

Utilizing Integrated Bioinformatics Analysis to Explore Potential Alterations in Mitochondrial Function Within Immune Cells Associated with Thoracic Aortic Aneurysms

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Screening Differentially Expressed Mitochondrial-Related Genes (DE-MRGs) Between TAA and Control
2.3. snRNA-seq Data Processing
2.4. Evaluation of the Mitoscore
2.5. Construction of the LASSO Model and the SVM-RFE Feature Selection Process
2.6. ROC Curve Analysis
2.7. Animal Studies
2.8. qRT-PCR Analysis
2.9. Statistical Analysis
3. Results
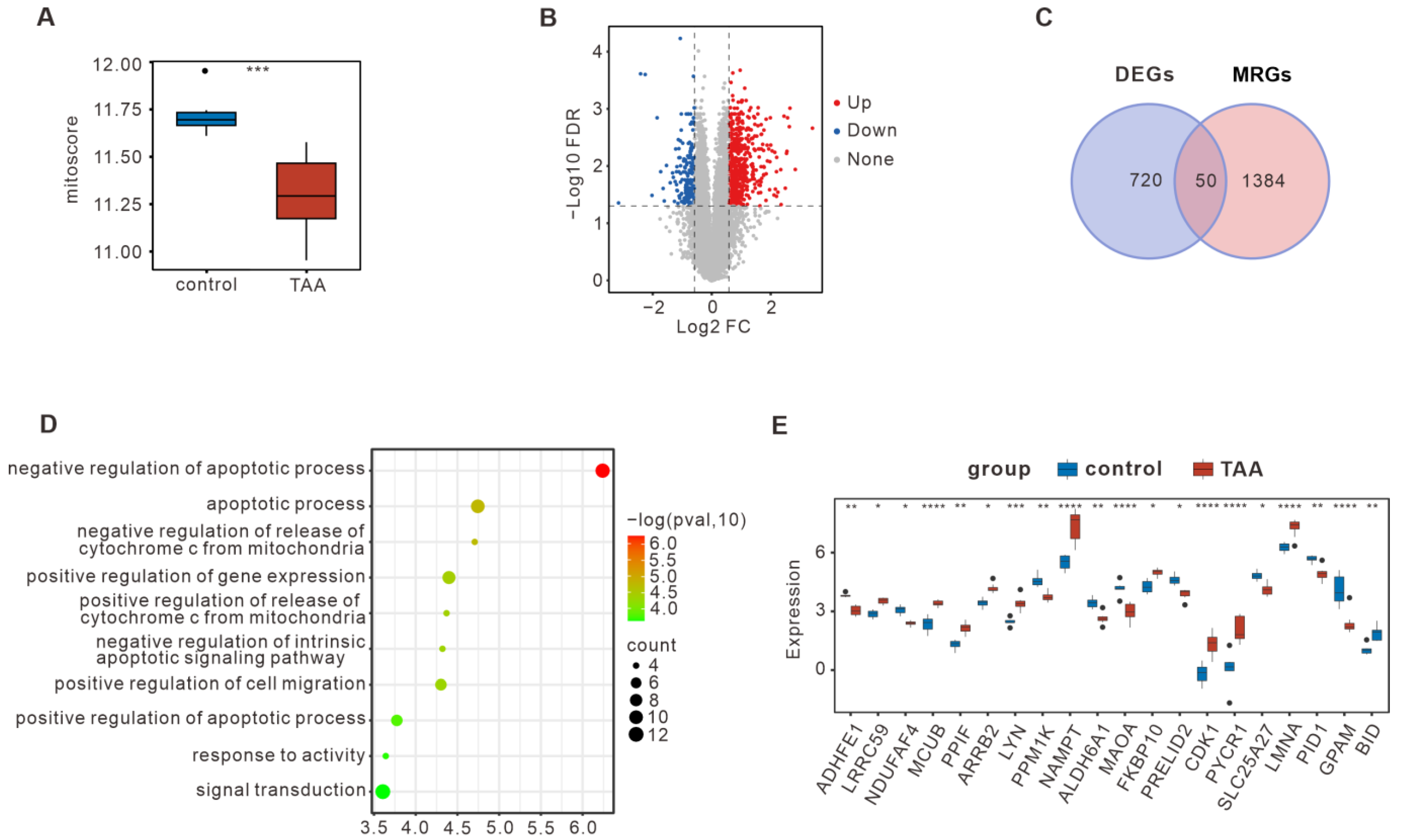
3.1. Bulk RNA-seq Summarized the DE-MRGs in TAA
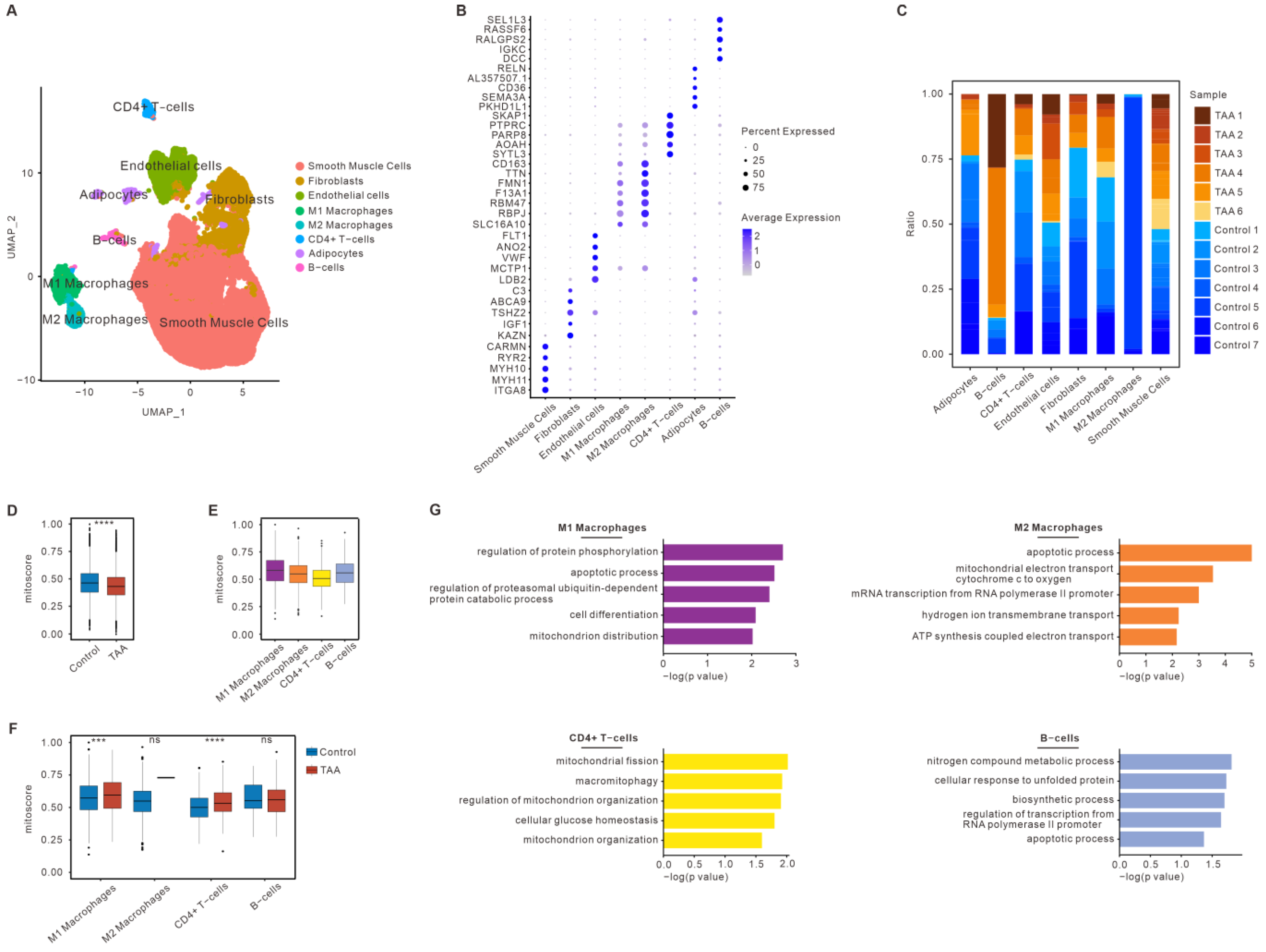
3.2. Mitochondrial Dysfunction of Immune Cells in TAA Based on snRNA-seq
3.3. Construction of a Diagnostic Model for TAA
3.4. Analysis of the Key DE-MRGs and Metabolic-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Isselbacher, E.M. 2022 ACC/AHA Guideline for the Diagnosis and Management of Aortic Disease: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022, 146, e334–e482. [Google Scholar] [CrossRef] [PubMed]
- Sidloff, D. Mortality from thoracic aortic diseases and associations with cardiovascular risk factors. Circulation 2014, 130, 2287–2294. [Google Scholar] [CrossRef]
- Bossone, E. Epidemiology and management of aortic disease: Aortic aneurysms and acute aortic syndromes. Nat. Rev. Cardiol. 2021, 18, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Yang, X. Targeting endothelial tight junctions to predict and protect thoracic aortic aneurysm and dissection. Eur. Heart J. 2023, 44, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J. Inherited Thoracic Aortic Disease: New Insights and Translational Targets. Circulation 2020, 141, 1570–1587. [Google Scholar] [CrossRef]
- Thakker, P.D. Cardiogenetics: Genetic testing in the diagnosis and management of patients with aortic disease. Heart 2021, 107, 619–626. [Google Scholar] [CrossRef]
- Quintana, R.A. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H. Aortic Aneurysms and Dissections Series. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e37–e46. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.H. Targeting mitochondrial function in macrophages: A novel treatment strategy for atherosclerotic cardiovascular disease? Pharmacol. Ther. 2023, 247, 108441. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Mitochondria spatially and temporally modulate VSMC phenotypes via interacting with cytoskeleton in cardiovascular diseases. Redox Biol. 2023, 64, 102778. [Google Scholar] [CrossRef] [PubMed]
- Hinton, A., Jr. Mitochondrial Structure and Function in Human Heart Failure. Circ. Res. 2024, 135, 372–396. [Google Scholar] [CrossRef]
- Oller, J. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- van der Pluijm, I. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef]
- Yu, E. Mitochondrial function in thoracic aortic aneurysms. Cardiovasc. Res. 2018, 114, 1696–1698. [Google Scholar] [CrossRef] [PubMed]
- Liu, X. Single-cell RNA sequencing identifies an Il1rn(+)/Trem1(+) macrophage subpopulation as a cellular target for mitigating the progression of thoracic aortic aneurysm and dissection. Cell Discov. 2022, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Chou, E.L. Aortic Cellular Diversity and Quantitative Genome-Wide Association Study Trait Prioritization Through Single-Nuclear RNA Sequencing of the Aneurysmal Human Aorta. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X. Single-cell sequencing reveals the impact of endothelial cell PIEZO1 expression on thoracic aortic aneurysm. J. Mol. Cell. Cardiol. 2024, 191, 63–75. [Google Scholar] [CrossRef]
- Slyper, M. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S. Single-cell and single-nucleus RNA-sequencing from paired normal-adenocarcinoma lung samples provide both common and discordant biological insights. PLoS Genet. 2024, 20, e1011301. [Google Scholar] [CrossRef]
- Garbincius, J.F. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992. [Google Scholar] [CrossRef]
- Huo, J. MCUb Induction Protects the Heart From Postischemic Remodeling. Circ. Res. 2020, 127, 379–390. [Google Scholar] [CrossRef]
- Lambert, J.P. MCUB Regulates the Molecular Composition of the Mitochondrial Calcium Uniporter Channel to Limit Mitochondrial Calcium Overload During Stress. Circulation 2019, 140, 1720–1733. [Google Scholar] [CrossRef]
- Liu, T. MCU Overexpression Rescues Inotropy and Reverses Heart Failure by Reducing SR Ca2+ Leak. Circ. Res. 2021, 128, 1191–1204. [Google Scholar] [CrossRef]
- Wang, Y. β-arrestin 2 mediates cardiac ischemia-reperfusion injury via inhibiting GPCR-independent cell survival signalling. Cardiovasc. Res. 2017, 113, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, N.M. ARRB2 (β-Arrestin-2) Deficiency Alters Fluid Homeostasis and Blood Pressure Regulation. Hypertension 2022, 79, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A. Beta-arrestin 2 mediates cardiac hypertrophy induced by thyroid hormones via AT1R. J. Cell. Physiol. 2021, 236, 4640–4654. [Google Scholar] [CrossRef]
- Wei, X. Myeloid beta-arrestin 2 depletion attenuates metabolic dysfunction-associated steatohepatitis via the metabolic reprogramming of macrophages. Cell Metab. 2024, 36, 2281–2297.e7. [Google Scholar] [CrossRef]
- Acín-Pérez, R. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat. Metab. 2020, 2, 974–988. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.J. Signalling through Src family kinase isoforms is not redundant in models of thrombo-inflammatory vascular disease. J. Cell. Mol. Med. 2018, 22, 4317–4327. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, C. TNAP as a therapeutic target for cardiovascular calcification: A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2022, 118, 84–96. [Google Scholar] [CrossRef]
- Tani, T. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model. J. Pathol. 2020, 250, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X. TNAP is a novel regulator of cardiac fibrosis after myocardial infarction by mediating TGF-β/Smads and ERK1/2 signaling pathways. EBioMedicine 2021, 67, 103370. [Google Scholar] [CrossRef] [PubMed]
- Gao, L. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways. Cell Death Dis. 2020, 11, 44. [Google Scholar] [CrossRef]
- Fang, W. Methionine restriction constrains lipoylation and activates mitochondria for nitrogenic synthesis of amino acids. Nat. Commun. 2023, 14, 2504. [Google Scholar] [CrossRef] [PubMed]
- Mucha, P. Vitamin B(12) Metabolism: A Network of Multi-Protein Mediated Processes. Int. J. Mol. Sci. 2024, 25, 8021. [Google Scholar] [CrossRef]
- Bassila, C. Methionine synthase and methionine synthase reductase interact with MMACHC and with MMADHC. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 103–112. [Google Scholar] [CrossRef]
- Mascarenhas, R. Cobalt-Sulfur Coordination Chemistry Drives B(12) Loading onto Methionine Synthase. J. Am. Chem. Soc. 2023, 299, S338. [Google Scholar]
- Wang, Q. Targeting metabolism in aortic aneurysm and dissection: From basic research to clinical applications. Int. J. Biol. Sci. 2023, 19, 3869–3891. [Google Scholar] [CrossRef] [PubMed]
- Elia, L. The involvement of epigenetics in vascular disease development. Int. J. Biochem. Cell Biol. 2019, 107, 27–31. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | ||
|---|---|---|
| LMNA | ARRB2 | ACADL |
| MCL1 | SREBF1 | DCN |
| PPP2R2B | MRPS6 | MAOA |
| IL6 | PLN | IGF1 |
| ALPL | TFRC | NAMPT |
| FGR | MCUB | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, C.; Chen, S.-X.; Huang, C.-L.; Du, Y.-P.; Wang, K.-H.; Li, P.-X.; Liu, S.-R.; Liu, Z.-Y.; Huang, Z. Utilizing Integrated Bioinformatics Analysis to Explore Potential Alterations in Mitochondrial Function Within Immune Cells Associated with Thoracic Aortic Aneurysms. Bioengineering 2025, 12, 197. https://doi.org/10.3390/bioengineering12020197
Guan C, Chen S-X, Huang C-L, Du Y-P, Wang K-H, Li P-X, Liu S-R, Liu Z-Y, Huang Z. Utilizing Integrated Bioinformatics Analysis to Explore Potential Alterations in Mitochondrial Function Within Immune Cells Associated with Thoracic Aortic Aneurysms. Bioengineering. 2025; 12(2):197. https://doi.org/10.3390/bioengineering12020197
Chicago/Turabian StyleGuan, Chang, Si-Xu Chen, Chun-Ling Huang, Yi-Peng Du, Kai-Hao Wang, Pei-Xin Li, Shen-Rong Liu, Zhao-Yu Liu, and Zheng Huang. 2025. "Utilizing Integrated Bioinformatics Analysis to Explore Potential Alterations in Mitochondrial Function Within Immune Cells Associated with Thoracic Aortic Aneurysms" Bioengineering 12, no. 2: 197. https://doi.org/10.3390/bioengineering12020197
APA StyleGuan, C., Chen, S.-X., Huang, C.-L., Du, Y.-P., Wang, K.-H., Li, P.-X., Liu, S.-R., Liu, Z.-Y., & Huang, Z. (2025). Utilizing Integrated Bioinformatics Analysis to Explore Potential Alterations in Mitochondrial Function Within Immune Cells Associated with Thoracic Aortic Aneurysms. Bioengineering, 12(2), 197. https://doi.org/10.3390/bioengineering12020197