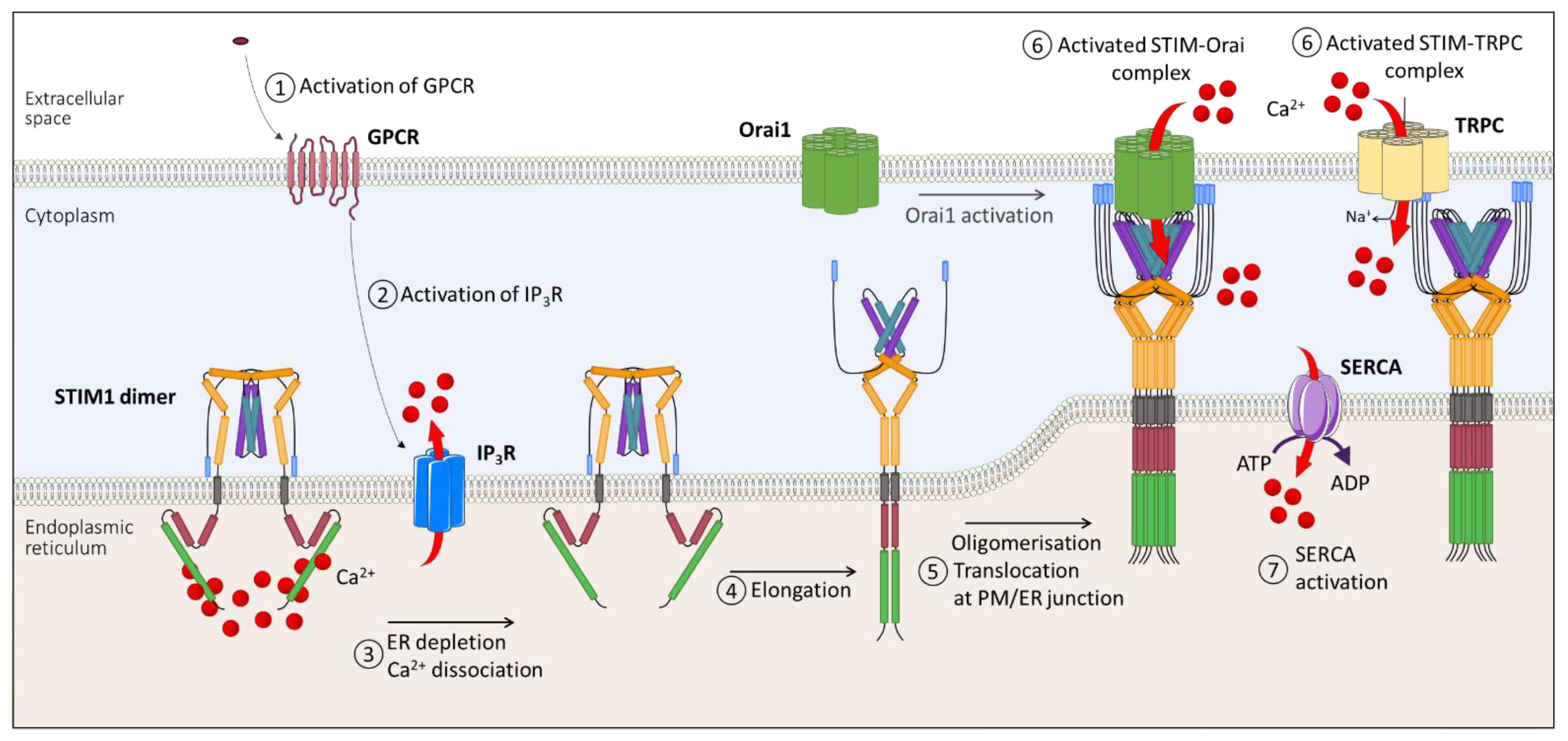
1. Overview of SOCE
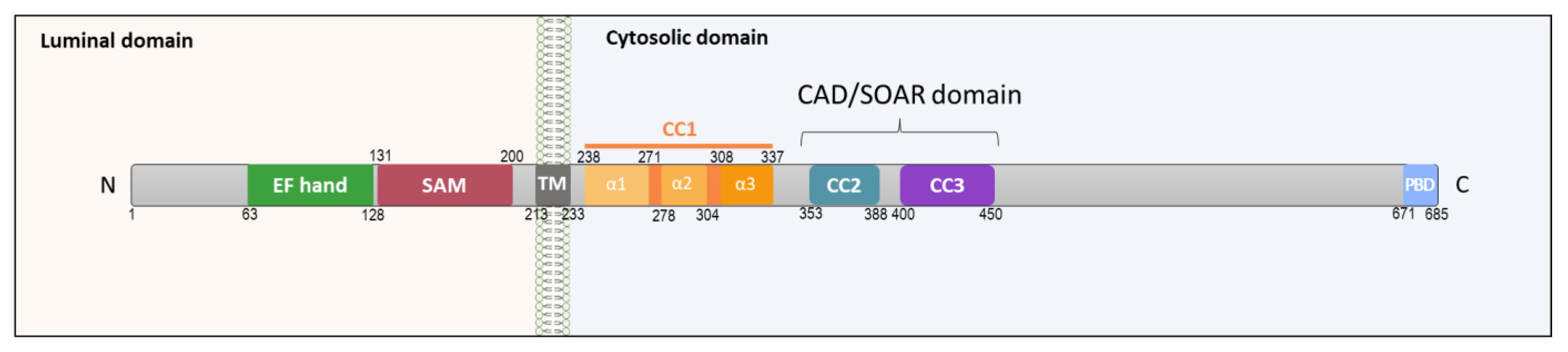
1.1. STIM1
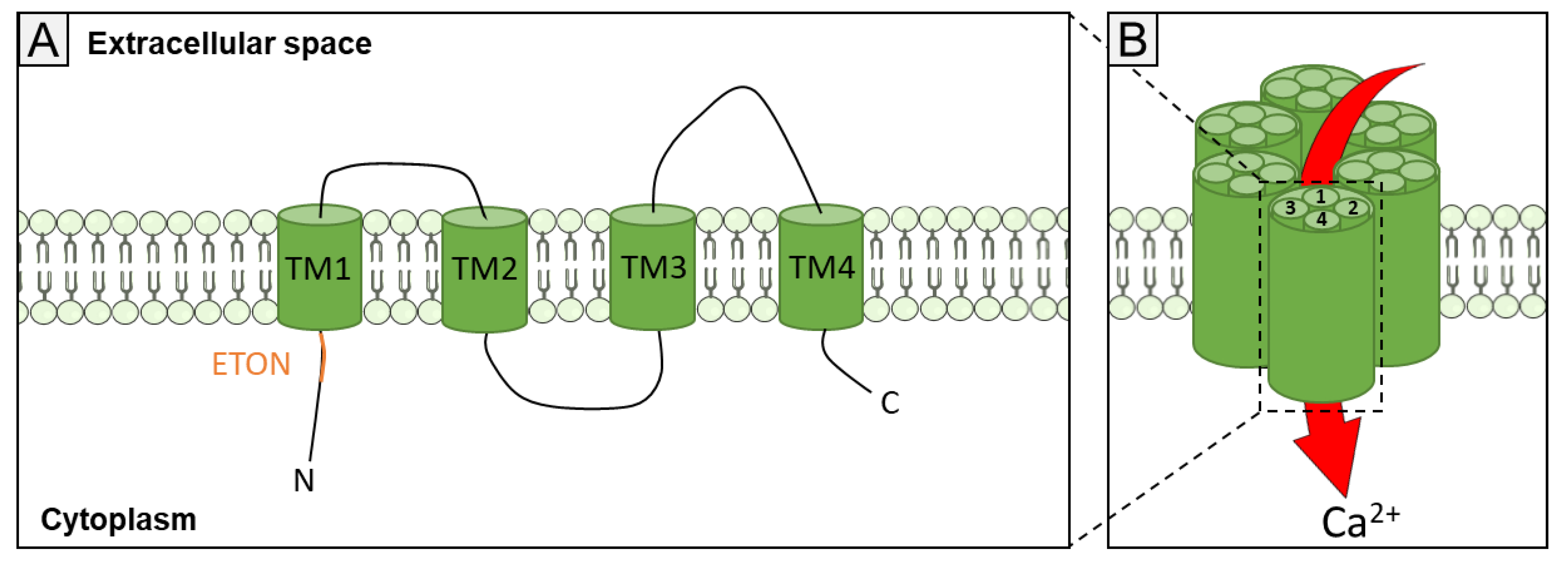
1.2. Orai Channels
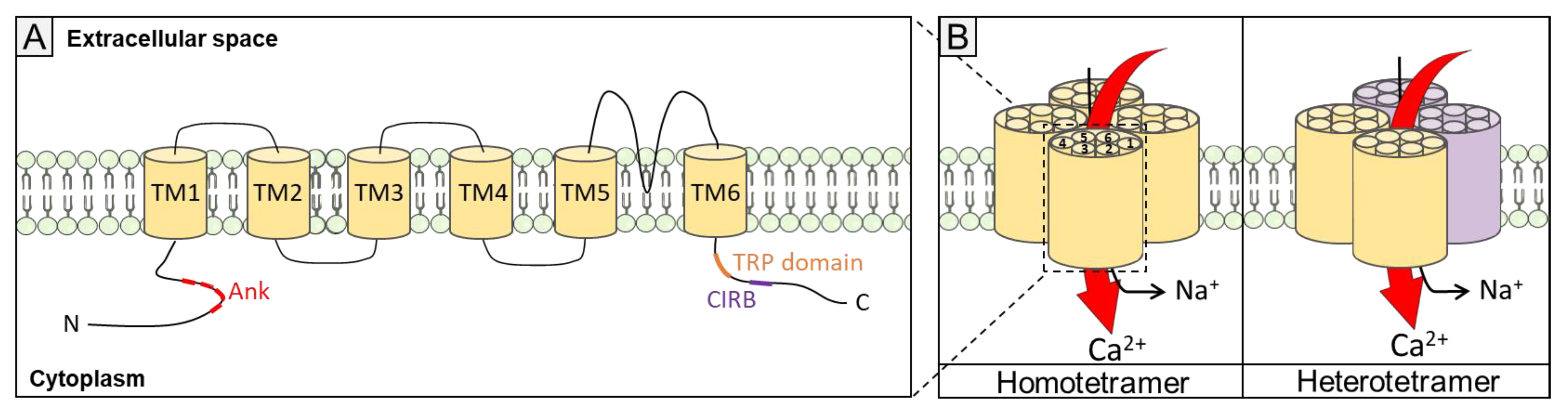
1.3. TRPC Channels
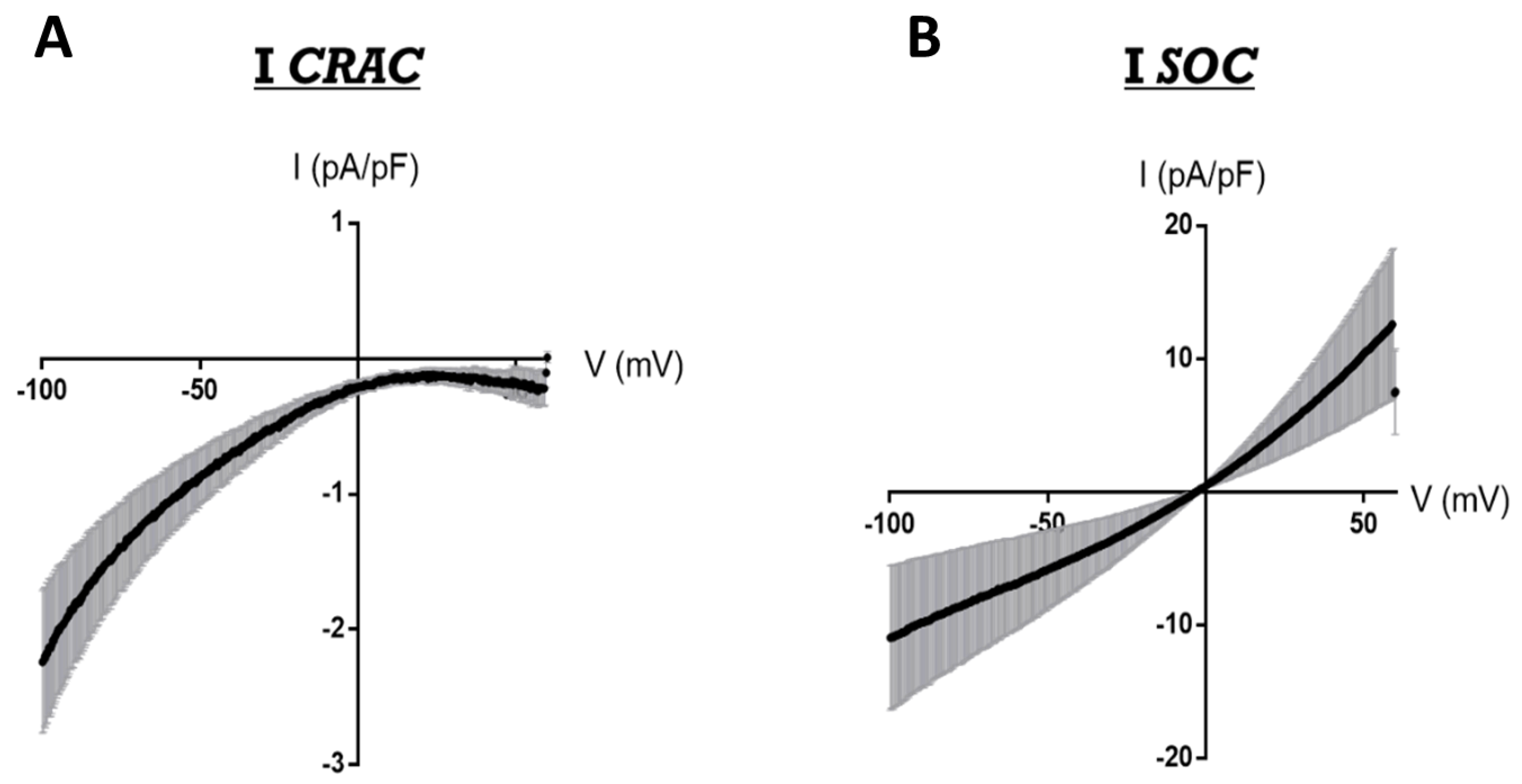
1.4. SOCE Mechanism
2. SOCE in PAH
2.1. Pulmonary Arterial Hypertension
2.2. Physiological Implication of SOCE in Control PASMC
2.3. Physiopathological Implication of SOCE in PAH PASMC
3. Targeted SOCE in PAH: A Novel Therapeutic Option?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bootman, M.D.; Collins, T.J.; Peppiatt, C.M.; Prothero, L.S.; MacKenzie, L.; De Smet, P.; Travers, M.; Tovey, S.C.; Seo, J.T.; Berridge, M.J.; et al. Calcium Signalling—an Overview. Semin. Cell Dev. Biol. 2001, 12, 3–10. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium Signalling: Dynamics, Homeostasis and Remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Putney, J.W. A Model for Receptor-Regulated Calcium Entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of Intracellular Calcium Stores Activates a Calcium Current in Mast Cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Zweifach, A.; Lewis, R.S. Slow Calcium-Dependent Inactivation of Depletion-Activated Calcium Current. Store-Dependent and -Independent Mechanisms. J. Biol. Chem. 1995, 270, 14445–14451. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B.; Penner, R. Store Depletion and Calcium Influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an Essential and Conserved Component of Store-Operated Ca2+ Channel Function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T. STIM Is a Ca2+ Sensor Essential for Ca2+-Store-Depletion-Triggered Ca2+ Influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A Mutation in Orai1 Causes Immune Deficiency by Abrogating CRAC Channel Function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R.; et al. Identification and Characterization of the STIM (Stromal Interaction Molecule) Gene Family: Coding for a Novel Class of Transmembrane Proteins. Biochem. J. 2001, 357, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Li, G.-Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ Depletion-Induced Oligomerization of Stromal Interaction Molecule 1 (STIM1) via the EF-SAM Region. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [PubMed]
- Spassova, M.A.; Soboloff, J.; He, L.-P.; Xu, W.; Dziadek, M.A.; Gill, D.L. STIM1 Has a Plasma Membrane Role in the Activation of Store-Operated Ca2+ Channels. Proc. Natl. Acad. Sci. USA 2006, 103, 4040–4045. [Google Scholar] [CrossRef]
- Mercer, J.C.; DeHaven, W.I.; Smyth, J.T.; Wedel, B.; Boyles, R.R.; Bird, G.S.; Putney, J.W. Large store-operated calcium-selective currents due to co-expression of orai1 or orai2 with the intracellular calcium sensor, stim1. J. Biol. Chem. 2006, 281, 24979–24990. [Google Scholar] [CrossRef] [PubMed]
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal Interaction Molecule (STIM) 1 and STIM2 Calcium Sensing Regions Exhibit Distinct Unfolding and Oligomerization Kinetics. J. Biol. Chem. 2009, 284, 728–732. [Google Scholar] [CrossRef]
- Kim, C.A.; Bowie, J.U. SAM Domains: Uniform Structure, Diversity of Function. Trends Biochem. Sci. 2003, 28, 625–628. [Google Scholar] [CrossRef]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ Signalling Prompted by STIM1 Conformational Switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 Coiled-Coil Interplay in the Regulation of Store-Operated Calcium Entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A Coiled-Coil Clamp Controls Both Conformation and Clustering of Stromal Interaction Molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 Couples to ORAI1 via an Intramolecular Transition into an Extended Conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef]
- Kim, M.S.; Zeng, W.; Yuan, J.P.; Shin, D.M.; Worley, P.F.; Muallem, S. Native Store-Operated Ca2+ Influx Requires the Channel Function of Orai1 and TRPC1. J. Biol. Chem. 2009, 284, 9733–9741. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.-J.; Worley, P.F.; Muallem, S. SOAR and the Polybasic STIM1 Domains Gate and Regulate Orai Channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Park, C.Y.; Shcheglovitov, A.; Dolmetsch, R. The CRAC Channel Activator STIM1 Binds and Inhibits L-Type Voltage-Gated Calcium Channels. Science 2010, 330, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Frischauf, I.; Muik, M.; Derler, I.; Bergsmann, J.; Fahrner, M.; Schindl, R.; Groschner, K.; Romanin, C. Molecular Determinants of the Coupling between STIM1 and Orai Channels: Differential Activation Of Orai1–3 Channels by a Stim1 Coiled-Coil Mutant. J. Biol. Chem. 2009, 284, 21696–21706. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Yuan, J.P.; Kim, M.S.; Choi, Y.J.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 Gates TRPC Channels but Not Orai1 by Electrostatic Interaction. Mol. Cell 2008, 32, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-Cell Imaging Reveals Sequential Oligomerization and Local Plasma Membrane Targeting of Stromal Interaction Molecule 1 after Ca2+ Store Depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 Is a Feedback Regulator That Stabilizes Basal Cytosolic and Endoplasmic Reticulum Ca2+ Levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Zhou, Y.; Mancarella, S.; Wang, Y.; Yue, C.; Ritchie, M.; Gill, D.L.; Soboloff, J. The Short N-Terminal Domains of STIM1 and STIM2 Control the Activation Kinetics of Orai1 Channels. J. Biol. Chem. 2009, 284, 19164–19168. [Google Scholar] [CrossRef]
- Darbellay, B.; Arnaudeau, S.; Bader, C.R.; Konig, S.; Bernheim, L. STIM1L Is a New Actin-Binding Splice Variant Involved in Fast Repetitive Ca2+ Release. J. Cell Biol. 2011, 194, 335–346. [Google Scholar] [CrossRef]
- Ramesh, G.; Jarzembowski, L.; Schwarz, Y.; Poth, V.; Konrad, M.; Knapp, M.L.; Schwär, G.; Lauer, A.A.; Grimm, M.O.W.; Alansary, D.; et al. A Short Isoform of STIM1 Confers Frequency-Dependent Synaptic Enhancement. Cell Rep. 2021, 34, 108844. [Google Scholar] [CrossRef]
- Miederer, A.-M.; Alansary, D.; Schwär, G.; Lee, P.-H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 Splice Variant Negatively Regulates Store-Operated Calcium Entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjö, S.; Park, C.Y.; Dolmetsch, R.E.; Lewis, R.S. Alternative Splicing Converts STIM2 from an Activator to an Inhibitor of Store-Operated Calcium Channels. J. Cell Biol. 2015, 209, 653–670. [Google Scholar] [CrossRef]
- Feske, S.; Prakriya, M.; Rao, A.; Lewis, R.S. A Severe Defect in CRAC Ca2+ Channel Activation and Altered K+ Channel Gating in T Cells from Immunodeficient Patients. J. Exp. Med. 2005, 202, 651–662. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.-F.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-Wide RNAi Screen of Ca2+ Influx Identifies Genes That Regulate Ca2+ Release-Activated Ca2+ Channel Activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef] [PubMed]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular Identification of the CRAC Channel by Altered Ion Selectivity in a Mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 Is a Plasma Membrane Protein Essential for Store-Operated Ca2+ Entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 Is an Essential Pore Subunit of the CRAC Channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal Structure of the Calcium Release-Activated Calcium Channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The Extended Transmembrane Orai1 N-Terminal (ETON) Region Combines Binding Interface and Gate for Orai1 Activation by STIM1. J. Biol. Chem. 2013, 288, 29025–29034. [Google Scholar] [CrossRef]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and Functional Characterization of Orai Proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.A.; Wissenbach, U.; Philipp, S.E.; Freichel, M.; Cavalié, A.; Flockerzi, V. Murine ORAI2 Splice Variants Form Functional Ca2+ Release-Activated Ca2+ (CRAC) Channels. J. Biol. Chem. 2007, 282, 19375–19384. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A Novel Native Store-Operated Calcium Channel Encoded by Orai3: Selective requirement of orai3 versus orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 Are Store-Operated Ca2+ Channels with Distinct Functional Properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef]
- Hoth, M.; Niemeyer, B.A. Chapter Ten—The Neglected CRAC Proteins: Orai2, Orai3, and STIM2. In Current Topics in Membranes; Prakriya, M., Ed.; Store-Operated Calcium Channels; Academic Press: Cambridge, MA, USA, 2013; Volume 71, pp. 237–271. [Google Scholar]
- Fukushima, M.; Tomita, T.; Janoshazi, A.; Putney, J.W. Alternative Translation Initiation Gives Rise to Two Isoforms of Orai1 with Distinct Plasma Membrane Mobilities. J. Cell Sci. 2012, 125, 4354–4361. [Google Scholar] [CrossRef]
- Desai, P.N.; Zhang, X.; Wu, S.; Janoshazi, A.; Bolimuntha, S.; Putney, J.W.; Trebak, M. Multiple Types of Calcium Channels Arising from Alternative Translation Initiation of the Orai1 Message. Sci. Signal. 2015, 8, ra74. [Google Scholar] [CrossRef]
- Montell, C. The History of TRP Channels, a Commentary and Reflection. Pflug. Arch.—Eur. J. Physiol. 2011, 461, 499–506. [Google Scholar] [CrossRef]
- Li, H. TRP Channel Classification. In Transient Receptor Potential Canonical Channels and Brain Diseases; Wang, Y., Ed.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2017; pp. 1–8. ISBN 978-94-024-1088-4. [Google Scholar]
- Owsianik, G.; Talavera, K.; Voets, T.; Nilius, B. Permeation and Selectivity of TRP Channels. Annu. Rev. Physiol. 2006, 68, 685–717. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W. The Mammalian TRPC Cation Channels. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2004, 1742, 21–36. [Google Scholar] [CrossRef]
- Salido, G.M.; Sage, S.O.; Rosado, J.A. TRPC Channels and Store-Operated Ca2+ Entry. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2009, 1793, 223–230. [Google Scholar] [CrossRef]
- Tang, Q.; Guo, W.; Zheng, L.; Wu, J.-X.; Liu, M.; Zhou, X.; Zhang, X.; Chen, L. Structure of the Receptor-Activated Human TRPC6 and TRPC3 Ion Channels. Cell Res. 2018, 28, 746–755. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, X.; Tian, J.; Xiao, Y.; Tian, T.; Xu, F.; Hong, X.; Zhu, M.X. TRPC Channels: Structure, Function, Regulation and Recent Advances in Small Molecular Probes. Pharmacol. Ther. 2020, 209, 107497. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.K.; Lussier, M.P.; Barajas-Martinez, H.; Bousquet, S.M.; Blanchard, A.P.; Francoeur, N.; Dumaine, R.; Boulay, G. Identification of Two Domains Involved in the Assembly of Transient Receptor Potential Canonical Channels. J. Biol. Chem. 2006, 281, 30356–30364. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, R. A Primer on Ankyrin Repeat Function in TRP Channels and Beyond. Mol. Biosyst. 2008, 4, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Brown, D.M.; Qin, N.; Jiang, M.; Dietrich, A.; Zhu, M.X.; Chen, Z.; Birnbaumer, M.; Mikoshiba, K.; Birnbaumer, L. Modulation of Ca2+ Entry by Polypeptides of the Inositol 1,4,5-Trisphosphate Receptor (IP3R) That Bind Transient Receptor Potential (TRP): Evidence for Roles of TRP and IP3R in Store Depletion-Activated Ca2+ Entry. Proc. Natl. Acad. Sci. USA 1999, 96, 14955–14960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tang, J.; Tikunova, S.; Johnson, J.D.; Chen, Z.; Qin, N.; Dietrich, A.; Stefani, E.; Birnbaumer, L.; Zhu, M.X. Activation of Trp3 by Inositol 1,4,5-Trisphosphate Receptors through Displacement of Inhibitory Calmodulin from a Common Binding Domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3168–3173. [Google Scholar] [CrossRef]
- Wedel, B.J.; Vazquez, G.; McKay, R.R.; Bird, G.S.T.; Putney, J.W. A Calmodulin/Inositol 1,4,5-Trisphosphate (IP3) Receptor-Binding Region Targets TRPC3 to the Plasma Membrane in a Calmodulin/IP3 Receptor-Independent Process. J. Biol. Chem. 2003, 278, 25758–25765. [Google Scholar] [CrossRef]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Lückhoff, A.; Schultz, G. Cloning and Functional Expression of a Human Ca2+-Permeable Cation Channel Activated by Calcium Store Depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. Trp, a Novel Mammalian Gene Family Essential for Agonist-Activated Capacitative Ca2+ Entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef]
- Liu, X.; Wang, W.; Singh, B.B.; Lockwich, T.; Jadlowiec, J.; O’Connell, B.; Wellner, R.; Zhu, M.X.; Ambudkar, I.S. Trp1, a Candidate Protein for the Store-Operated Ca2+ Influx Mechanism in Salivary Gland Cells. J. Biol. Chem. 2000, 275, 3403–3411. [Google Scholar] [CrossRef]
- Mehta, D.; Ahmmed, G.U.; Paria, B.C.; Holinstat, M.; Voyno-Yasenetskaya, T.; Tiruppathi, C.; Minshall, R.D.; Malik, A.B. RhoA Interaction with Inositol 1,4,5-Trisphosphate Receptor and Transient Receptor Potential Channel-1 Regulates Ca2+ Entry: Role in signaling increased endothelial permeability. J. Biol. Chem. 2003, 278, 33492–33500. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Koenig, S.; Bernheim, L.; Frieden, M. During Post-Natal Human Myogenesis, Normal Myotube Size Requires TRPC1- and TRPC4-Mediated Ca2+ Entry. J. Cell Sci. 2013, 126, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Lamiche, C.; Vandebrouck, A.; Magaud, C.; Rivet, J.; Cognard, C.; Bourmeyster, N.; Constantin, B. Regulation of TRPC1 and TRPC4 Cation Channels Requires an Alpha1-Syntrophin-Dependent Complex in Skeletal Mouse Myotubes. J. Biol. Chem. 2009, 284, 36248–36261. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Sabourin, J.; Saüc, S.; Bernheim, L.; Koenig, S.; Frieden, M. TRPC1 and TRPC4 Channels Functionally Interact with STIM1L to Promote Myogenesis and Maintain Fast Repetitive Ca2+ Release in Human Myotubes. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 806–813. [Google Scholar] [CrossRef]
- Brownlow, S.L.; Harper, A.G.S.; Harper, M.T.; Sage, S.O. A Role for HTRPC1 and Lipid Raft Domains in Store-Mediated Calcium Entry in Human Platelets. Cell Calcium 2004, 35, 107–113. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, K.T.; Bandyopadhyay, B.C.; Pani, B.; Dietrich, A.; Paria, B.C.; Swaim, W.D.; Beech, D.; Yildrim, E.; Singh, B.B.; et al. Attenuation of Store-Operated Ca2+ Current Impairs Salivary Gland Fluid Secretion in TRPC1(−/−) Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 17542–17547. [Google Scholar] [CrossRef]
- Hong, J.H.; Li, Q.; Kim, M.S.; Shin, D.M.; Feske, S.; Birnbaumer, L.; Cheng, K.T.; Ambudkar, I.S.; Muallem, S. Polarized but Differential Localization and Recruitment of STIM1/Orai1 and STIM1/TRPC Channels in Secretory Cells. Traffic 2011, 12, 232–245. [Google Scholar] [CrossRef]
- Ma, X.; Cheng, K.-T.; Wong, C.-O.; O’Neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 Channels Contribute to Store-Operated Ca2+ Entry in Vascular Endothelial Cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Trebak, M.; Bird, G.S.J.; Putney, J.W. Expression Level of the Canonical Transient Receptor Potential 3 (TRPC3) Channel Determines Its Mechanism of Activation. J. Biol. Chem. 2003, 278, 21649–21654. [Google Scholar] [CrossRef]
- Wu, X.; Zagranichnaya, T.K.; Gurda, G.T.; Eves, E.M.; Villereal, M.L. A TRPC1/TRPC3-Mediated Increase in Store-Operated Calcium Entry Is Required for Differentiation of H19-7 Hippocampal Neuronal Cells. J. Biol. Chem. 2004, 279, 43392–43402. [Google Scholar] [CrossRef]
- Antigny, F.; Jousset, H.; König, S.; Frieden, M. Thapsigargin Activates Ca2+ Entry Both by Store-Dependent, STIM1/Orai1-Mediated, and Store-Independent, TRPC3/PLC/PKC-Mediated Pathways in Human Endothelial Cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 Heteromultimerizes TRPC Channels to Determine Their Function as Store-Operated Channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Hong, J.H.; Li, Q.; Shin, D.M.; Abramowitz, J.; Birnbaumer, L.; Muallem, S. Deletion of TRPC3 in Mice Reduces Store-Operated Ca2+ Influx and the Severity of Acute Pancreatitis. Gastroenterology 2009, 137, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, K.P.; Yang, D.; Shin, D.M.; Abramowitz, J.; Kiyonaka, S.; Birnbaumer, L.; Mori, Y.; Muallem, S. Genetic and Pharmacological Inhibition of the Ca2+ Influx Channel TRPC3 Protects Secretory Epithelia from Ca2+-Dependent Toxicity. Gastroenterology 2011, 140, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Akaike, A.; Satoh, M.; Kaneko, S. Positive Regulation of Capacitative Ca2+ Entry by Intracellular Ca2+ in Xenopus Oocytes Expressing Rat TRP4. Cell Calcium 2000, 28, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Trost, C.; Warnat, J.; Rautmann, J.; Himmerkus, N.; Schroth, G.; Kretz, O.; Nastainczyk, W.; Cavalie, A.; Hoth, M.; et al. TRP4 (CCE1) Protein Is Part of Native Calcium Release-Activated Ca2+-like Channels in Adrenal Cells. J. Biol. Chem. 2000, 275, 23965–23972. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mergler, S.; Sun, X.; Wang, Z.; Lu, L.; Bonanno, J.A.; Pleyer, U.; Reinach, P.S. TRPC4 Knockdown Suppresses Epidermal Growth Factor-Induced Store-Operated Channel Activation and Growth in Human Corneal Epithelial Cells. J. Biol. Chem. 2005, 280, 32230–32237. [Google Scholar] [CrossRef] [PubMed]
- Fatherazi, S.; Presland, R.B.; Belton, C.M.; Goodwin, P.; Al-Qutub, M.; Trbic, Z.; Macdonald, G.; Schubert, M.M.; Izutsu, K.T. Evidence That TRPC4 Supports the Calcium Selective I(CRAC)-like Current in Human Gingival Keratinocytes. Pflug. Arch. 2007, 453, 879–889. [Google Scholar] [CrossRef]
- Sabourin, J.; Bartoli, F.; Antigny, F.; Gomez, A.M.; Benitah, J.-P. Transient Receptor Potential Canonical (TRPC)/Orai1-Dependent Store-Operated Ca2+ Channels: New targets of aldosterone in cardiomyocytes. J. Biol. Chem. 2016, 291, 13394–13409. [Google Scholar] [CrossRef]
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca2+ Sensor Stromal Interaction Molecule 1 (STIM1) Is Necessary and Sufficient for the Store-Operated Ca2+ Entry Function of Transient Receptor Potential Canonical (TRPC) 1 and 4 Channels in Endothelial Cells. Mol. Pharmacol. 2012, 81, 510–526. [Google Scholar] [CrossRef]
- Freichel, M.; Suh, S.H.; Pfeifer, A.; Schweig, U.; Trost, C.; Weißgerber, P.; Biel, M.; Philipp, S.; Freise, D.; Droogmans, G.; et al. Lack of an Endothelial Store-Operated Ca2+ Current Impairs Agonist-Dependent Vasorelaxation in TRP4−/− Mice. Nat. Cell Biol. 2001, 3, 121–127. [Google Scholar] [CrossRef]
- Tiruppathi, C.; Freichel, M.; Vogel, S.M.; Paria, B.C.; Mehta, D.; Flockerzi, V.; Malik, A.B. Impairment of Store-Operated Ca2+ Entry in TRPC4−/− Mice Interferes With Increase in Lung Microvascular Permeability. Circ. Res. 2002, 91, 70–76. [Google Scholar] [CrossRef]
- Xu, S.-Z.; Boulay, G.; Flemming, R.; Beech, D.J. E3-Targeted Anti-TRPC5 Antibody Inhibits Store-Operated Calcium Entry in Freshly Isolated Pial Arterioles. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H2653–H2659. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Rodríguez, A.; Mayoral-Gonzalez, I.; Avila-Medina, J.; de Rojas-de Pedro, E.S.; Calderón-Sánchez, E.; Díaz, I.; Hmadcha, A.; Castellano, A.; Rosado, J.A.; Benitah, J.-P.; et al. Urocortin-2 Prevents Dysregulation of Ca2+ Homeostasis and Improves Early Cardiac Remodeling After Ischemia and Reperfusion. Front. Physiol. 2018, 9, 813. [Google Scholar] [CrossRef] [PubMed]
- Bréchard, S.; Melchior, C.; Plançon, S.; Schenten, V.; Tschirhart, E.J. Store-Operated Ca2+ Channels Formed by TRPC1, TRPC6 and Orai1 and Non-Store-Operated Channels Formed by TRPC3 Are Involved in the Regulation of NADPH Oxidase in HL-60 Granulocytes. Cell Calcium 2008, 44, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Jardín, I.; Redondo, P.C.; Salido, G.M.; Rosado, J.A. Phosphatidylinositol 4,5-Bisphosphate Enhances Store-Operated Calcium Entry through HTRPC6 Channel in Human Platelets. Biochim. Biophys. Acta 2008, 1783, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Jardin, I.; Gómez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic Interaction of HTRPC6 with the Orai1-STIM1 Complex or HTRPC3 Mediates Its Role in Capacitative or Non-Capacitative Ca2+ Entry Pathways. Biochem. J. 2009, 420, 267–276. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Galan, C.; Dionisio, N.; Gomez, L.J.; Salido, G.M.; Rosado, J.A. Capacitative and Non-Capacitative Signaling Complexes in Human Platelets. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 1242–1251. [Google Scholar] [CrossRef]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative Calcium Entry and Transient Receptor Potential Canonical 6 Expression Control Human Hepatoma Cell Proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef]
- DeHaven, W.I.; Jones, B.F.; Petranka, J.G.; Smyth, J.T.; Tomita, T.; Bird, G.S.; Putney, J.W. TRPC Channels Function Independently of STIM1 and Orai1. J. Physiol. 2009, 587, 2275–2298. [Google Scholar] [CrossRef]
- Zhang, X.; Trebak, M. Transient Receptor Potential Canonical 7 (TRPC7): A Diacylglycerol-Activated Non-Selective Cation Channel. Handb. Exp. Pharmacol. 2014, 222, 189–204. [Google Scholar] [CrossRef]
- Lemonnier, L.; Trebak, M.; Putney, J.W. Complex Regulation of the TRPC3,6,7 Channel Subfamily by Diacylglycerol and Phosphatidylinositol 4,5-Bisphosphate. Cell Calcium 2008, 43, 506–514. [Google Scholar] [CrossRef]
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 Forms the Stretch-Activated Cation Channel in Vertebrate Cells. Nat. Cell. Biol. 2005, 7, 179–185. [Google Scholar] [CrossRef]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a Human Homolog of a Drosophila Store-Operated Channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar] [CrossRef] [PubMed]
- Lucas, P.; Ukhanov, K.; Leinders-Zufall, T.; Zufall, F. A Diacylglycerol-Gated Cation Channel in Vomeronasal Neuron Dendrites Is Impaired in TRPC2 Mutant Mice: Mechanism of Pheromone Transduction. Neuron 2003, 40, 551–561. [Google Scholar] [CrossRef]
- Zitt, C.; Obukhov, A.G.; Strübing, C.; Zobel, A.; Kalkbrenner, F.; Lückhoff, A.; Schultz, G. Expression of TRPC3 in Chinese Hamster Ovary Cells Results in Calcium-Activated Cation Currents Not Related to Store Depletion. J. Cell Biol. 1997, 138, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct Activation of Human TRPC6 and TRPC3 Channels by Diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Kamouchi, M.; Philipp, S.; Flockerzi, V.; Wissenbach, U.; Mamin, A.; Raeymaekers, L.; Eggermont, J.; Droogmans, G.; Nilius, B. Properties of Heterologously Expressed HTRP3 Channels in Bovine Pulmonary Artery Endothelial Cells. J. Physiol. 1999, 518, 345–358. [Google Scholar] [CrossRef]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-Mediated Regulation of the Nonselective Cation Channels TRPC4 and TRPC5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar] [CrossRef]
- Kew, J.N.C.; Davies, C.H. Ion Channels: From Structure to Function; Oxford University Press: Oxford, UK, 2010; ISBN 978-0-19-929675-0. [Google Scholar]
- Feng, S. TRPC Channel Structure and Properties. Adv. Exp. Med. Biol. 2017, 976, 9–23. [Google Scholar] [CrossRef]
- Jung, S.; Mühle, A.; Schaefer, M.; Strotmann, R.; Schultz, G.; Plant, T.D. Lanthanides Potentiate TRPC5 Currents by an Action at Extracellular Sites Close to the Pore Mouth. J. Biol. Chem. 2003, 278, 3562–3571. [Google Scholar] [CrossRef]
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A Common Mechanism Underlies Stretch Activation and Receptor Activation of TRPC6 Channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591. [Google Scholar] [CrossRef]
- Okada, T.; Inoue, R.; Yamazaki, K.; Maeda, A.; Kurosaki, T.; Yamakuni, T.; Tanaka, I.; Shimizu, S.; Ikenaka, K.; Imoto, K.; et al. Molecular and Functional Characterization of a Novel Mouse Transient Receptor Potential Protein Homologue TRP7. J. Biol. Chem. 1999, 274, 27359–27370. [Google Scholar] [CrossRef]
- Berridge, M.J.; Irvine, R.F. Inositol Trisphosphate, a Novel Second Messenger in Cellular Signal Transduction. Nature 1984, 312, 315–321. [Google Scholar] [CrossRef]
- Streb, H.; Irvine, R.F.; Berridge, M.J.; Schulz, I. Release of Ca2+ from a Nonmitochondrial Intracellular Store in Pancreatic Acinar Cells by Inositol-1,4,5-Trisphosphate. Nature 1983, 306, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Korzeniowski, M.K.; Manjarrés, I.M.; Varnai, P.; Balla, T. Activation of STIM1-ORAI1 involves an intramolecular switching mechanism. Sci. Signal. 2010, 3, ra82. [Google Scholar] [CrossRef] [PubMed]
- Korzeniowski, M.K.; Wisniewski, E.; Baird, B.; Holowka, D.A.; Balla, T. Molecular Anatomy of the Early Events in STIM1 Activation—Oligomerization or Conformational Change? J. Cell Sci. 2017, 130, 2821–2832. [Google Scholar] [CrossRef]
- Luik, R.M.; Wang, B.; Prakriya, M.; Wu, M.M.; Lewis, R.S. Oligomerization of STIM1 Couples ER Calcium Depletion to CRAC Channel Activation. Nature 2008, 454, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Hoover, P.J.; Lewis, R.S. Stoichiometric Requirements for Trapping and Gating of Ca2+ Release-Activated Ca2+ (CRAC) Channels by Stromal Interaction Molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2011, 108, 13299–13304. [Google Scholar] [CrossRef]
- Huang, G.N.; Zeng, W.; Kim, J.Y.; Yuan, J.P.; Han, L.; Muallem, S.; Worley, P.F. STIM1 Carboxyl-Terminus Activates Native SOC, I Crac and TRPC1 Channels. Nat. Cell Biol. 2006, 8, 1003–1010. [Google Scholar] [CrossRef]
- Lee, K.P.; Yuan, J.P.; So, I.; Worley, P.F.; Muallem, S. STIM1-Dependent and STIM1-Independent Function of Transient Receptor Potential Canonical (TRPC) Channels Tunes Their Store-Operated Mode. J. Biol. Chem. 2010, 285, 38666–38673. [Google Scholar] [CrossRef]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ Entry Via Orai1 Regulates Plasma Membrane Recruitment of TRPC1 and Controls Cytosolic Ca2+ Signals Required for Specific Cell Functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef]
- de Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Fast Endocytic Recycling Determines TRPC1-STIM1 Clustering in ER-PM Junctions and Plasma Membrane Function of the Channel. Biochim. Biophys. Acta 2015, 1853, 2709–2721. [Google Scholar] [CrossRef]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Ca2+ Selectivity and Fatty Acid Specificity of the Noncapacitative, Arachidonate-Regulated Ca2+ (ARC) Channels. J. Biol. Chem. 2003, 278, 10174–10181. [Google Scholar] [CrossRef] [PubMed]
- González-Cobos, J.C.; Zhang, X.; Zhang, W.; Ruhle, B.; Motiani, R.K.; Schindl, R.; Muik, M.; Spinelli, A.M.; Bisaillon, J.M.; Shinde, A.V.; et al. Store-Independent Orai1/3 Channels Activated by Intracrine LeukotrieneC4: Role in Neointimal Hyperplasia. Circ. Res. 2013, 112, 1013–1025. [Google Scholar] [CrossRef]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. Both Orai1 and Orai3 Are Essential Components of the Arachidonate-Regulated Ca2+-Selective (ARC) Channels. J. Physiol. 2008, 586, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, W.; González-Cobos, J.C.; Jardin, I.; Romanin, C.; Matrougui, K.; Trebak, M. Complex Role of STIM1 in the Activation of Store-Independent Orai1/3 Channels. J. Gen. Physiol. 2014, 143, 345–359. [Google Scholar] [CrossRef]
- Mignen, O.; Thompson, J.L.; Shuttleworth, T.J. STIM1 Regulates Ca2+ Entry via Arachidonate-Regulated Ca2+-Selective (ARC) Channels without Store Depletion or Translocation to the Plasma Membrane. J. Physiol. 2007, 579, 703–715. [Google Scholar] [CrossRef]
- Thompson, J.L.; Shuttleworth, T.J. Molecular Basis of Activation of the Arachidonate-Regulated Ca2+ (ARC) Channel, a Store-Independent Orai Channel, by Plasma Membrane STIM1. J. Physiol. 2013, 591, 3507–3523. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and Pathobiology of Pulmonary Hypertension: State of the Art and Research Perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic Definitions and Updated Clinical Classification of Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Noordegraaf, A.V.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and Treatment of Pulmonary Arterial Hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Farber, H.W.; Miller, D.P.; Poms, A.D.; Badesch, D.B.; Frost, A.E.; Muros-Le Rouzic, E.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-Year Outcomes of Patients Enrolled in the REVEAL Registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Gomberg-Maitland, M.; Granton, J.; Lewis, M.I.; Mathai, S.C.; Rainisio, M.; Stockbridge, N.L.; Wilkins, M.R.; Zamanian, R.T.; Rubin, L.J. Clinical Trial Design and New Therapies for Pulmonary Arterial Hypertension. Eur. Respir. J. 2019, 53, 1801908. [Google Scholar] [CrossRef]
- Antigny, F.; Girardin, N.; Frieden, M. Transient Receptor Potential Canonical Channels Are Required for in Vitro Endothelial Tube Formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef]
- Antigny, F.; Mercier, O.; Humbert, M.; Sabourin, J. Excitation-Contraction Coupling and Relaxation Alteration in Right Ventricular Remodelling Caused by Pulmonary Arterial Hypertension. Arch. Cardiovasc. Dis. 2020, 113, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.-M.; Benitah, J.-P.; Perros, F.; Humbert, M.; Antigny, F. Ca2+ Handling Remodeling and STIM1L/Orai1/TRPC1/TRPC4 Upregulation in Monocrotaline-Induced Right Ventricular Hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224. [Google Scholar] [CrossRef]
- Sankhe, S.; Manousakidi, S.; Antigny, F.; Arthur Ataam, J.; Bentebbal, S.; Ruchon, Y.; Lecerf, F.; Sabourin, J.; Price, L.; Fadel, E.; et al. T-Type Ca2+ Channels Elicit pro-Proliferative and Anti-Apoptotic Responses through Impaired PP2A/Akt1 Signaling in PASMCs from Patients with Pulmonary Arterial Hypertension. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2017, 1864, 1631–1641. [Google Scholar] [CrossRef]
- Ng, L.C.; Ramduny, D.; Airey, J.A.; Singer, C.A.; Keller, P.S.; Shen, X.-M.; Tian, H.; Valencik, M.; Hume, J.R. Orai1 Interacts with STIM1 and Mediates Capacitative Ca2+ Entry in Mouse Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol.-Cell Physiol. 2010, 299, C1079–C1090. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Orai1, 2, 3 and STIM1 Promote Store-Operated Calcium Entry in Pulmonary Arterial Smooth Muscle Cells. Cell Death Discov. 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, R.A.; Wan, J.; Song, S.; Smith, K.A.; Gu, Y.; Tauseef, M.; Tang, H.; Makino, A.; Mehta, D.; Yuan, J.X.-J. Upregulated Expression of STIM2, TRPC6, and Orai2 Contributes to the Transition of Pulmonary Arterial Smooth Muscle Cells from a Contractile to Proliferative Phenotype. Am. J. Physiol.-Cell Physiol. 2015, 308, C581–C593. [Google Scholar] [CrossRef] [PubMed]
- Song, M.Y.; Makino, A.; Yuan, J.X.-J. STIM2 Contributes to Enhanced Store-Operated Ca2+ Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension. Pulm. Circ. 2011, 1, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Firth, A.L.; Smith, K.A.; Maliakal, M.V.; Yuan, J.X.-J. PDGF Enhances Store-Operated Ca2+ Entry by Upregulating STIM1/Orai1 via Activation of Akt/MTOR in Human Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol.-Cell Physiol. 2011, 302, C405–C411. [Google Scholar] [CrossRef]
- Chen, T.-X.; Xu, X.-Y.; Zhao, Z.; Zhao, F.-Y.; Gao, Y.-M.; Yan, X.-H.; Wan, Y. Hydrogen Peroxide Is a Critical Regulator of the Hypoxia-Induced Alterations of Store-Operated Ca2+ Entry into Rat Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L477–L487. [Google Scholar] [CrossRef]
- Smith, K.A.; Voiriot, G.; Tang, H.; Fraidenburg, D.R.; Song, S.; Yamamura, H.; Yamamura, A.; Guo, Q.; Wan, J.; Pohl, N.M.; et al. Notch Activation of Ca2+ Signaling in the Development of Hypoxic Pulmonary Vasoconstriction and Pulmonary Hypertension. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 355–367. [Google Scholar] [CrossRef]
- Hou, X.; Chen, J.; Luo, Y.; Liu, F.; Xu, G.; Gao, Y. Silencing of STIM1 Attenuates Hypoxia-Induced PASMCs Proliferation via Inhibition of the SOC/Ca2+/NFAT Pathway. Respir. Res. 2013, 14, 2. [Google Scholar] [CrossRef]
- Zhang, S.; Remillard, C.V.; Fantozzi, I.; Yuan, J.X.-J. ATP-Induced Mitogenesis Is Mediated by Cyclic AMP Response Element-Binding Protein-Enhanced TRPC4 Expression and Activity in Human Pulmonary Artery Smooth Muscle Cells. Am. J. Physiol.-Cell Physiol. 2004, 287, C1192–C1201. [Google Scholar] [CrossRef]
- Kunichika, N.; Yu, Y.; Remillard, C.V.; Platoshyn, O.; Zhang, S.; Yuan, J.X.-J. Overexpression of TRPC1 Enhances Pulmonary Vasoconstriction Induced by Capacitative Ca2+ Entry. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 287, L962–L969. [Google Scholar] [CrossRef]
- Golovina, V.A.; Platoshyn, O.; Bailey, C.L.; Wang, J.; Limsuwan, A.; Sweeney, M.; Rubin, L.J.; Yuan, J.X.-J. Upregulated TRP and Enhanced Capacitative Ca2+ Entry in Human Pulmonary Artery Myocytes during Proliferation. Am. J. Physiol.-Heart Circ. Physiol. 2001, 280, H746–H755. [Google Scholar] [CrossRef]
- Sweeney, M.; Yu, Y.; Platoshyn, O.; Zhang, S.; McDaniel, S.S.; Yuan, J.X.-J. Inhibition of Endogenous TRP1 Decreases Capacitative Ca2+ Entry and Attenuates Pulmonary Artery Smooth Muscle Cell Proliferation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2002, 283, L144–L155. [Google Scholar] [CrossRef]
- Lin, M.-J.; Leung, G.P.H.; Zhang, W.-M.; Yang, X.-R.; Yip, K.-P.; Tse, C.-M.; Sham, J.S.K. Chronic Hypoxia-Induced Upregulation of Store-Operated and Receptor-Operated Ca2+ Channels in Pulmonary Arterial Smooth Muscle Cells: A Novel Mechanism of Hypoxic Pulmonary Hypertension. Circ. Res. 2004, 95, 496–505. [Google Scholar] [CrossRef]
- Yu, Y.; Fantozzi, I.; Remillard, C.V.; Landsberg, J.W.; Kunichika, N.; Platoshyn, O.; Tigno, D.D.; Thistlethwaite, P.A.; Rubin, L.J.; Yuan, J.X.-J. Enhanced Expression of Transient Receptor Potential Channels in Idiopathic Pulmonary Arterial Hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.C.; Gurney, A.M. Store-Operated Channels Mediate Ca2+ Influx and Contraction in Rat Pulmonary Artery. Circ. Res. 2001, 89, 923–929. [Google Scholar] [CrossRef]
- Yu, Y.; Sweeney, M.; Zhang, S.; Platoshyn, O.; Landsberg, J.; Rothman, A.; Yuan, J.X.-J. PDGF Stimulates Pulmonary Vascular Smooth Muscle Cell Proliferation by Upregulating TRPC6 Expression. Am. J. Physiol.-Cell Physiol. 2003, 284, C316–C330. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, S.S.; Platoshyn, O.; Wang, J.; Yu, Y.; Sweeney, M.; Krick, S.; Rubin, L.J.; Yuan, J.X.-J. Capacitative Ca2+ Entry in Agonist-Induced Pulmonary Vasoconstriction. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2001, 280, L870–L880. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shimoda, L.A.; Sylvester, J.T. Capacitative Calcium Entry and TRPC Channel Proteins Are Expressed in Rat Distal Pulmonary Arterial Smooth Muscle. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L848–L858. [Google Scholar] [CrossRef]
- Ng, L.C.; O’Neill, K.G.; French, D.; Airey, J.A.; Singer, C.A.; Tian, H.; Shen, X.-M.; Hume, J.R. TRPC1 and Orai1 Interact with STIM1 and Mediate Capacitative Ca2+ Entry Caused by Acute Hypoxia in Mouse Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol.-Cell Physiol. 2012, 303, C1156–C1172. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, X.-R.; Fu, Z.; Paudel, O.; Abramowitz, J.; Birnbaumer, L.; Sham, J.S.K. TRPC1 and TRPC6 Contribute to Hypoxic Pulmonary Hypertension through Differential Regulation of Pulmonary Vascular Functions RR. Hypertension 2014, 63, 173–180. [Google Scholar] [CrossRef]
- Malczyk, M.; Veith, C.; Fuchs, B.; Hofmann, K.; Storch, U.; Schermuly, R.T.; Witzenrath, M.; Ahlbrecht, K.; Fecher-Trost, C.; Flockerzi, V.; et al. Classical Transient Receptor Potential Channel 1 in Hypoxia-Induced Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2013, 188, 1451–1459. [Google Scholar] [CrossRef]
- Lu, W.; Ran, P.; Zhang, D.; Lai, N.; Zhong, N.; Wang, J. Bone Morphogenetic Protein 4 Enhances Canonical Transient Receptor Potential Expression, Store-Operated Ca2+ Entry, and Basal [Ca2+]i in Rat Distal Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol.-Cell Physiol. 2010, 299, C1370–C1378. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, Z.; Su, D. BMP4 Protects Rat Pulmonary Arterial Smooth Muscle Cells from Apoptosis by PI3K/AKT/Smad1/5/8 Signaling. Int. J. Mol. Sci. 2014, 15, 13738–13754. [Google Scholar] [CrossRef]
- Weissmann, N.; Dietrich, A.; Fuchs, B.; Kalwa, H.; Ay, M.; Dumitrascu, R.; Olschewski, A.; Storch, U.; Schnitzler, M.M.Y.; Ghofrani, A.; et al. Classical Transient Receptor Potential Channel 6 (TRPC6) Is Essential for Hypoxic Pulmonary Vasoconstriction and Alveolar Gas Exchange. Proc. Natl. Acad. Sci. USA 2006, 103, 19093–19098. [Google Scholar] [CrossRef]
- Wang, C.; Li, J.-F.; Zhao, L.; Liu, J.; Wan, J.; Wang, Y.X.; Wang, J.; Wang, C. Inhibition of SOC/Ca2+/NFAT Pathway Is Involved in the Anti-Proliferative Effect of Sildenafil on Pulmonary Artery Smooth Muscle Cells. Respir. Res. 2009, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Ingueneau, C.; Huynh-Do, U.; Marcheix, B.; Athias, A.; Gambert, P.; Nègre-Salvayre, A.; Salvayre, R.; Vindis, C. TRPC1 Is Regulated by Caveolin-1 and Is Involved in Oxidized LDL-induced Apoptosis of Vascular Smooth Muscle Cells. J. Cell. Mol. Med. 2009, 13, 1620–1631. [Google Scholar] [CrossRef]
- Ng, L.C.; Airey, J.A.; Hume, J.R. The Contribution of TRPC1 and STIM1 to Capacitative Ca2+ Entry in Pulmonary Artery. Adv. Exp. Med. Biol. 2010, 661, 123–135. [Google Scholar] [CrossRef]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.-Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 Channel in Vascular Injury In Vivo and Its Role in Human Neointimal Hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Watanabe, H.; Murakami, M.; Ono, K.; Munehisa, Y.; Koyama, T.; Nobori, K.; Iijima, T.; Ito, H. Functional Role of Stromal Interaction Molecule 1 (STIM1) in Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2007, 361, 934–940. [Google Scholar] [CrossRef]
- Li, J.; McKeown, L.; Ojelabi, O.; Stacey, M.; Foster, R.; O’Regan, D.; Porter, K.E.; Beech, D.J. Nanomolar Potency and Selectivity of a Ca2+ Release-Activated Ca2+ Channel Inhibitor against Store-Operated Ca2+ Entry and Migration of Vascular Smooth Muscle Cells. Br. J. Pharmacol. 2011, 164, 382–393. [Google Scholar] [CrossRef]
- Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Abdullaev, I.F.; Bisaillon, J.M.; Singer, H.A.; Trebak, M. Evidence for STIM1- and Orai1-Dependent Store-Operated Calcium Influx through ICRAC in Vascular Smooth Muscle Cells: Role in Proliferation and Migration. FASEB J. 2009, 23, 2425–2437. [Google Scholar] [CrossRef]
- Guo, R.; Yang, L.; Li, M.; Pan, X.; Liu, B.; Deng, Y. Stim1- and Orai1-Mediated Store-Operated Calcium Entry Is Critical for Angiotensin II-Induced Vascular Smooth Muscle Cell Proliferation. Cardiovasc. Res. 2012, 93, 360–370. [Google Scholar] [CrossRef]
- Rodríguez-Moyano, M.; Díaz, I.; Dionisio, N.; Zhang, X.; Ávila-Medina, J.; Calderón-Sánchez, E.; Trebak, M.; Rosado, J.A.; Ordóñez, A.; Smani, T. Urotensin-II Promotes Vascular Smooth Muscle Cell Proliferation through Store-Operated Calcium Entry and EGFR Transactivation. Cardiovasc. Res. 2013, 100, 297–306. [Google Scholar] [CrossRef]
- Bisaillon, J.M.; Motiani, R.K.; Gonzalez-Cobos, J.C.; Potier, M.; Halligan, K.E.; Alzawahra, W.F.; Barroso, M.; Singer, H.A.; Jourd’heuil, D.; Trebak, M. Essential Role for STIM1/Orai1-Mediated Calcium Influx in PDGF-Induced Smooth Muscle Migration. Am. J. Physiol.-Cell Physiol. 2010, 298, C993–C1005. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Stevenson, R.J.; Zhang, X.; Meizoso-Huesca, A.; Xin, P.; Johnson, M.; Flanagan, J.U.; Chalmers, S.B.; Yoast, R.E.; Kapure, J.S.; et al. A New Selective Pharmacological Enhancer of the Orai1 Ca2+ Channel Reveals Roles for Orai1 in Smooth and Skeletal Muscle Functions. ACS Pharmacol. Transl. Sci. 2020, 3, 135–147. [Google Scholar] [CrossRef]
- Yang, H.; Chen, X.-Y.; Kuang, S.-J.; Zhou, M.-Y.; Zhang, L.; Zeng, Z.; Liu, L.; Wu, F.-L.; Zhang, M.-Z.; Mai, L.-P.; et al. Abnormal Ca2+ Handling Contributes to the Impairment of Aortic Smooth Muscle Contractility in Zucker Diabetic Fatty Rats. J. Mol. Cell. Cardiol. 2020, 141, 82–92. [Google Scholar] [CrossRef]
- Trebak, M. STIM/Orai Signalling Complexes in Vascular Smooth Muscle. J. Physiol. 2012, 590, 4201–4208. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordoñez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef]
- Kwan, H.-Y.; Shen, B.; Ma, X.; Kwok, Y.-C.; Huang, Y.; Man, Y.-B.; Yu, S.; Yao, X. TRPC1 Associates With BKCa Channel to Form a Signal Complex in Vascular Smooth Muscle Cells. Circ. Res. 2009, 104, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Patel, H.H.; Murray, F.; Remillard, C.V.; Schach, C.; Thistlethwaite, P.A.; Insel, P.A.; Yuan, J.X.-J. Pulmonary Artery Smooth Muscle Cells from Normal Subjects and IPAH Patients Show Divergent CAMP-Mediated Effects on TRPC Expression and Capacitative Ca2+ Entry. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 292, L1202–L1210. [Google Scholar] [CrossRef]
- He, X.; Song, S.; Ayon, R.J.; Balisterieri, A.; Black, S.M.; Makino, A.; Wier, W.G.; Zang, W.-J.; Yuan, J.X.-J. Hypoxia Selectively Upregulates Cation Channels and Increases Cytosolic [Ca2+] in Pulmonary, but Not Coronary, Arterial Smooth Muscle Cells. Am. J. Physiol.-Cell Physiol. 2018, 314, C504–C517. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An Abnormal Mitochondrial–Hypoxia Inducible Factor-1α–Kv Channel Pathway Disrupts Oxygen Sensing and Triggers Pulmonary Arterial Hypertension in Fawn Hooded Rats. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Dai, Z.; Li, M.; Wharton, J.; Zhu, M.M.; Zhao, Y.-Y. Prolyl-4 Hydroxylase 2 (PHD2) Deficiency in Endothelial Cells and Hematopoietic Cells Induces Obliterative Vascular Remodeling and Severe Pulmonary Arterial Hypertension in Mice and Humans Through Hypoxia-Inducible Factor-2α. Circulation 2016, 133, 2447–2458. [Google Scholar] [CrossRef]
- Lei, W.; He, Y.; Shui, X.; Li, G.; Yan, G.; Zhang, Y.; Huang, S.; Chen, C.; Ding, Y. Expression and Analyses of the HIF-1 Pathway in the Lungs of Humans with Pulmonary Arterial Hypertension. Mol. Med. Rep. 2016, 14, 4383–4390. [Google Scholar] [CrossRef]
- Chen, J.; Sysol, J.R.; Singla, S.; Zhao, S.; Yamamura, A.; Valdez-Jasso, D.; Abbasi, T.; Shioura, K.M.; Sahni, S.; Reddy, V.; et al. Nicotinamide Phosphoribosyltransferase Promotes Pulmonary Vascular Remodeling and Is a Therapeutic Target in Pulmonary Arterial Hypertension. Circulation 2017, 135, 1532–1546. [Google Scholar] [CrossRef]
- Lu, W.; Wang, J.; Shimoda, L.A.; Sylvester, J.T. Differences in STIM1 and TRPC Expression in Proximal and Distal Pulmonary Arterial Smooth Muscle Are Associated with Differences in Ca2+ Responses to Hypoxia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 295, L104–L113. [Google Scholar] [CrossRef]
- Giachini, F.R.C.; Chiao, C.-W.; Carneiro, F.S.; Lima, V.V.; Carneiro, Z.N.; Dorrance, A.M.; Tostes, R.C.; Webb, R.C. CHBPR: Increased Activation of STIM-1/Orai-1 in Aorta from Hypertensive Rats: A Novel Insight into Vascular Dysfunction. Hypertension 2009, 53, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Galán, S.; Arenas, G.A.; Reyes, R.V.; Krause, B.J.; Iturriaga, R. Stim-Activated TRPC-ORAI Channels in Pulmonary Hypertension Induced by Chronic Intermittent Hypoxia. Pulm. Circ. 2020, 10, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Galan, S.; Arenas, G.; Krause, B.; Iturriaga, R. Late Breaking Abstract—Contribution of STIM-Activated TRPC-ORAI Channels to the Intermittent Hypoxia-Induced Pulmonary Hypertension. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Perros, F.; Montani, D.; Dorfmüller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-Derived Growth Factor Expression and Function in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of Experimental Pulmonary Hypertension by PDGF Inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, V.; Le Cras, T.D.; Ivy, D.D.; Grover, T.R.; Kinsella, J.P.; Abman, S.H. Role of Platelet-Derived Growth Factor in Vascular Remodeling during Pulmonary Hypertension in the Ovine Fetus. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 284, L826–L833. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-K.; Zhen, Y.-Y.; Lu, H.-I.; Sung, P.-H.; Chang, L.-T.; Tsai, T.-H.; Sheu, J.-J.; Chen, Y.-L.; Chua, S.; Chang, H.-W.; et al. Reducing TRPC1 Expression through Liposome-Mediated SiRNA Delivery Markedly Attenuates Hypoxia-Induced Pulmonary Arterial Hypertension in a Murine Model. Stem Cells Int. 2014, 2014, e316214. [Google Scholar] [CrossRef]
- Liu, X.-R.; Zhang, M.-F.; Yang, N.; Liu, Q.; Wang, R.-X.; Cao, Y.-N.; Yang, X.-R.; Sham, J.S.K.; Lin, M.-J. Enhanced Store-Operated Ca2+ Entry and TRPC Channel Expression in Pulmonary Arteries of Monocrotaline-Induced Pulmonary Hypertensive Rats. Am. J. Physiol.-Cell Physiol. 2012, 302, C77–C87. [Google Scholar] [CrossRef]
- Alzoubi, A.; Almalouf, P.; Toba, M.; O’Neill, K.; Qian, X.; Francis, M.; Taylor, M.S.; Alexeyev, M.; McMurtry, I.F.; Oka, M.; et al. TRPC4 Inactivation Confers a Survival Benefit in Severe Pulmonary Arterial Hypertension. Am. J. Pathol. 2013, 183, 1779–1788. [Google Scholar] [CrossRef]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.-Y.; et al. A Functional Single-Nucleotide Polymorphism in the TRPC6 Gene Promoter Associated With Idiopathic Pulmonary Arterial Hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Pousada, G.; Baloira, A.; Valverde, D. Molecular and Clinical Analysis of TRPC6 and AGTR1 Genes in Patients with Pulmonary Arterial Hypertension. Orphanet J. Rare Dis. 2015, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Linde, C.I.; Karashima, E.; Raina, H.; Zulian, A.; Wier, W.G.; Hamlyn, J.M.; Ferrari, P.; Blaustein, M.P.; Golovina, V.A. Increased Arterial Smooth Muscle Ca2+ Signaling, Vasoconstriction, and Myogenic Reactivity in Milan Hypertensive Rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H611–H620. [Google Scholar] [CrossRef]
- Jain, P.P.; Lai, N.; Xiong, M.; Chen, J.; Babicheva, A.; Zhao, T.; Parmisano, S.; Zhao, M.; Paquin, C.; Matti, M.; et al. TRPC6, a Therapeutic Target for Pulmonary Hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021. [Google Scholar] [CrossRef]
- Deng, Z.; Morse, J.H.; Slager, S.L.; Cuervo, N.; Moore, K.J.; Venetos, G.; Kalachikov, S.; Cayanis, E.; Fischer, S.G.; Barst, R.J.; et al. Familial Primary Pulmonary Hypertension (Gene PPH1) Is Caused by Mutations in the Bone Morphogenetic Protein Receptor-II Gene. Am. J. Hum. Genet. 2000, 67, 737–744. [Google Scholar] [CrossRef]
- Evans, J.D.W.; Girerd, B.; Montani, D.; Wang, X.-J.; Galiè, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grünig, E.; Yan, Y.; et al. BMPR2 Mutations and Survival in Pulmonary Arterial Hypertension: An Individual Participant Data Meta-Analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary Pulmonary Hypertension Is Associated with Reduced Pulmonary Vascular Expression of Type II Bone Morphogenetic Protein Receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Long, L.; Southwood, M.; Rudarakanchana, N.; Upton, P.D.; Jeffery, T.K.; Atkinson, C.; Chen, H.; Trembath, R.C.; Morrell, N.W. Dysfunctional Smad Signaling Contributes to Abnormal Smooth Muscle Cell Proliferation in Familial Pulmonary Arterial Hypertension. Circ. Res. 2005, 96, 1053–1063. [Google Scholar] [CrossRef]
- Yang, J.; Li, X.; Li, Y.; Southwood, M.; Ye, L.; Long, L.; Al-Lamki, R.S.; Morrell, N.W. Id Proteins Are Critical Downstream Effectors of BMP Signaling in Human Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L312–L321. [Google Scholar] [CrossRef] [PubMed]
- Orriols, M.; Gomez-Puerto, M.C.; ten Dijke, P. BMP Type II Receptor as a Therapeutic Target in Pulmonary Arterial Hypertension. Cell. Mol. Life Sci. 2017, 74, 2979–2995. [Google Scholar] [CrossRef]
- Anderson, L.; Lowery, J.W.; Frank, D.B.; Novitskaya, T.; Jones, M.; Mortlock, D.P.; Chandler, R.L.; de Caestecker, M.P. Bmp2 and Bmp4 Exert Opposing Effects in Hypoxic Pulmonary Hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R833–R842. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, W.; Yang, K.; Xu, L.; Lai, N.; Tian, L.; Jiang, Q.; Duan, X.; Chen, M.; Wang, J. Bone Morphogenetic Protein 2 Decreases TRPC Expression, Store-Operated Ca2+ Entry, and Basal [Ca2+]i in Rat Distal Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2013, 304, C833–C843. [Google Scholar] [CrossRef] [PubMed]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 Mediate SOCE and Contribute to Apoptotic Resistance of Pancreatic Adenocarcinoma. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef]
- Liang, S.-J.; Zeng, D.-Y.; Mai, X.-Y.; Shang, J.-Y.; Wu, Q.-Q.; Yuan, J.-N.; Yu, B.-X.; Zhou, P.; Zhang, F.-R.; Liu, Y.-Y.; et al. Inhibition of Orai1 Store-Operated Calcium Channel Prevents Foam Cell Formation and Atherosclerosis. Arter. Thromb. Vasc. Biol. 2016, 36, 618–628. [Google Scholar] [CrossRef]
- Vacher, P.; Vacher, A.-M.; Pineau, R.; Latour, S.; Soubeyran, I.; Pangault, C.; Tarte, K.; Soubeyran, P.; Ducret, T.; Bresson-Bepoldin, L. Localized Store-Operated Calcium Influx Represses CD95-Dependent Apoptotic Effects of Rituximab in Non-Hodgkin B Lymphomas. J. Immunol. 2015, 195, 2207–2215. [Google Scholar] [CrossRef]
- Weigand, L.; Foxson, J.; Wang, J.; Shimoda, L.A.; Sylvester, J.T. Inhibition of Hypoxic Pulmonary Vasoconstriction by Antagonists of Store-Operated Ca2+ and Nonselective Cation Channels. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2005, 289, L5–L13. [Google Scholar] [CrossRef][Green Version]
- Ward, J.P.T.; Robertson, T.P.; Aaronson, P.I. Capacitative Calcium Entry: A Central Role in Hypoxic Pulmonary Vasoconstriction? Am. J. Physiol.-Lung Cell. Mol. Physiol. 2005, 289, L2–L4. [Google Scholar] [CrossRef]
- Shibata, A.; Uchida, K.; Kodo, K.; Miyauchi, T.; Mikoshiba, K.; Takahashi, T.; Yamagishi, H. Type 2 Inositol 1,4,5-Trisphosphate Receptor Inhibits the Progression of Pulmonary Arterial Hypertension via Calcium Signaling and Apoptosis. Heart Vessels 2019, 34, 724–734. [Google Scholar] [CrossRef]
- Kiselyov, K.; Mignery, G.A.; Zhu, M.X.; Muallem, S. The N-Terminal Domain of the IP3 Receptor Gates Store-Operated HTrp3 Channels. Mol. Cell 1999, 4, 423–429. [Google Scholar] [CrossRef]
- Tang, J.; Lin, Y.; Zhang, Z.; Tikunova, S.; Birnbaumer, L.; Zhu, M.X. Identification of Common Binding Sites for Calmodulin and Inositol 1,4,5-Trisphosphate Receptors on the Carboxyl Termini of Trp Channels. J. Biol. Chem. 2001, 276, 21303–21310. [Google Scholar] [CrossRef]
- Yang, Z.; Song, T.; Truong, L.; Reyes-García, J.; Wang, L.; Zheng, Y.-M.; Wang, Y.-X. Important Role of Sarcoplasmic Reticulum Ca2+ Release via Ryanodine Receptor-2 Channel in Hypoxia-Induced Rieske Iron-Sulfur Protein-Mediated Mitochondrial Reactive Oxygen Species Generation in Pulmonary Artery Smooth Muscle Cells. Antioxid. Redox Signal. 2020, 32, 447–462. [Google Scholar] [CrossRef]
- Kaßmann, M.; Szijártó, I.A.; García-Prieto, C.F.; Fan, G.; Schleifenbaum, J.; Anistan, Y.-M.; Tabeling, C.; Shi, Y.; le Noble, F.; Witzenrath, M.; et al. Role of Ryanodine Type 2 Receptors in Elementary Ca2+ Signaling in Arteries and Vascular Adaptive Responses. J. Am. Heart Assoc. 2019, 8, e010090. [Google Scholar] [CrossRef]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.-P.; Quignard, J.-F. Stretch-Induced Ca2+ Signalling in Vascular Smooth Muscle Cells Depends on Ca2+ Store Segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Dahan, D.; Ducret, T.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Estève, E. Implication of the Ryanodine Receptor in TRPV4-Induced Calcium Response in Pulmonary Arterial Smooth Muscle Cells from Normoxic and Chronically Hypoxic Rats. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 303, L824–L833. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Humbert, M.; Jaïs, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-Term Response to Calcium Channel Blockers in Idiopathic Pulmonary Arterial Hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef]
- The Effect of High Doses of Calcium-Channel Blockers on Survival in Primary Pulmonary Hypertension|NEJM. Available online: https://www.nejm.org/doi/full/10.1056/NEJM199207093270203 (accessed on 6 October 2021).
- He, L.-P.; Hewavitharana, T.; Soboloff, J.; Spassova, M.A.; Gill, D.L. A Functional Link between Store-Operated and TRPC Channels Revealed by the 3,5-Bis(Trifluoromethyl)Pyrazole Derivative, BTP2. J. Biol. Chem. 2005, 280, 10997–11006. [Google Scholar] [CrossRef]
- Takezawa, R.; Cheng, H.; Beck, A.; Ishikawa, J.; Launay, P.; Kubota, H.; Kinet, J.-P.; Fleig, A.; Yamada, T.; Penner, R. A Pyrazole Derivative Potently Inhibits Lymphocyte Ca2 Influx and Cytokine Production by Facilitating Transient Receptor Potential Melastatin 4 Channel Activity. Mol. Pharmacol. 2006, 69, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, J.; Ohga, K.; Yoshino, T.; Takezawa, R.; Ichikawa, A.; Kubota, H.; Yamada, T. A Pyrazole Derivative, YM-58483, Potently Inhibits Store-Operated Sustained Ca2+ Influx and IL-2 Production in T Lymphocytes. J. Immunol. 2003, 170, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Zitt, C.; Strauss, B.; Schwarz, E.C.; Spaeth, N.; Rast, G.; Hatzelmann, A.; Hoth, M. Potent Inhibition of Ca2+ Release-Activated Ca2+ Channels and T-Lymphocyte Activation by the Pyrazole Derivative BTP2. J. Biol. Chem. 2004, 279, 12427–12437. [Google Scholar] [CrossRef] [PubMed]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.; Kappe, C.; Romanin, C.; Groschner, K. Novel Pyrazole Compounds for Pharmacological Discrimination between Receptor-Operated and Store-Operated Ca2+ Entry Pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct Pharmacological Profiles of ORAI1, ORAI2, and ORAI3 Channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The Action of Selective CRAC Channel Blockers Is Affected by the Orai Pore Geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef]
- Shawer, H.; Norman, K.; Cheng, C.W.; Foster, R.; Beech, D.J.; Bailey, M.A. ORAI1 Ca2+ Channel as a Therapeutic Target in Pathological Vascular Remodelling. Front. Cell Dev. Biol. 2021, 9, 686. [Google Scholar] [CrossRef]
- Waldherr, L.; Tiffner, A.; Mishra, D.; Sallinger, M.; Schober, R.; Frischauf, I.; Schmidt, T.; Handl, V.; Sagmeister, P.; Köckinger, M.; et al. Blockage of Store-Operated Ca2+ Influx by Synta66 Is Mediated by Direct Inhibition of the Ca2+ Selective Orai1 Pore. Cancers 2020, 12, 2876. [Google Scholar] [CrossRef]
- Ng, S.W.; di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained Activation of the Tyrosine Kinase Syk by Antigen in Mast Cells Requires Local Ca2+ Influx through Ca2+ Release-Activated Ca2+ Channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar] [CrossRef]
- Sabatino, A.D.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A.B.; Wilde, J.I.; Scott, L.; et al. Targeting Gut T Cell Ca2+ Release-Activated Ca2+ Channels Inhibits T Cell Cytokine Production and T-Box Transcription Factor T-Bet in Inflammatory Bowel Disease. J. Immunol. 2009, 183, 3454–3462. [Google Scholar] [CrossRef]
- Bartoli, F.; Bailey, M.A.; Rode, B.; Mateo, P.; Antigny, F.; Bedouet, K.; Gerbaud, P.; Gosain, R.; Plante, J.; Norman, K.; et al. Orai1 Channel Inhibition Preserves Left Ventricular Systolic Function and Normal Ca 2+ Handling After Pressure Overload. Circulation 2020, 141, 199–216. [Google Scholar] [CrossRef]
- Trebak, M.; Earley, S. Signal Transduction and Smooth Muscle; CRC Press: Boca Raton, FL, USA, 2018; ISBN 978-1-4987-7423-9. [Google Scholar]
- Sadaghiani, A.M.; Lee, S.M.; Odegaard, J.I.; Leveson-Gower, D.B.; McPherson, O.M.; Novick, P.; Kim, M.R.; Koehler, A.N.; Negrin, R.; Dolmetsch, R.E.; et al. Identification of Orai1 Channel Inhibitors by Using Minimal Functional Domains to Screen Small Molecule Microarrays. Chem. Biol. 2014, 21, 1278–1292. [Google Scholar] [CrossRef]
- Kim, K.-D.; Srikanth, S.; Tan, Y.-V.; Yee, K.; Jew, M.; Damoiseaux, R.; Jung, M.; Shimizu, S.; An, D.; Ribalet, B.; et al. Calcium Signaling via Orai1 Is Essential for Induction of the Nuclear Orphan Receptor Pathway To Drive Th17 Differentiation. J. Immunol. 2013, 192, 110–122. [Google Scholar] [CrossRef]
- Smyth, J.T.; DeHaven, W.I.; Bird, G.S.; Putney, J.W., Jr. Ca2+-Store-Dependent and -Independent Reversal of Stim1 Localization and Function. J. Cell Sci. 2008, 121, 762–772. [Google Scholar] [CrossRef]
- Kiyonaka, S.; Kato, K.; Nishida, M.; Mio, K.; Numaga, T.; Sawaguchi, Y.; Yoshida, T.; Wakamori, M.; Mori, E.; Numata, T.; et al. Selective and Direct Inhibition of TRPC3 Channels Underlies Biological Activities of a Pyrazole Compound. Proc. Natl. Acad. Sci. USA 2009, 106, 5400–5405. [Google Scholar] [CrossRef]
- Miller, M.; Shi, J.; Zhu, Y.; Kustov, M.; Tian, J.; Stevens, A.; Wu, M.; Xu, J.; Long, S.; Yang, P.; et al. Identification of ML204, a Novel Potent Antagonist That Selectively Modulates Native TRPC4/C5 Ion Channels. J. Biol. Chem. 2011, 286, 33436–33446. [Google Scholar] [CrossRef]
- Bauer, C.C.; Minard, A.; Pickles, I.B.; Simmons, K.J.; Chuntharpursat-Bon, E.; Burnham, M.P.; Kapur, N.; Beech, D.J.; Muench, S.P.; Wright, M.H.; et al. Xanthine-Based Photoaffinity Probes Allow Assessment of Ligand Engagement by TRPC5 Channels. RSC Chem. Biol. 2020, 1, 436–448. [Google Scholar] [CrossRef]
- Just, S.; Chenard, B.L.; Ceci, A.; Strassmaier, T.; Chong, J.A.; Blair, N.T.; Gallaschun, R.J.; del Camino, D.; Cantin, S.; D’Amours, M.; et al. Treatment with HC-070, a Potent Inhibitor of TRPC4 and TRPC5, Leads to Anxiolytic and Antidepressant Effects in Mice. PLoS ONE 2018, 13, e0191225. [Google Scholar] [CrossRef]
- Rubaiy, H.N.; Ludlow, M.J.; Henrot, M.; Gaunt, H.J.; Miteva, K.; Cheung, S.Y.; Tanahashi, Y.; Hamzah, N.; Musialowski, K.E.; Blythe, N.M.; et al. Picomolar, Selective, and Subtype-Specific Small-Molecule Inhibition of TRPC1/4/5 Channels. J. Biol. Chem. 2017, 292, 8158–8173. [Google Scholar] [CrossRef]
- Minard, A.; Bauer, C.C.; Wright, D.J.; Rubaiy, H.N.; Muraki, K.; Beech, D.J.; Bon, R.S. Remarkable Progress with Small-Molecule Modulation of TRPC1/4/5 Channels: Implications for Understanding the Channels in Health and Disease. Cells 2018, 7, 52. [Google Scholar] [CrossRef]
- Yu, M.; Ledeboer, M.W.; Daniels, M.; Malojcic, G.; Tibbitts, T.T.; Coeffet-Le Gal, M.; Pan-Zhou, X.-R.; Westerling-Bui, A.; Beconi, M.; Reilly, J.F.; et al. Discovery of a Potent and Selective TRPC5 Inhibitor, Efficacious in a Focal Segmental Glomerulosclerosis Model. ACS Med. Chem. Lett. 2019, 10, 1579–1585. [Google Scholar] [CrossRef]
- Vinayagam, D.; Quentin, D.; Yu-Strzelczyk, J.; Sitsel, O.; Merino, F.; Stabrin, M.; Hofnagel, O.; Yu, M.; Ledeboer, M.W.; Nagel, G.; et al. Structural Basis of TRPC4 Regulation by Calmodulin and Pharmacological Agents. eLife 2020, 9, e60603. [Google Scholar] [CrossRef]
- Maier, T.; Follmann, M.; Hessler, G.; Kleemann, H.-W.; Hachtel, S.; Fuchs, B.; Weissmann, N.; Linz, W.; Schmidt, T.; Löhn, M.; et al. Discovery and Pharmacological Characterization of a Novel Potent Inhibitor of Diacylglycerol-Sensitive TRPC Cation Channels. Br. J. Pharmacol. 2015, 172, 3650–3660. [Google Scholar] [CrossRef]
- Merritt, J.E.; Armstrong, W.P.; Benham, C.D.; Hallam, T.J.; Jacob, R.; Jaxa-Chamiec, A.; Leigh, B.K.; McCarthy, S.A.; Moores, K.E.; Rink, T.J. SK&F 96365, a Novel Inhibitor of Receptor-Mediated Calcium Entry. Biochem. J. 1990, 271, 515–522. [Google Scholar] [CrossRef]
- Washburn, D.G.; Holt, D.A.; Dodson, J.; McAtee, J.J.; Terrell, L.R.; Barton, L.; Manns, S.; Waszkiewicz, A.; Pritchard, C.; Gillie, D.J.; et al. The Discovery of Potent Blockers of the Canonical Transient Receptor Channels, TRPC3 and TRPC6, Based on an Anilino-Thiazole Pharmacophore. Bioorg. Med. Chem. Lett. 2013, 23, 4979–4984. [Google Scholar] [CrossRef]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.; Jo, S.-H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 Blockade by Selective Small-Molecule or Genetic Deletion Inhibits Pathological Cardiac Hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In Vivo Selective Inhibition of TRPC6 by Antagonist BI 749327 Ameliorates Fibrosis and Dysfunction in Cardiac and Renal Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef]
- Häfner, S.; Burg, F.; Kannler, M.; Urban, N.; Mayer, P.; Dietrich, A.; Trauner, D.; Broichhagen, J.; Schaefer, M. A (+)-Larixol Congener with High Affinity and Subtype Selectivity toward TRPC6. ChemMedChem 2018, 13, 1028–1035. [Google Scholar] [CrossRef]
- Bai, Y.; Yu, X.; Chen, H.; Horne, D.; White, R.; Wu, X.; Lee, P.; Gu, Y.; Ghimire-Rijal, S.; Lin, D.C.-H.; et al. Structural Basis for Pharmacological Modulation of the TRPC6 Channel. eLife 2020, 9, e53311. [Google Scholar] [CrossRef]
- Wen, L.; Voronina, S.; Javed, M.A.; Awais, M.; Szatmary, P.; Latawiec, D.; Chvanov, M.; Collier, D.; Huang, W.; Barrett, J.; et al. Inhibitors of ORAI1 Prevent Cytosolic Calcium-Associated Injury of Human Pancreatic Acinar Cells and Acute Pancreatitis in 3 Mouse Models. Gastroenterology 2015, 149, 481–492. [Google Scholar] [CrossRef]
- Bruen, C.; Miller, J.; Wilburn, J.; Mackey, C.; Bollen, T.L.; Stauderman, K.; Hebbar, S. Auxora for the Treatment of Patients With Acute Pancreatitis and Accompanying Systemic Inflammatory Response Syndrome. Pancreas 2021, 50, 537–543. [Google Scholar] [CrossRef]
- Miller, J.; Bruen, C.; Schnaus, M.; Zhang, J.; Ali, S.; Lind, A.; Stoecker, Z.; Stauderman, K.; Hebbar, S. Auxora versus Standard of Care for the Treatment of Severe or Critical COVID-19 Pneumonia: Results from a Randomized Controlled Trial. Crit. Care 2020, 24, 502. [Google Scholar] [CrossRef]
- Barde, P.J.; Viswanadha, S.; Veeraraghavan, S.; Vakkalanka, S.V.; Nair, A. A First-in-Human Study to Evaluate the Safety, Tolerability and Pharmacokinetics of RP3128, an Oral Calcium Release-Activated Calcium (CRAC) Channel Modulator in Healthy Volunteers. J. Clin. Pharm. Ther. 2021, 46, 677–687. [Google Scholar] [CrossRef]
- PRCL Research Inc. A Phase 2a Study to Evaluate Safety, Tolerability, and Efficacy of PRCL-02 in Patients with Moderate to Severe Chronic Plaque Psoriasis; Clinicaltrials.gov: 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03614078 (accessed on 26 November 2021).
- Walsh, L.; Reilly, J.F.; Cornwall, C.; Gaich, G.A.; Gipson, D.S.; Heerspink, H.J.L.; Johnson, L.; Trachtman, H.; Tuttle, K.R.; Farag, Y.M.K.; et al. Safety and Efficacy of GFB-887, a TRPC5 Channel Inhibitor, in Patients With Focal Segmental Glomerulosclerosis, Treatment-Resistant Minimal Change Disease, or Diabetic Nephropathy: TRACTION-2 Trial Design. Kidney Int. Rep. 2021, 6, 2575–2584. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Song, X.; Liu, R.; Zhang, J.; Li, Z. The Structure of TRPC Ion Channels. Cell Calcium 2019, 80, 25–28. [Google Scholar] [CrossRef]
- Zhao, Y.; McVeigh, B.M.; Moiseenkova-Bell, V.Y. Structural Pharmacology of TRP Channels. J. Mol. Biol. 2021, 433, 166914. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel | Selectivity PCa/PNa | Conductance (pS) | Activation Mechanism | References |
|---|---|---|---|---|
| TRPC1 | 1 | 16 | Store depletion, GPCR–PLC pathway, membrane stretching, | [60,95,96] |
| TRPC2 | 2.7 | 42 | DAG | [97] |
| TRPC3 | 1.6 | 66 | DAG, store depletion, PKC phosphorylation, membrane stretching | [73,94,98,99,100] |
| TRPC4 | 1.1–7.7 | 42 | Store depletion, GPCR–PLC pathway | [101,102,103] |
| TRPC5 | 1.8–9.5 | 63 | Store depletion, GPCR–PLC pathway | [101,103,104] |
| TRPC6 | 5 | 35 | DAG, membrane stretching | [77,97,105] |
| TRPC7 | 1.9 | 25–50 | DAG, store depletion | [50,94,106] |
| TRPC1 | TRPC3 | TRPC4 | TRPC5 | TRPC6 | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Expression | mRNA | Protein | mRNA | Protein | mRNA | Protein | mRNA | Protein | mRNA | Protein | |
| Human | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | - | ✓ | ✓ | [141,142,143,144,145,146] |
| Rat | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | - | ✓ | ✓ | [147,148,149,150] |
| Mouse | ✓ | ✓ | ✓ | - | ✓ | ✓ | - | - | ✓ | ✓ | [133,151,152,153,154,155,156] |
| mRNA | Protein | SOCE | hPASMC | Rodents Models | Consequences | References | |
|---|---|---|---|---|---|---|---|
| STIM1 | - | ↑ | - | Hypoxia | Increased proliferation of rat PASMC Increased NFATc3 nuclear translocation | [140,173] | |
| ↑ | ↑ | - | Rat CH Distal PA | ||||
| STIM2 | - | ↑ | ↑ | PAH | Increased proliferation of iPAH hPASMC | [136,173] | |
| - | ↑ | - | Hypoxia | ||||
| Orai1 | - | ↑ | - | Hypoxia | [134,173] | ||
| ↑ | ↑ | ↑ | Rat CH PA Rat CH PASMC | ||||
| Orai2 | - | ↑ | - | Hypoxia | [134,173] | ||
| ↑ | ↑ | ↑ | Rat CH PA Rat CH PASMC | ||||
| TRPC1 | ↑ | ↑ | ↑ | Rat CH/MCT PA Rat CH PASMC | Reduced CH-induced PH phenotype and PASMC proliferation in trpc1−/− mice Pharmacological inhibition normalized vascular tone in PAs of CH-induced PH rats Increased murine PASMC proliferation | [145,152,153,185,186] | |
| ↑ | - | - | Mouse PASMC exposed to hypoxia | ||||
| TRPC3 | ↑ | ↑ | ↑ | PAH | [173] | ||
| TRPC4 | ↑ | ↑ | ↑ | Rat SU/Hx PA Rat SU/Hx PASMC | trpc4 gene deletion reduces PH | [186,187] | |
| TRPC6 | ↑ | ↑ | - | PAH | hPASMC proliferation trpc6 gene deletion reduces CH-induced PH in mice Pharmacological inhibition normalized vascular tone in PAs of CH-induced PH rats | [145,146,152,173] | |
| - | ↑ | - | Hypoxia | ||||
| ↑ | ↑ | ↑ | Rat CH PA and PASMC | ||||
| SOCE | - | - | ↑ | PAH | Increased STIM1–Orai1–TRPC1 interaction in hypoxic mouse PASMC | [151,157,172] | |
| - | - | ↑ | Mouse PASMC exposed to hypoxia |
| Compound | Mode of Action | IC50 | Side Effects | References |
|---|---|---|---|---|
| Orai1 Inhibitor | ||||
| YM-58483 (BTP2 or Pyr2) | - | 10–590 nM | Inhibits TRPC3 and -C6 (IC50: 0.3 µM) Activates TRPM4 channels (EC50: 8 nM) Inhibit Orai2 and Orai3 at 10 µM | [214,215,216,217,218,219] |
| GSK7975A and GSK-5503A | Potentially allosteric effect on the selectivity filter of Orai | 4 µM | Orai2 and Orai3 at 10 µM, L-type Ca2+ (IC50: 8 µM), and TRPV6 channels | [219,220] |
| Synta-66 | Binds TM1 and TM3 helices and the extracellular loop segments | 26 nM-3 µM | Potentiate Orai2 at 10 µM | [219,221,222,223,224] |
| JPIII | - | 244 nM | - | [225] |
| Gd3+ or La3+ | Binds extracellular loop of Orai1 | 200 nM | Inhibit Orai2/3 | [219,226] |
| AnCoA4 | Binds the C-terminus of Orai1 | 880 nM | - | [227] |
| 5J-4 | - | 10 µM | - | [228] |
| STIM1 Inhibitor | ||||
| ML-9 | Inhibit STIM1 puncta formation | 10 µM | Inhibit Myosin light chain kinase | [229] |
| TRPC3 Inhibitor | ||||
| Pyr3 | Direct binding | 0.7 μM | - | [230] |
| Pyr10 | - | 0.72 µM | - | [218] |
| TRPC4 Inhibitor | ||||
| ML-204 | - | 1–3 μM | Inhibit TRPC5 and weakly TRPC6 | [231] |
| HC-070 | Direct binding | 9.3 nM | Inhibit TRPC4 (IC50:46 nM) and TRPC3 (IC50: 1 µM) | [232,233] |
| HC-608 (Pico145) | - | 0.35 nM | Inhibit TRPC5 (IC50: 1.3 nM), TRPC1-4 complex (IC50: 0.03 nM) and TRPC1-5 complex (IC50: 0.2 nM) | [234] |
| TRPC5 Inhibitor | ||||
| AC1903 | - | 13.6 μM | Inhibit TRPC4 (IC50 > 100 µM) | [235] |
| GFB-8438 | Direct binding | 0.18 µM | Inhibit TRPC4 (IC50: 0.29 µM) | [236,237] |
| TRPC6 Inhibitor | ||||
| SAR7334 | - | 9.5 nM | Inhibit TRPC3 (IC50: 282 nM) and TRPC7 (IC50: 226 nM) | [238] |
| SKF-96365 | - | 10 µM | - | [239] |
| GSK2833503 | - | 3 nM | Inhibit TRPC3 (IC50: 21 nM) | [240,241] |
| BI 749327 | - | 19 nM | - | [242] |
| SH045 | - | 7.9 nM | Inhibit TRPC3 (IC50: 282 nM) and TRPC7 (IC50: 226 nM) | [243] |
| AM-1473 | Direct binding | 0.22 nM | - | [244] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masson, B.; Montani, D.; Humbert, M.; Capuano, V.; Antigny, F. Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension. Biomolecules 2021, 11, 1781. https://doi.org/10.3390/biom11121781
Masson B, Montani D, Humbert M, Capuano V, Antigny F. Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension. Biomolecules. 2021; 11(12):1781. https://doi.org/10.3390/biom11121781
Chicago/Turabian StyleMasson, Bastien, David Montani, Marc Humbert, Véronique Capuano, and Fabrice Antigny. 2021. "Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension" Biomolecules 11, no. 12: 1781. https://doi.org/10.3390/biom11121781
APA StyleMasson, B., Montani, D., Humbert, M., Capuano, V., & Antigny, F. (2021). Role of Store-Operated Ca2+ Entry in the Pulmonary Vascular Remodeling Occurring in Pulmonary Arterial Hypertension. Biomolecules, 11(12), 1781. https://doi.org/10.3390/biom11121781