
No Effect of Hypercholesterolemia on Elastase-Induced Experimental Abdominal Aortic Aneurysm Progression

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Induction of Hypercholesterolemia and Definition of Model-Specific Control Mice
2.2. Experimental AAA Creation
2.3. Imaging AAA Formation and Progression
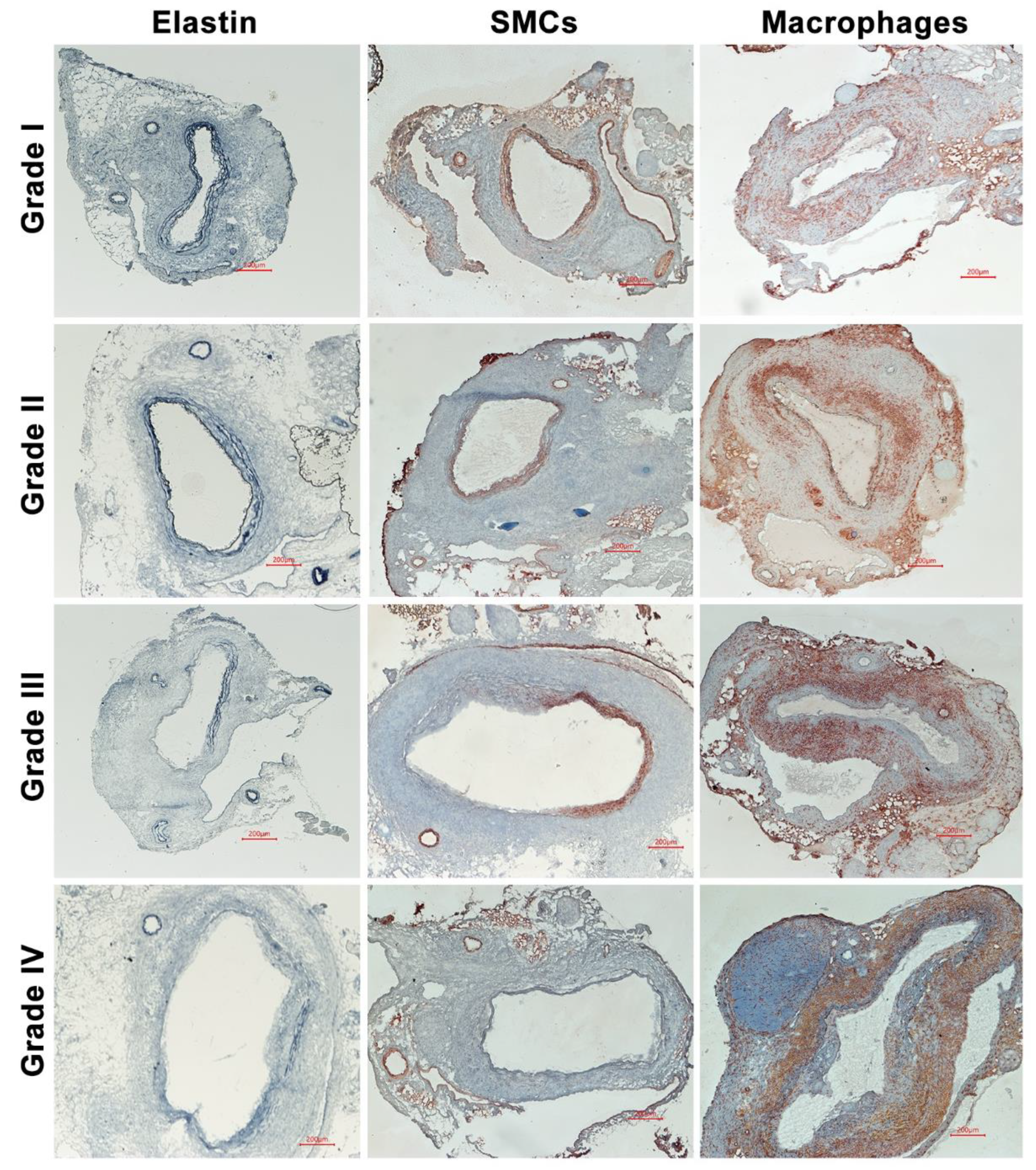
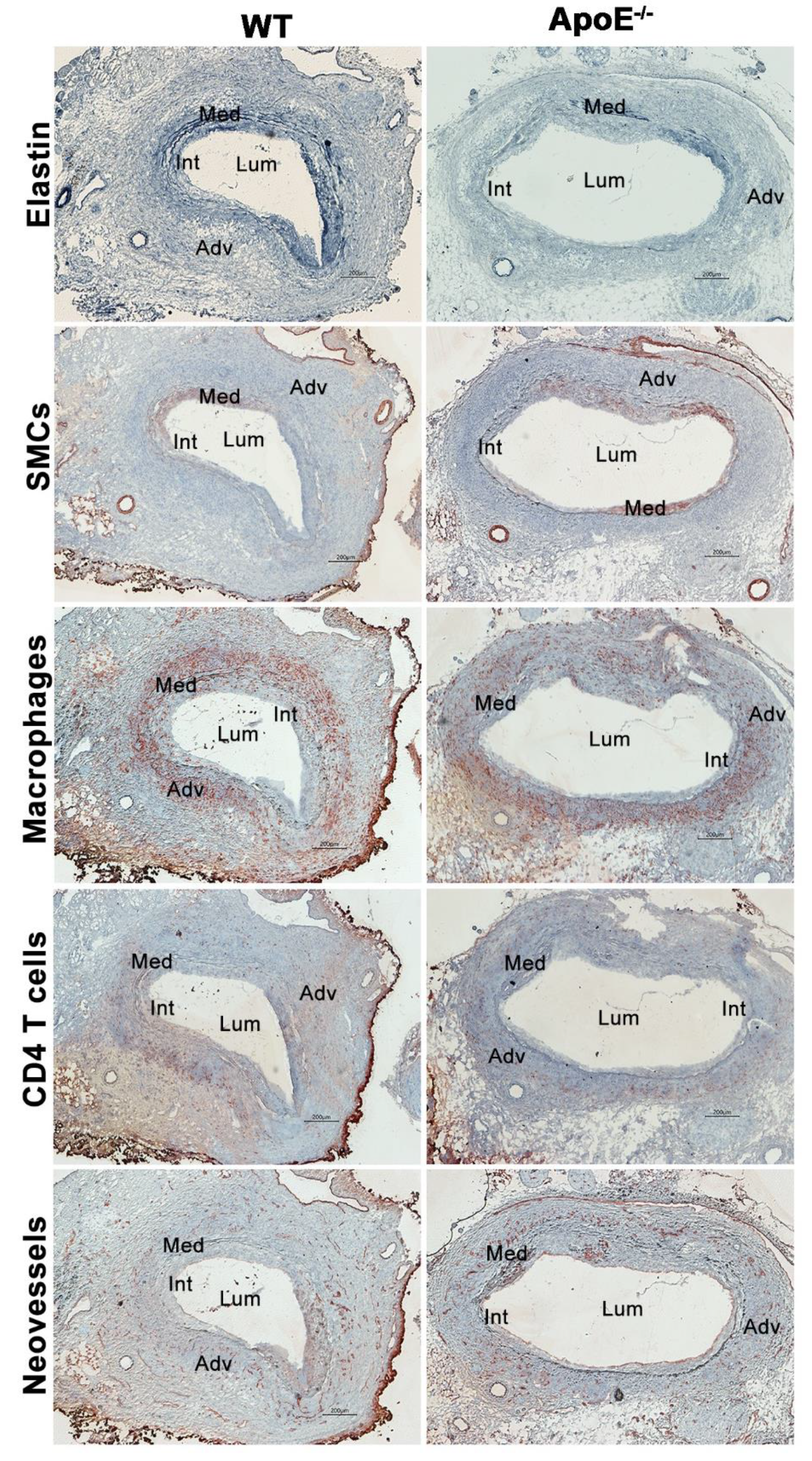
2.4. Histological Analyses
2.5. Statistical Analyses
3. Result
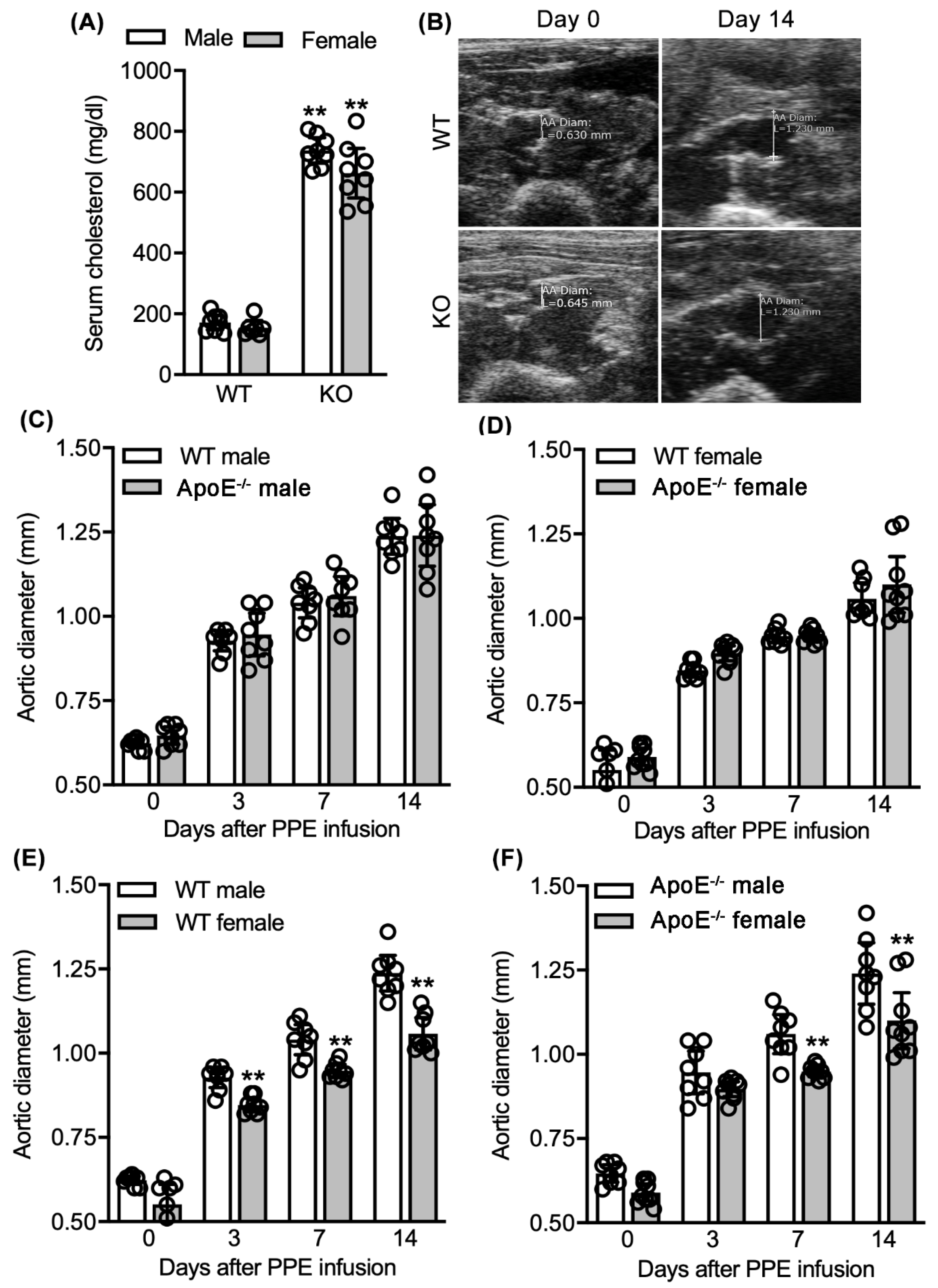
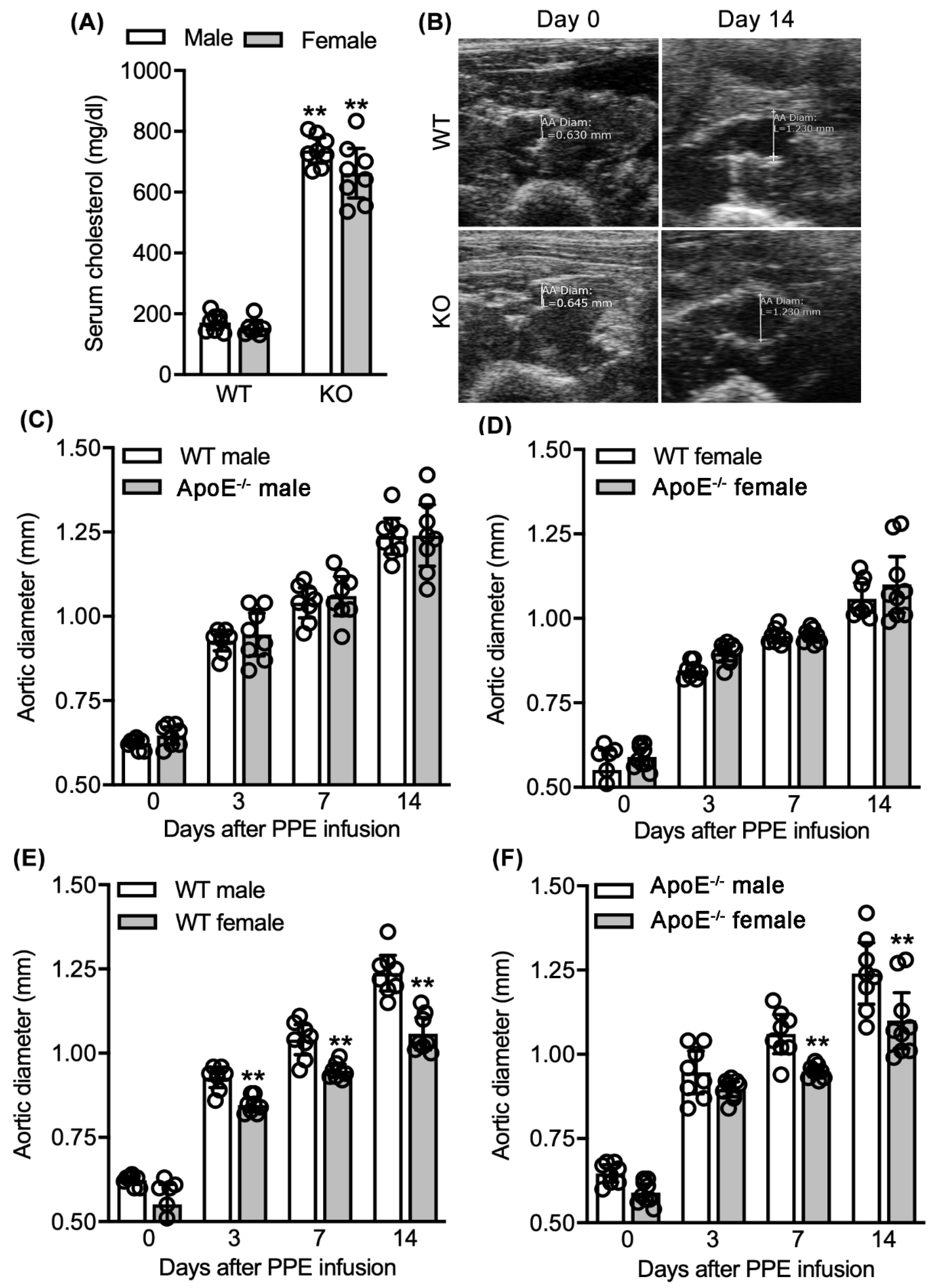
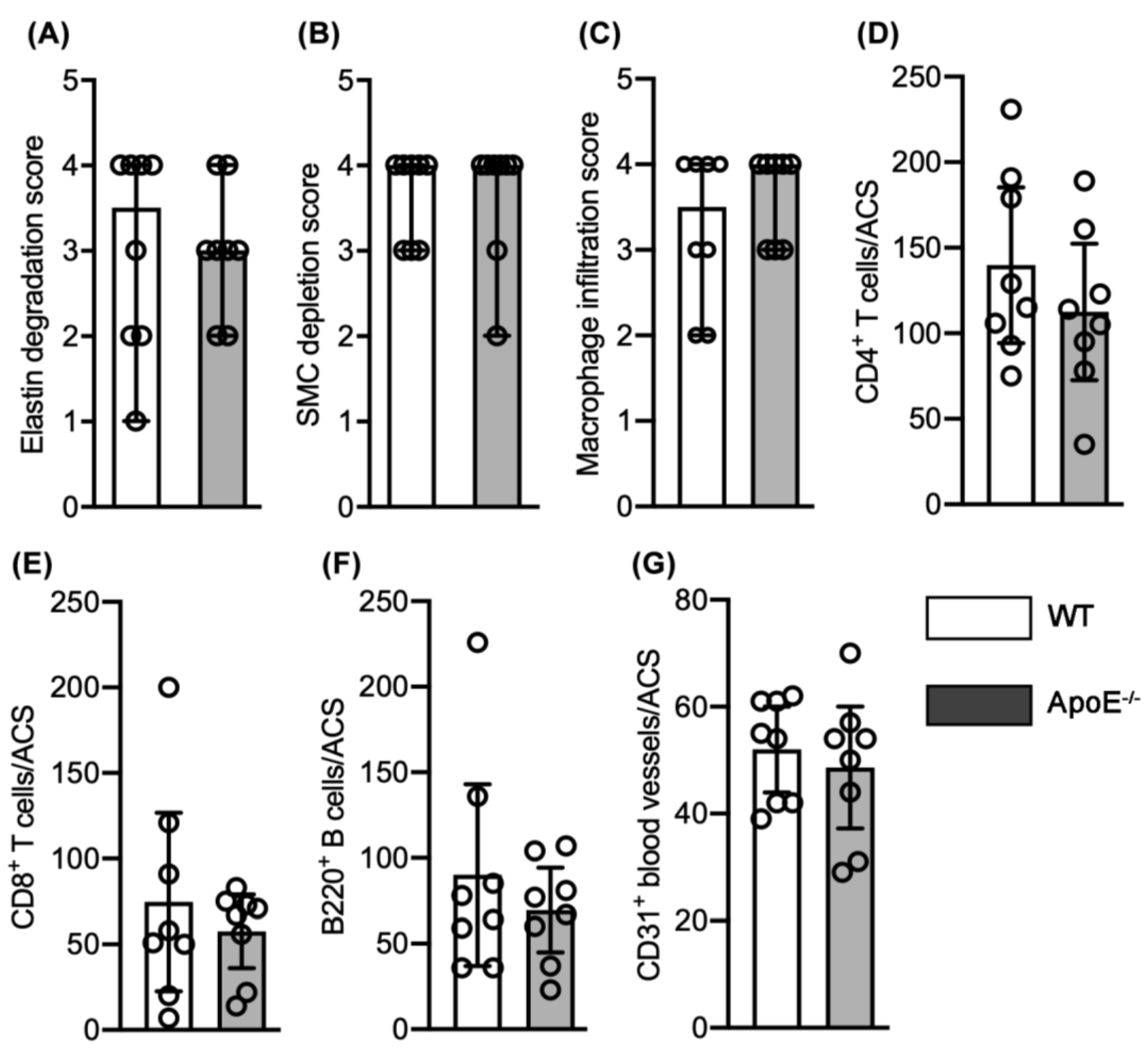
3.1. No Effect of Spontaneous Hypercholesterolemia on Experimental AAA Diameter
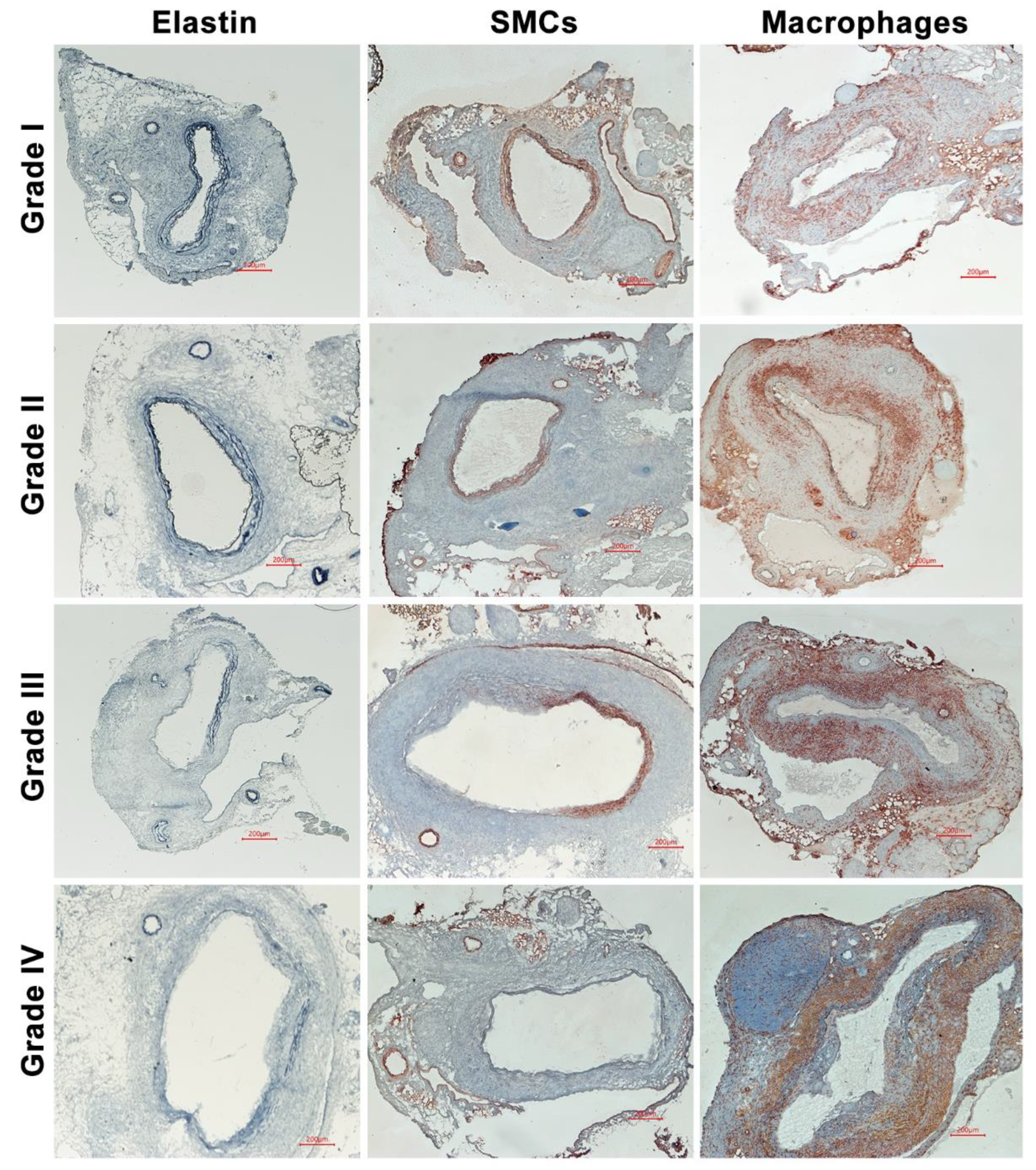
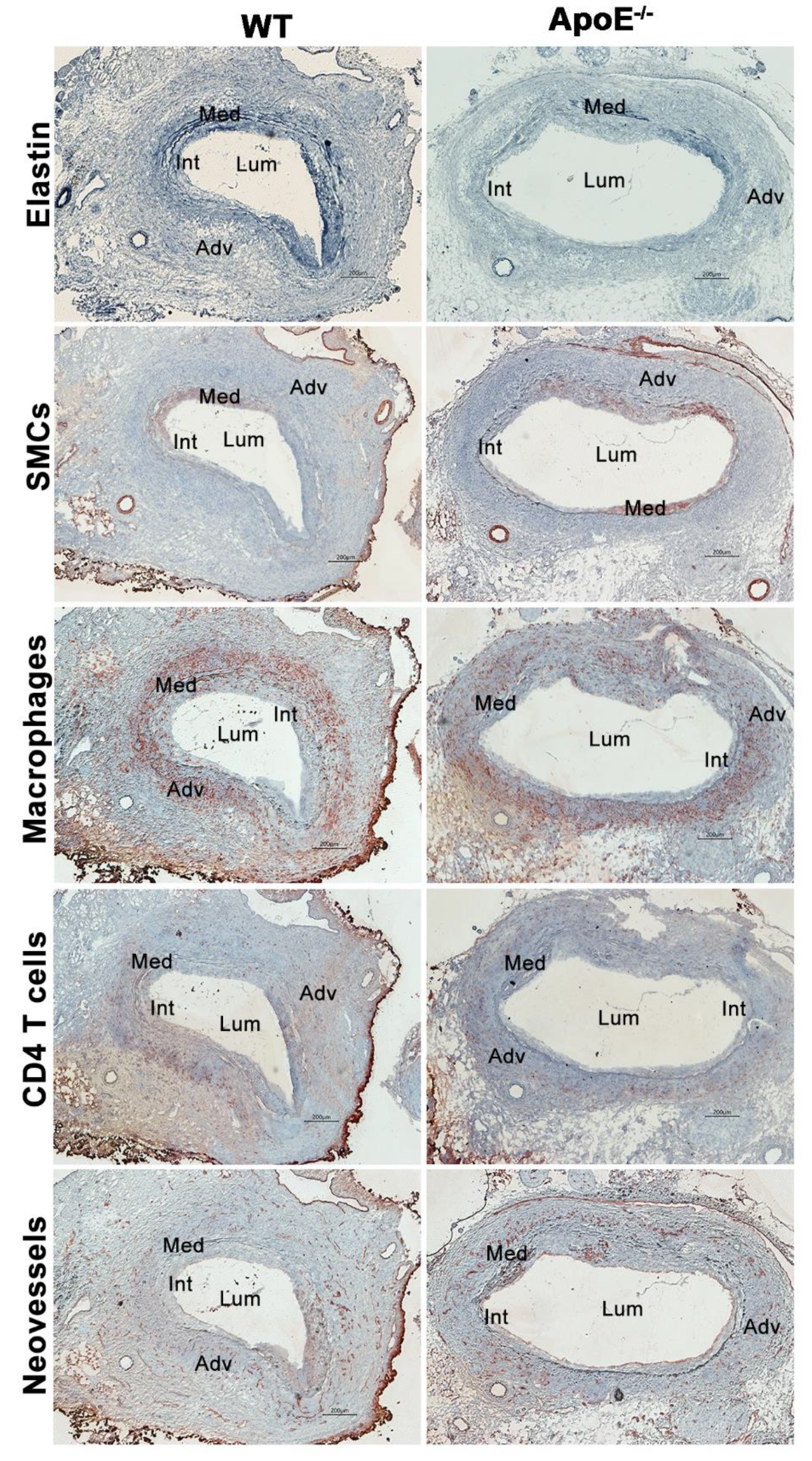
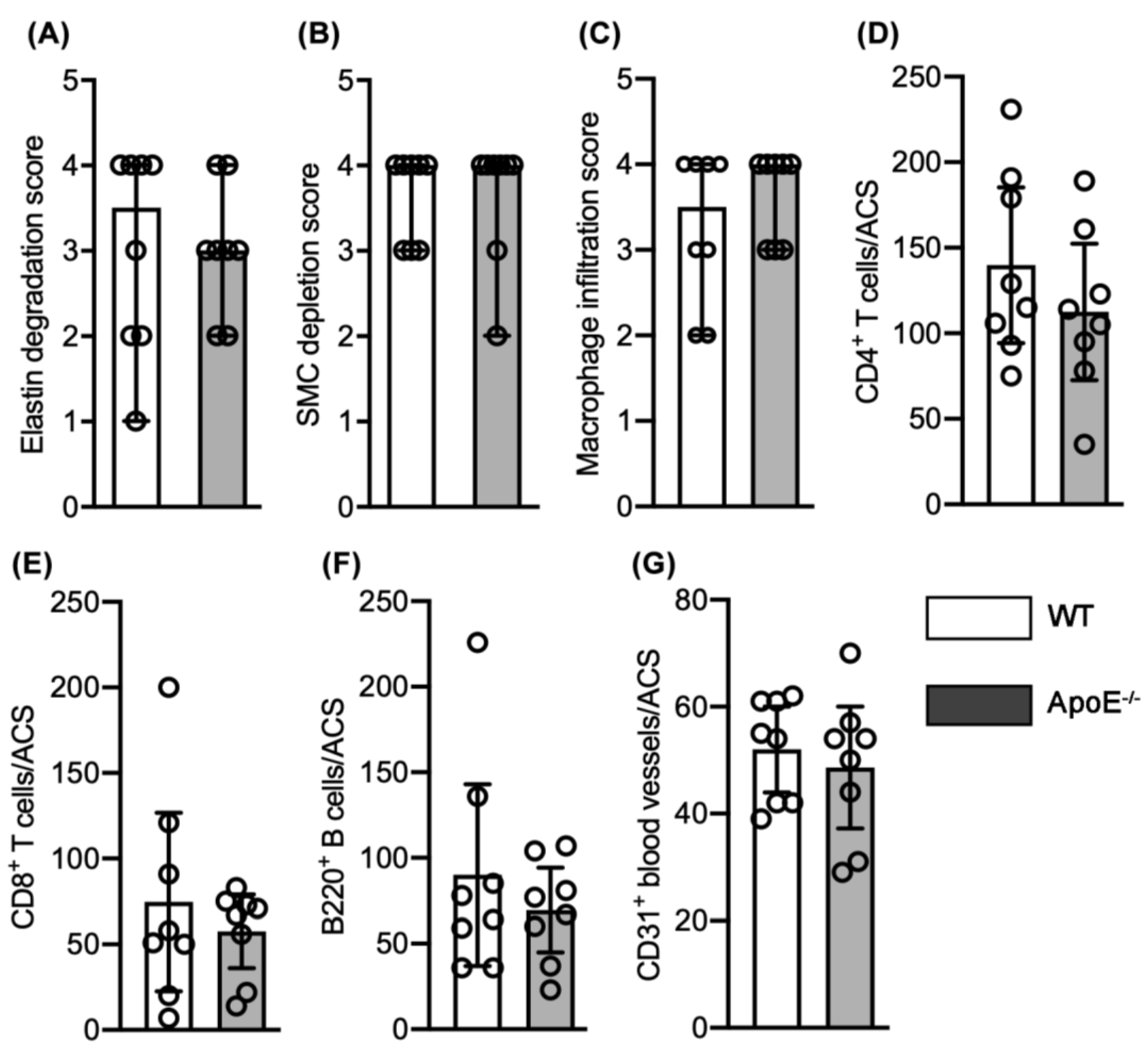
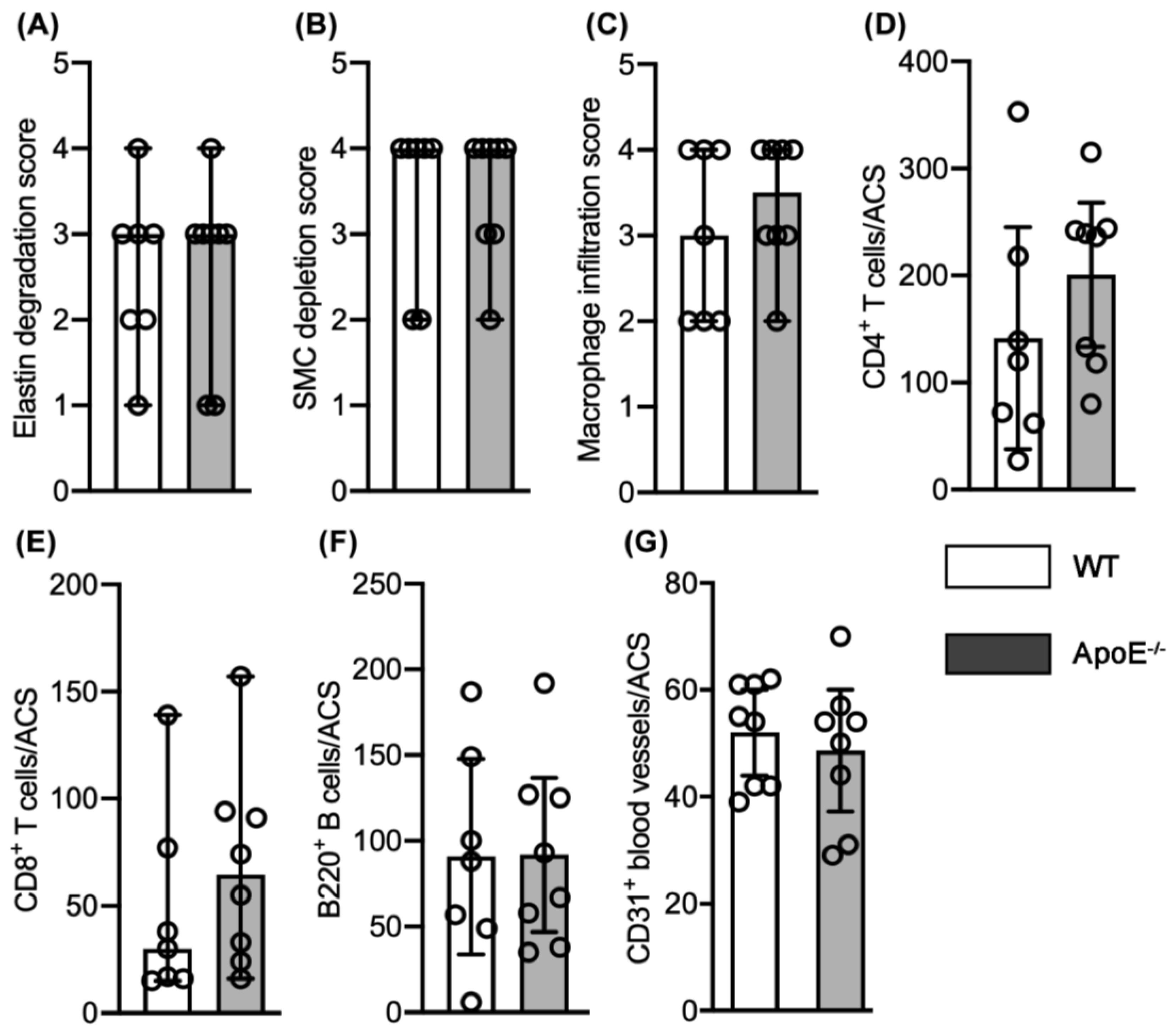
3.2. No Effect of Spontaneous Hypercholesterolemia on AAA Histopathology
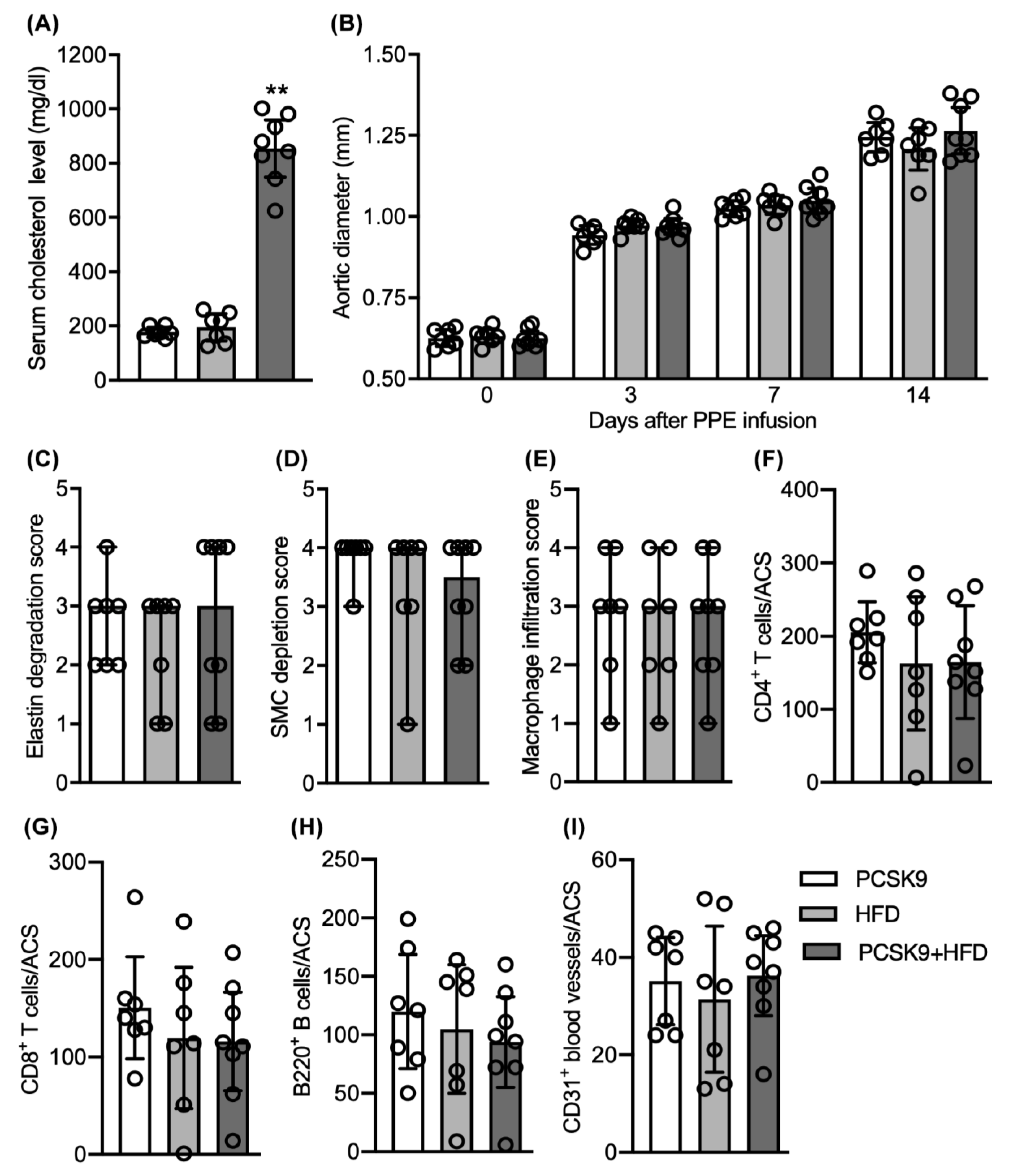
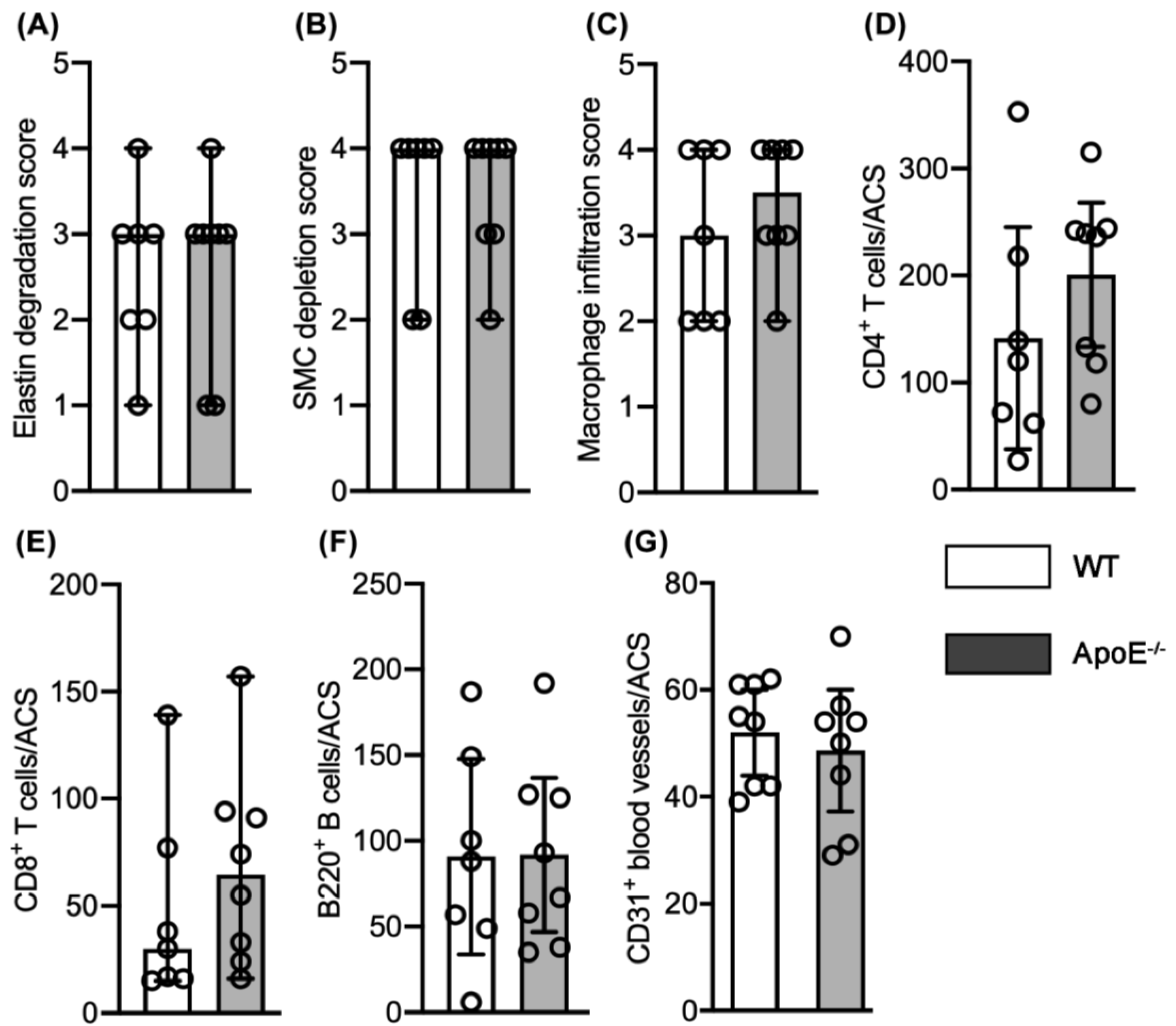
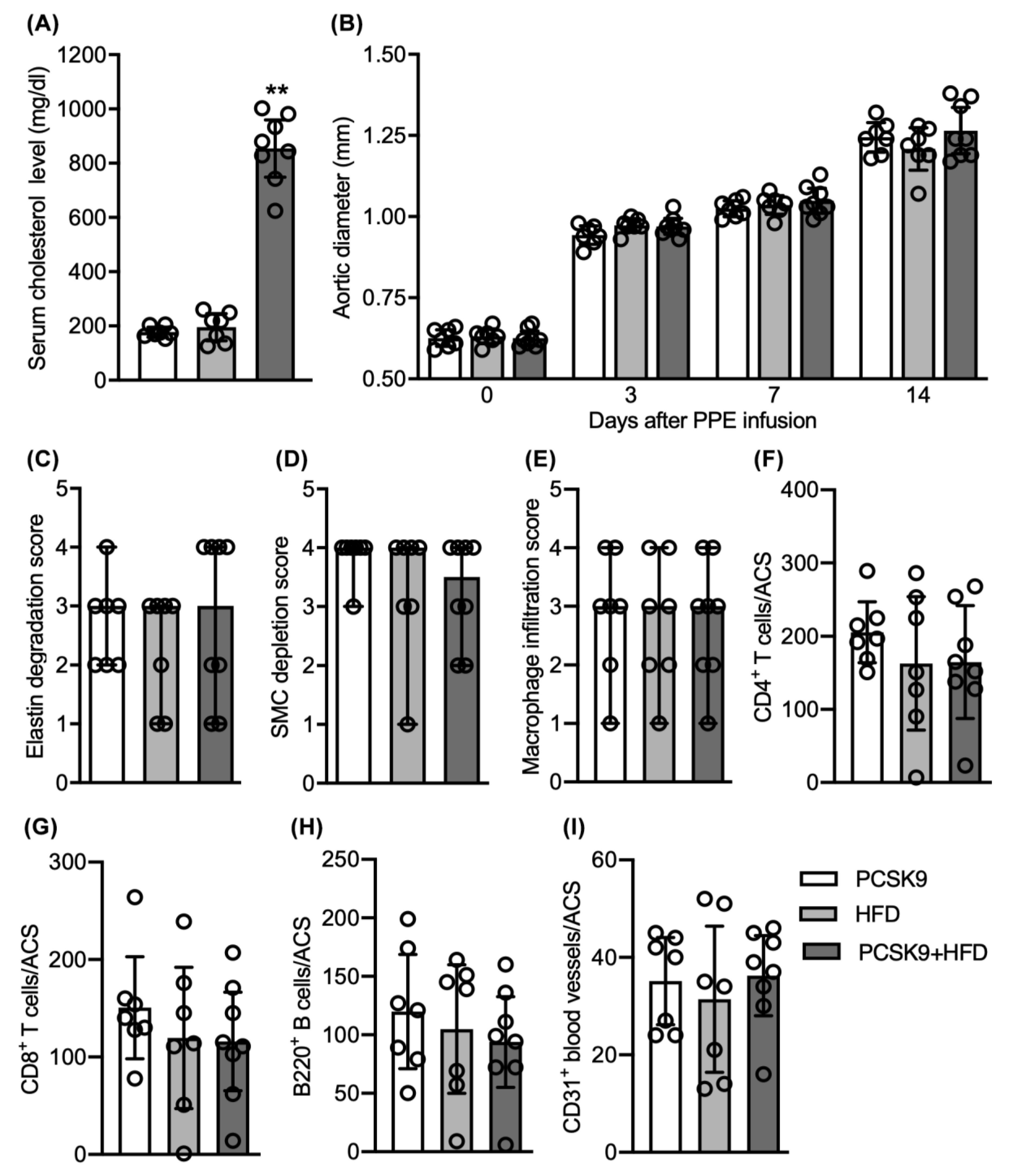
3.3. No Differences in Aneurysmal Progresssion and Histopathologies Noted between Normocholesterolemia and “Acquired” Hypercholesterolemia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Lederle, F.A.; Johnson, G.R.; Wilson, S.E.; Chute, E.P.; Hye, R.J.; Makaroun, M.S.; Barone, G.W.; Bandyk, D.; Moneta, G.L.; Makhoul, R.G. The aneurysm detection and management study screening program: Validation cohort and final results. Aneurysm detection and management veterans affairs cooperative study investigators. Arch. Intern. Med. 2000, 160, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.C.; Zwolak, R.M.; Egorova, N.N.; Riles, T.S.; Manganaro, A.; Moskowitz, A.; Gelijns, A.C.; Greco, G. Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J. Vasc. Surg. 2010, 52, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klarin, D.; Damrauer, S.M.; Cho, K.; Sun, Y.V.; Teslovich, T.M.; Honerlaw, J.; Gagnon, D.R.; DuVall, S.L.; Li, J.; Peloso, G.M.; et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the million veteran program. Nat. Genet. 2018, 50, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Cui, H.; Wu, N.; Zhang, H. Effect of Statin Therapy on Abdominal Aortic Aneurysm Growth Rate and Mortality: A Systematic Review and Meta-analysis. Ann. Vasc. Surg. 2020, 67, 503–510. [Google Scholar] [CrossRef]
- Schouten, O.; Van Laanen, J.; Boersma, E.; Vidakovic, R.; Feringa, H.; Dunkelgrün, M.; Bax, J.; Koning, J.; Van Urk, H.; Poldermans, D. Statins are Associated with a Reduced Infrarenal Abdominal Aortic Aneurysm Growth. Eur. J. Vasc. Endovasc. Surg. 2006, 32, 21–26. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Yamamoto, H.; Iwata, K.; Goto, S.; Umemoto, T. Effects of Statin Therapy on Abdominal Aortic Aneurysm Growth: A Meta-analysis and Meta-regression of Observational Comparative Studies. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 287–292. [Google Scholar] [CrossRef] [Green Version]
- Salata, K.; Syed, M.; Hussain, M.A.; de Mestral, C.; Greco, E.; Mamdani, M.; Tu, J.V.; Forbes, T.L.; Bhatt, D.L.; Verma, S.; et al. Statins reduce abdominal aortic aneurysm growth, rupture, and perioperative mortality: A systematic review and meta-analysis. J. Am. Heart Assoc. 2018, 7, e008657. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.D.; Clancy, P.; Bourke, B.; Walker, P.J.; Dear, A.; Buckenham, T.; Norman, P.; Golledge, J. Association of statin prescription with small abdominal aortic aneurysm progression. Am. Heart J. 2010, 159, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Twine, C.; Williams, I.M. Systematic review and meta-analysis of the effects of statin therapy on abdominal aortic aneurysms. J. Br. Surg. 2010, 98, 346–353. [Google Scholar] [CrossRef]
- Zhang, Y.; Naggar, J.C.; Welzig, C.M.; Beasley, D.; Moulton, K.S.; Park, H.J.; Galper, J.B. Simvastatin inhibits angiotensin ii-induced abdominal aortic aneurysm formation in apolipoprotein e-knockout mice: Possible role of erk. Arter. Thromb. Vasc. Biol. 2009, 29, 1764–1771. [Google Scholar] [CrossRef] [Green Version]
- Golledge, J.; Cullen, B.; Moran, C.; Rush, C. Efficacy of Simvastatin in Reducing Aortic Dilatation in Mouse Models of Abdominal Aortic Aneurysm. Cardiovasc. Drugs Ther. 2010, 24, 373–378. [Google Scholar] [CrossRef]
- Takahashi, K.; Matsumoto, Y.; Doe, Z.; Kanazawa, M.; Satoh, K.; Shimizu, T.; Sato, A.; Fukumoto, Y.; Shimokawa, H. Combination therapy with atorvastatin and amlodipine suppresses angiotensin ii-induced aortic aneurysm formation. PLoS ONE 2013, 8, e72558. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Xu, B.; Schultz, G.M.; Chow, V.; White, J.J.; Sulaimon, S.; Hezi-Yamit, A.; Peterson, S.R.; Dalman, R.L. Efficacy and Mechanism of Angiotensin II Receptor Blocker Treatment in Experimental Abdominal Aortic Aneurysms. PLoS ONE 2012, 7, e49642. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Xu, B.; Xuan, H.; Glover, K.J.; Tanaka, H.; Hu, X.; Fujimura, N.; Wang, W.; Schultz, J.R.; Turner, C.R.; et al. Peptide Inhibitor of CXCL4–CCL5 Heterodimer Formation, MKEY, Inhibits Experimental Aortic Aneurysm Initiation and Progression. Arter. Thromb. Vasc. Biol. 2013, 33, 718–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, A.; Cassis, L. Chronic Angiotensin II Infusion Promotes Atherogenesis in Low Density Lipoprotein Receptor -/- Mice. Ann. N. Y. Acad. Sci. 1999, 892, 108–118. [Google Scholar] [CrossRef]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E–deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Howatt, D.A.; Balakrishnan, A.; Graham, M.J.; Mullick, A.E.; Daugherty, A. Hypercholesterolemia Induced by a PCSK9 Gain-of-Function Mutation Augments Angiotensin II–Induced Abdominal Aortic Aneurysms in C57BL/6 Mice—Brief Report. Arter. Thromb. Vasc. Biol. 2016, 36, 1753–1757. [Google Scholar] [CrossRef] [Green Version]
- Pyo, R.; Lee, J.K.; Shipley, J.M.; Curci, J.A.; Mao, D.; Ziporin, S.J.; Ennis, T.L.; Shapiro, S.D.; Senior, R.M.; Thompson, R.W. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J. Clin. Investig. 2000, 105, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, N.; Xiong, J.; Kettler, E.; Xuan, H.; Glover, K.J.; Mell, M.W.; Xu, B.; Dalman, R.L. Metformin treatment status and abdominal aortic aneurysm disease progression. J. Vasc. Surg. 2016, 64, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, E.F.; Buckley, C.; Shames, M.L.; Ennis, T.L.; Vanvickle-Chavez, S.J.; Mao, D.; Goeddel, L.A.; Hawkins, C.J.; Thompson, R.W. Treatment With Simvastatin Suppresses the Development of Experimental Abdominal Aortic Aneurysms in Normal and Hypercholesterolemic Mice. Ann. Surg. 2005, 241, 92–101. [Google Scholar] [CrossRef]
- Wang, Y.; Krishna, S.M.; Moxon, J.; Dinh, T.N.; Jose, R.J.; Yu, H.; Golledge, J. Influence of apolipoprotein E, age and aortic site on calcium phosphate induced abdominal aortic aneurysm in mice. Atherosclerosis 2014, 235, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Niederhoffer, N.; Lartaud-Idjouadiene, I.; Giummelly, P.; Duvivier, C.; Peslin, R.; Atkinson, J. Calcification of medial elastic fibers and aortic elasticity. Hypertension 1997, 29, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Hildreth, C.M.; Phillips, J.K.; Avolio, A.P. Aortic stiffness is associated with vascular calcification and remodeling in a chronic kidney disease rat model. Am. J. Physiol. Physiol. 2011, 300, F1431–F1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, O.M.; Yuan, X.; Cain, C.; Salloum, F.; Li, P. Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: A ceramide-mediated vasculopathy. J. Cell. Mol. Med. 2019, 24, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenhäuser, M.U.; Schellinger, I.N.; Yoshino, T.; Toyama, K.; Kayama, Y.; Deng, A.; Guenther, S.P.; Petzold, A.; Mulorz, J.; Mulorz, P.; et al. Chronic nicotine exposure induces murine aortic remodeling and stiffness segmentation-implications for abdominal aortic aneurysm susceptibility. Front. Physiol. 2018, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- Raaz, U.; Zöllner, A.M.; Schellinger, I.N.; Toh, R.; Nakagami, F.; Brandt, M.; Emrich, F.C.; Kayama, Y.; Eken, S.; Adam, M.; et al. Segmental Aortic Stiffening Contributes to Experimental Abdominal Aortic Aneurysm Development. Circulation 2015, 131, 1783–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopacz, A.; Werner, E.; Grochot-Przęczek, A.; Klóska, D.; Hajduk, K.; Neumayer, C.; Józkowicz, A.; Piechota-Polanczyk, A. Simvastatin Attenuates Abdominal Aortic Aneurysm Formation Favoured by Lack of Nrf2 Transcriptional Activity. Oxidative Med. Cell. Longev. 2020, 2020, 6340190. [Google Scholar] [CrossRef]
- Mastoraki, S.T.; Toumpoulis, I.K.; Anagnostopoulos, C.E.; Tiniakos, D.; Papalois, A.; Chamogeorgakis, T.P.; Angouras, D.C.; Rokkas, C.K. Treatment With Simvastatin Inhibits the Formation of Abdominal Aortic Aneurysms in Rabbits. Ann. Vasc. Surg. 2012, 26, 250–258. [Google Scholar] [CrossRef]
- Kalyanasundaram, A.; Elmore, J.R.; Manazer, J.R.; Golden, A.; Franklin, D.P.; Galt, S.W.; Zakhary, E.M.; Carey, D.J. Simvastatin suppresses experimental aortic aneurysm expansion. J. Vasc. Surg. 2006, 43, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Katsuki, S.; Koga, J.-I.; Matoba, T.; Umezu, R.; Nakashiro, S.; Nakano, K.; Tsutsui, H.; Egashira, K. Nanoparticle-Mediated Delivery of Pitavastatin to Monocytes/Macrophages Inhibits Angiotensin II-Induced Abdominal Aortic Aneurysm Formation in ApoE−/− Mice. J. Atheroscler. Thromb. 2021, 54379. [Google Scholar] [CrossRef]
- Fukuhara, N.; Honda, Y.; Ukita, N.; Matsui, M.; Miura, Y.; Hoshina, K. Efficient Suppression of Abdominal Aortic Aneurysm Expansion in Rats through Systemic Administration of Statin-Loaded Nanomedicine. Int. J. Mol. Sci. 2020, 21, 8702. [Google Scholar] [CrossRef] [PubMed]
- Xuan, H.; Xu, B.; Wang, W.; Tanaka, H.; Fujimura, N.; Miyata, M.; Michie, S.A.; Dalman, R.L. Inhibition or deletion of angiotensin II type 1 receptor suppresses elastase-induced experimental abdominal aortic aneurysms. J. Vasc. Surg. 2017, 67, 573–584.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Xuan, H.; Iida, Y.; Miyata, M.; Dalman, R.L. Pathogenic and Therapeutic Significance of Angiotensin II Type I Receptor in Abdominal Aortic Aneurysms. Curr. Drug Targets 2018, 19, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Cassis, L.A.; Rateri, D.L.; Lu, H.; Daugherty, A. Bone Marrow Transplantation Reveals That Recipient AT1a Receptors Are Required to Initiate Angiotensin II–Induced Atherosclerosis and Aneurysms. Arter. Thromb. Vasc. Biol. 2007, 27, 380–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanematsu, Y.; Kanematsu, M.; Kurihara, C.; Tsou, T.-L.; Nuki, Y.; Liang, E.I.; Makino, H.; Hashimoto, T. Pharmacologically Induced Thoracic and Abdominal Aortic Aneurysms in Mice. Hypertension 2010, 55, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, H.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Sorci-Thomas, M.; Cassis, L.A.; Daugherty, A. Associations of apoai and apob-containing lipoproteins with angii-induced abdominal aortic aneurysms in mice. Arter. Thromb. Vasc. Biol. 2015, 35, 1826–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sawada, H.; Howatt, D.A.; Moorleghen, J.J.; Vsevolozhskaya, O.; Daugherty, A.; Lu, H.S. Hypercholesterolemia Accelerates Both the Initiation and Progression of Angiotensin II-induced Abdominal Aortic Aneurysms. Ann. Vasc. Med. Res. 2020, 6, 1099. [Google Scholar]
- Angelov, S.N.; Hu, J.H.; Wei, H.; Airhart, N.; Shi, M.; Dichek, D.A. TGF-β (Transforming Growth Factor-β) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arter. Thromb. Vasc. Biol. 2017, 37, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Police, S.B.; Thatcher, S.; Charnigo, R.; Daugherty, A.; Cassis, L.A. Obesity Promotes Inflammation in Periaortic Adipose Tissue and Angiotensin II-Induced Abdominal Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2009, 29, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; Hara, H.; Fujiwara, T.; Kanaya, T.; Maemura, S.; Komuro, I. Tgf-beta signaling-related genes and thoracic aortic aneurysms and dissections. Int. J. Mol. Sci. 2018, 19, 2125. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadiere, C.; Renia, L.; et al. Tgf-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin ii-infused mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Cassis, L.A.; Daugherty, A. Tgf-β neutralization enhances angii-induced aortic rupture and aneurysm in both thoracic and abdominal regions. PLoS ONE 2016, 11, e0153811. [Google Scholar] [CrossRef]
- Risum, Ø.; Sandven, I.; Sundhagen, J.O.; Abdelnoor, M. Editor’s Choice–Effect of Statins on Total Mortality in Abdominal Aortic Aneurysm Repair: A Systematic Review and Meta-analysis. Eur. J. Vasc. Endovasc. Surg. 2020, 61, 114–120. [Google Scholar] [CrossRef] [PubMed]
- AlShaikh, H.N.; Bohsali, F.; Gani, F.; Nejim, B.; Malas, M. Statin intensity and postoperative mortality following open repair of intact abdominal aortic aneurysm. BJS Open 2018, 2, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Søgaard, R.; Lindholt, J. Pharmacological Preventive Potential among Attenders at Vascular Screening: Findings from the VIVA Trial. Eur. J. Vasc. Endovasc. Surg. 2020, 59, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Hołda, M.K.; Iwaszczuk, P.; Wszołek, K.; Chmiel, J.; Brzychczy, A.; Trystuła, M.; Misztal, M. Coexistence and management of abdominal aortic aneurysm and coronary artery disease. Cardiol. J. 2020, 27, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Bobryshev, Y.V.; Lord, R.S. Vascular-associated lymphoid tissue (valt) involvement in aortic aneurysm. Atherosclerosis 2001, 154, 15–21. [Google Scholar] [CrossRef]
- Srikakulapu, P.; Hu, D.; Yin, C.; Mohanta, S.K.; Bontha, S.V.; Peng, L.; Beer, M.; Weber, C.; McNamara, C.A.; Grassia, G.; et al. Artery Tertiary Lymphoid Organs Control Multilayered Territorialized Atherosclerosis B-Cell Responses in Aged ApoE−/− Mice. Arter. Thromb. Vasc. Biol. 2016, 36, 1174–1185. [Google Scholar] [CrossRef] [Green Version]
- Mohanta, S.; Yin, C.; Peng, L.; Srikakulapu, P.; Bontha, V.; Hu, D.; Weih, F.; Weber, C.; Gerdes, N.; Habenicht, A.J. Artery Tertiary Lymphoid Organs Contribute to Innate and Adaptive Immune Responses in Advanced Mouse Atherosclerosis. Circ. Res. 2014, 114, 1772–1787. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Manufacture | Catalog # | Clone # | Working Solution (μg/mL) | Incubation Time at Room Temperature (Minutes) |
|---|---|---|---|---|---|
| Rat anti-mouse-B220 mAb | Biolegend | 103201 | RA3-6B2 | 2.5 | 60 |
| Rat-anti-mouse CD4 mAb | Biolegend | 100402 | GK1.5 | 2.5 | 60 |
| Rat anti-mouse CD8 mAb | Biolegend | 100702 | 53–6.7 | 2.5 | 60 |
| Rat anti-mouse CD68 mAb | Biolegend | 137002 | FA-11 | 2.5 | 60 |
| Biotin-mouse anti-mouse SMC α actin mAb | Thermo Fisher Scientific | MA5-11544 | 1A4 | 1.0 | 60 |
| Biotin-goat anti-rat IgG antibody | Jackson ImmunoResearch Laboratories, Inc. | 112-065-062 | N/A | 4.0 | 30 |
| Peroxidase-streptavidin conjugate | Jackson ImmunoResearch Laboratories, Inc. | 016-030-084 | N/A | 5.0 | 30 |
| AEC peroxidase substrate Kit | Vector Laboratories, Inc. | SK-4200 | N/A | N/A | 10–20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikezoe, T.; Shoji, T.; Guo, J.; Shen, F.; Lu, H.S.; Daugherty, A.; Nunokawa, M.; Kubota, H.; Miyata, M.; Xu, B.; et al. No Effect of Hypercholesterolemia on Elastase-Induced Experimental Abdominal Aortic Aneurysm Progression. Biomolecules 2021, 11, 1434. https://doi.org/10.3390/biom11101434
Ikezoe T, Shoji T, Guo J, Shen F, Lu HS, Daugherty A, Nunokawa M, Kubota H, Miyata M, Xu B, et al. No Effect of Hypercholesterolemia on Elastase-Induced Experimental Abdominal Aortic Aneurysm Progression. Biomolecules. 2021; 11(10):1434. https://doi.org/10.3390/biom11101434
Chicago/Turabian StyleIkezoe, Toru, Takahiro Shoji, Jia Guo, Fanru Shen, Hong S. Lu, Alan Daugherty, Masao Nunokawa, Hiroshi Kubota, Masaaki Miyata, Baohui Xu, and et al. 2021. "No Effect of Hypercholesterolemia on Elastase-Induced Experimental Abdominal Aortic Aneurysm Progression" Biomolecules 11, no. 10: 1434. https://doi.org/10.3390/biom11101434