
The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection
by
 , , , and
, , , and
Miriam Mikušová
† ,
,
Karolína Tomčíková
†,
Katarína Briestenská
,
František Kostolanský
and
Eva Varečková
* Biomedical Research Center of the Slovak Academy of Sciences, Institute of Virology, Dúbravská Cesta 9, 845 05 Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Viruses 2022, 14(5), 1064; https://doi.org/10.3390/v14051064
Submission received: 6 April 2022
/
Revised: 12 May 2022
/
Accepted: 12 May 2022
/
Published: 16 May 2022
(This article belongs to the Special Issue State-of-the-Art Virology Research in Slovakia)
Abstract
:A severe course of acute respiratory disease caused by influenza A virus (IAV) infection is often linked with subsequent bacterial superinfection, which is difficult to cure. Thus, synergistic influenza–bacterial co-infection represents a serious medical problem. The pathogenic changes in the infected host are accelerated as a consequence of IAV infection, reflecting its impact on the host immune response. IAV infection triggers a complex process linked with the blocking of innate and adaptive immune mechanisms required for effective antiviral defense. Such disbalance of the immune system allows for easier initiation of bacterial superinfection. Therefore, many new studies have emerged that aim to explain why viral–bacterial co-infection can lead to severe respiratory disease with possible fatal outcomes. In this review, we discuss the key role of several IAV proteins—namely, PB1-F2, hemagglutinin (HA), neuraminidase (NA), and NS1—known to play a role in modulating the immune defense of the host, which consequently escalates the development of secondary bacterial infection, most often caused by Streptococcus pneumoniae. Understanding the mechanisms leading to pathological disorders caused by bacterial superinfection after the previous viral infection is important for the development of more effective means of prevention; for example, by vaccination or through therapy using antiviral drugs targeted at critical viral proteins.
1. Introduction
Lower respiratory tract infections annually cause millions of human deaths worldwide [1]. A substantial portion of these deaths is attributable to seasonal influenza virus infections, due to the constant emergence of new variants of influenza A viruses (IAV) and synergistic infections with other viruses or bacteria [2,3]. The development of severe disease is often associated with the ability of primary viral infection to alter the host immune response, resulting in the promotion of the secondary infection. Such an infection can cause a potentially lethal disease associated with the systemic inflammatory response of the body [4,5,6].
Secondary bacterial infections can be caused by several bacterial species, such as Acinetobacter baumannii, Haemophilus influenzae, Klebsiella pneumoniae, Mycobacterium tuberculosis, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pneumoniae, or Streptococcus pyogenes [6,7,8,9]. The most common is the bacterium Streptococcus pneumoniae, which can persist in the human nasopharynx in a dormant state from early childhood to adulthood [6]. The dormant form of S. pneumoniae is non-invasive but can be reactivated and cause invasive infection after influenza infection [6,10,11]. Both IAV and S. pneumoniae are considered to be among the most important pathogens of the respiratory tract. An example of their synergistic relationship is the Spanish flu pandemic in 1918. It has been shown that the high mortality of this pandemic was not caused by the virus itself, but was instead the result of co-infection by these two pathogens, which led to synergistic pathologic disorders with devastating impacts [12]. Analyses of influenza-associated bacterial infections in mouse models have revealed higher morbidity and mortality rates in comparison to infection with the individual pathogens [13,14]. In the last three years, influenza viruses have been replaced by the coronavirus SARS-CoV-2 as the prevailing respiratory infection agents. Published data have shown that SARS-CoV-2 is also capable of making patients vulnerable to secondary bacterial infection [15,16,17]. Therefore, exploring and identifying the underlying mechanisms and the role of influenza virus proteins in establishing consequent bacterial infection would provide a new perspective on the prevention through vaccination and treatment, not only by antibacterial drugs, but preferentially by drugs against viral infections [15,18,19].
2. Pathogenesis of Co-Infection by IAV and Streptococcus pneumoniae
2.1. Viral Influenza Infection
Infection with influenza A viruses occurs in the upper airway epithelium. In the human population, the influenza virus spreads by droplets, and the manifestation of airway infection may be asymptomatic or with only very mild symptoms of uncomplicated upper respiratory tract infection. However, IAVs can also trigger complicated disease associated with severe pneumonia, leading to multi-organ failure or the worsening of existing health conditions, especially in individuals with immunodeficiency or chronic lung or heart disease [20,21,22]. The initial sites of influenza A infection are the pseudostratified columnar cells of the respiratory epithelium in the trachea, nasal cavity, and sub-mucosal nodes, as well as the pneumocytes in pulmonary alveoli. The virus further spreads and infects surrounding epithelial as well as immune cells, such as macrophages, dendritic cells, and NK cells [23,24].
IAV infection activates the immune response in the respiratory tract by triggering the innate defense mechanisms. IAV must first overcome the physical barrier, consisting of mucosal surface fluid containing antimicrobial peptides, neutralizing secretory antibodies, IgA, mucus, and a protective layer of basal and ciliated epithelial cells. The integrity of the airway epithelium is also under the constant surveillance of leukocytes [25,26]. Homeostasis is maintained through the release of cytokines, chemokines, and growth factors. When IAV enters cells, a signal is transmitted along the interferon signaling pathway and the antiviral response is activated. After pattern-recognizing receptors (PRRs) recognize virus-specific nucleic acids, they activate the transcription of interferon (IFN) genes and the secretion of proinflammatory cytokines. Secreted type I IFNs trigger the expression of IFN-stimulated genes in infected cells, as well as in adjacent uninfected cells, which encode a variety of antiviral proteins, thus protecting them from viral invasion [27]. IFNs are major cytokines with antiviral, antiproliferative, and immunomodulatory functions against viral and bacterial infections and represent the first line of defense against IAV infection [28]. When the IFN response and leukocytes do not prevent IAV replication, the pro-inflammatory response and apoptosis are activated. The presence of the virus also triggers the adaptive immune response, which is mediated by T-cells and B-cells, and the production of IgM and IgG antibodies, which can interfere with different stages of the viral life cycle [25]. However, IAV proteins can inhibit the functions of IFN at all levels, helping viruses to avoid the antiviral response [27,29]. The impaired antiviral response and activity of type I and type II interferons as a result of IAV infection inhibit the function of neutrophils and macrophages, as well as the response of T-cells and NK cells [30,31,32,33,34].
Influenza viruses contain eight gene segments that, together, encode nine structural proteins and at least nine non-structural proteins with regulatory functions [35,36]. The most abundant envelope protein of the IAV is hemagglutinin (HA), which is incorporated into the lipid bilayer derived from the infected cell [37,38]. It is a glycoprotein responsible for the attachment of the virus to the host cell receptor—sialic acid—and the fusion of viral and endosomal membranes, enabling the viral genome to be released into the cytoplasm [23,39]. During uncoating, the matrix protein 2 (M2), with ion channel activity, plays an essential role [40]. The inner side of the virion is lined by matrix protein 1 (M1), which determines the shape of the viral particles and surrounds the eight segments of the ribonucleoprotein (RNP) complex comprising the nucleoprotein (NP) and negative-sense RNA. Each of the eight segments is bound by its own polymerase complex, composed of three proteins (PB1, PB2, and PA) responsible for the replication and transcription of the genome segments [41]. The viral replication and suppression of immune response against IAV infection are mediated mainly by the multi-functional non-structural protein 1 (NS1) [42,43]. Viral RNA is synthetized de novo in the nucleus and is exported to the cytoplasm by the nuclear export protein (NEP), also known as non-structural protein 2 (NS2), which is found freely in the mature virion [44,45]. Newly translated proteins are transported to the cell membrane, where virus particles are completed and released from the infected cell with help of the exo-sialidase activity of second immunodominant protein, neuraminidase (NA) [41]. Novel accessory proteins of IAV, including PB1-F2, PB1-N40, PA-X, PB2-S1, matrix protein M42, and NS3, have recently been discovered. These are products of alternative reading frames within IAV gene segments, and they provide a replication advantage to the virus or emerge as a consequence of IAV adaptation to a new host [23,24,39,46].
The pathogenesis of IAV infection is characterized by two phases. The first phase lasts approximately 1–3 days and determines the maximum level of viral titer and the extent of the associated inflammatory response. Depending on these two parameters, the second phase may result in the acquisition of control over the virus transmission or end up as a widespread disease associated with acute respiratory distress syndrome and death. The clinical course of the infection and the outcome of the pathogenetic processes of the viral infection are determined by both viral and host factors [22,47,48]. The viral factors influencing the course of infection include the proteins encoded by the IAV [49,50,51,52]. The most variable viral proteins are HA and NA. At present, 18 different HA sub-types and 11 NA sub-types are known, which together can create more than 130 IAV sub-type combinations [53]. This means that there is a possibility of more potentially dangerous variants emerging in the future. Here, we discuss the roles of particular IAV proteins in modulating viral infection, viral–bacterial co-infection, and immune response.
2.2. Bacterial Infection with Streptococcus pneumoniae
The human upper respiratory tract is a suitable environment for colonization by the various micro-organisms constituting the human microbiome [54,55,56]. A frequently occurring bacterial species in the upper respiratory tract is S. pneumoniae, which is a Gram-positive extracellular pathogen belonging to a group of approximately 100 different serotypes [57] depending on an important virulence factor—the polysaccharide capsule [58,59,60]. S. pneumoniae form a biofilm, a highly organized multi-cellular community of one or more bacterial types that produce an extracellular matrix and adhere to abiotic and biological surfaces such as the human nasopharynx. The main characteristic of this biofilm is high antibiotic resistance and the presence of a protective matrix on the surface of the biofilm, which enables the avoidance of immune surveillance by the host and helps to maintain the persistence and dissemination of bacteria in the host organism [11,61,62,63,64]. The nasopharyngeal environment, low temperatures (34 °C), and limited nutrient supply are essential for the formation of a bacterial biofilm [65,66]. Depending on the part of the human body where the bacterium enters, such as the spinal cord and brain, heart, middle ear, sinuses, or lungs, it can cause various diseases, including bacterial meningitis [67], endocarditis [68], otitis media [69], sinusitis [70,71], and pneumococcal pneumonia [59], respectively. The ability of the bacteria to enter the body is limited by the maturity of the immune system; therefore, young children, the elderly, and immunocompromised individuals are the most vulnerable groups [58]. The mechanisms responsible for the disease-related transition of bacterial colonization from an asymptomatic biofilm in the nasopharynx to an invasive phenotype are not yet known [63,72,73,74,75].
2.3. Co-Pathogenesis of IAV and Streptococcus pneumoniae
Infection with IAV paves the way for S. pneumoniae invasion by causing respiratory tract damage, manifested by impaired integrity of the epithelial barrier, inflammation, and elevated glucose availability [76]. Several very serious complications can occur in the lung, such as a decrease or loss of gas exchange function, oedema, or pleurisy [7,77], which appear mostly in immuno-compromised patients, individuals with underlying genetic conditions, children under the age of five, and elderly adults above the age of 65 years. Extensive tissue damage in the alveolar space also results in a loss of the repair ability [78,79,80,81]. Loss of healing response to lung damage and also the loss of basal epithelial cells required for airway epithelial regeneration (including alveolar epithelial cells type I and II) are associated with an increased number of cellular receptors for bacterial adhesion, the attachment of bacteria to the surface of these cells, and their apoptosis. Defective tissue integrity promotes the development of complications ranging from bacteremia to sepsis [76,82,83,84]. The clearance of bacteria is slowed by decreased mucociliary velocity [7,85] and by the disruption of immune mechanisms. The depletion of alveolar macrophages and neutrophils, along with the increased production of anti-inflammatory cytokines such as IL-10, TGF-β, type I interferons, and IFN-γ, creates suitable conditions for bacteria dispersing from the biofilm (see Figure 1) [54,61,63,72,76,86,87,88].
2.3.1. Disruption of Innate Immunity and Inflammatory Response during IAV and Bacterial Co-Infection
The first line of defense against respiratory pathogens includes the alveolar macrophages, which represent more than 90% of the immune cells found in the bronchoalveolar lavage fluid of a healthy individual [7]. These immune cells are specifically targeted by IAV, causing their depletion. As the proliferation and differentiation of new alveolar macrophages takes up to two weeks, the lung tissue is not sufficiently protected for this period of time, thus establishing a niche for the development of pneumococcal superinfection [89,90,91,92,93].
A crucial step in the development of a secondary bacterial infection is the disruption of the innate immunity and the inflammatory response. At the time of primary viral infection, when the viral titer decreases—usually 7 days after IAV infection—the anti-inflammatory response is very strongly induced, mainly by IL-10 or TGF-β produced by macrophages [94,95,96,97].
At this stage, IAV induces inhibition of the Th17 pathway, which suppresses bacterial clearance through NADPH-oxidase-dependent phagocytosis [31,98]. Previous recognition of viral dsRNA by the toll-like receptors (TLRs) of remaining alveolar macrophages causes an inability of the TLRs to recognize foreign bacterial products [90,91,99,100] and also triggers the production of IFN-γ by T-cells, preventing the clearance of S. pneumoniae by neutrophils and alveolar macrophages [32,101,102]. This step ensures that homeostasis is established but also reduces the ability of the immune system to recognize pathogens and to effectively defend itself [7,103,104].
In addition to the apoptosis of macrophages during IAV infection, the apoptosis of neutrophils [105,106], human dendritic cells [107], and NK cells [108] has also been observed. NK cells and T-cells produce immunomodulatory cytokines and mediate the cytotoxic response to viral infection; however, imbalances in their function during influenza infection can lead to an excessive inflammatory response and lung tissue damage [80,109,110,111,112].
2.3.2. Autophagy and Apoptosis Mediated by Influenza Infection
Influenza viruses are able to activate, as well as inhibit, the host-cell apoptotic process, depending on the phase of infection [113,114,115,116]. Apoptosis appears to be linked with autophagy during IAV infection [117,118,119,120], which is important for maintaining cellular homeostasis [121,122,123,124]. Interactions among several IAV proteins, such as PB1-F2, NA, HA, NS1, nucleoprotein (NP), matrix protein 1 (M1), and matrix protein 2 (M2), manipulate autophagy and apoptosis. Recent in vivo and in vitro studies have shown that autophagy is viral-strain-dependent [116,118,125,126,127,128].
IAV alters autophagy and apoptosis in favor of viral replication and the release of new viral particles. In the early phase of infection (see Figure 1), viral NS1 protein up-regulates the synthesis of HA and M2, thereby indirectly promoting the formation of autophagosomes [115,117,119,120,129,130,131,132]. Autophagosomes are transient double-membrane vesicles, which are usually fused by lysosomes in the process known as autophagosome maturation. During the terminal stage of IAV infection, M2 protein interfere with the formation of autophagolysosomes [129,133,134,135]. Accumulated viral antigens enwrapped in autophagosomes avoid recognition by the immune system and antiviral response. At this phase of IAV infection, M2–dependent inhibition of autophagy promotes apoptosis for effective replication [117,118,119,120,135] and, consequently, induces damage to lung epithelial cell as well as the production of anti-inflammatory cytokines. All of these processes triggered by IAV contribute to the development of secondary bacterial infection by S. pneumoniae [93,102,118,136].
3. Role of IAV Proteins in Viral and Bacterial Co-Infection
Many studies have shown that, during primary infection, the influenza virus proteins facilitate bacterial infection, colonization, and disease development by S. pneumoniae in individuals of all ages [6,86,137]. Different IAV proteins are involved in the progression of bacterial superinfection through various mechanisms (Figure 1). NA facilitates access to receptors and nutrients for S. pneumoniae, which promotes the development of bacterial infection [23,138,139]. NS1 and PB1-F2 affect the regulation of the interferon response, the disruption of the inflammatory response, and/or the manipulation of the processes of autophagy and apoptosis [35,140,141]. Other IAV proteins have not yet been shown to play a direct role in the development of bacterial co-infection, but M1 [142], NP [143,144], and HA [119,129] facilitate the manipulation of apoptosis. M2 protein is required for the activation of inflammasomes in macrophages and dendritic cells [145], which indirectly helps in secondary bacterial infection development (Table 1) [35,146,147].
3.1. Characterization of PB1-F2 Protein
PB1-F2 is a small accessory non-structural protein found in some influenza A virus strains, encoded by the a + 1 alternative open reading frame of the PB1 segment [133]. The length of the protein varies according to the host specificity. The original avian PB1-F2 is composed of 87–90 amino acids [187], in contrast to 11 amino acids in human and swine isolates [51]. Zoonotic IAV strains represent a reservoir of full-length PB1-F2 sequences and were introduced into the human population during the pandemics of IAV strains H1N1 (1918), H2N2 (1957), and H3N2 (1968), characterized by an increased incidence of secondary bacterial infections [188]. Full-length PB1-F2 is a phosphoprotein with two helical domains, a C-terminus formed by an extended α-helix, and an N-terminus formed by two short α-helices, connected by a flexible and unstructured hinge region [189]. The PB1-F2 molecule can form oligomeric structures and membrane pores in the lipid bilayer [190,191]. The protein is mainly localized in mitochondria, but it can also be present in the nucleus and cytoplasm of infected cells [133,192,193]. Several virulence-associated amino acid residues and motifs have been identified within PB1-F2. These motifs gave PB1-F2 both intracellular and extracellular functions, such as the strain-specific regulation of polymerase activity [194] or the exacerbation of viral pathogenesis in animal models, direct or indirect induction of apoptosis, and the modulation of innate immune responses. To a large extent, PB1-F2 also participates in the activation of neutrophils, alveolar macrophages, and dendritic cells and in their recruitment into the airways [146,156,195]. The effect of PB1-F2 seems to be cell-type-dependent. With respect to these properties, PB1-F2 protein is able to influence the course of infection and enhance not only viral infection [51,146,147,195,196] but also subsequent secondary bacterial infections on several levels.
3.1.1. Apoptosis and Cytotoxicity Mediated by PB1-F2
PB1-F2 itself increases IAV virulence by causing the cell death of the alveolar macrophages located in the lungs [197]. Their apoptosis leads to decreased antigen presentation, reduced initial viral clearance, and disrupted communication between alveolar macrophages and T-helper CD4+ cells. Consequently, the downstream effector functions of CD4+ cells, including the activation of cytotoxic T-lymphocytes (CTL), the production of antibodies, and the inflammatory response, are impaired [198]. Just one amino acid mutation at position 66 (N66S) can inhibit inflammasome activation and the IFN response induction, and increase the virulence of the virus [155]. PB1-F2 protein localized in mitochondria is able to promote apoptosis through mechanisms mediated by the mitochondrial targeting sequence at PB1-F2 C-terminus [199,200]. PB1-F2 localized in the outer mitochondrial membrane interacts with two mitochondrial membrane proteins involved in the formation of mitochondrial permeability transition pores, the voltage-dependent anion channel 1 (VDAC-1) and adenine nucleotide translocator 3 (ANT3). Their interaction causes the permeabilization of the mitochondrial outer membrane (MOMP), allowing cytochrome c efflux and resulting in apoptosis [150,201]. Moreover, PB1-F2 can form amyloid fibers and β-amyloid pore structures, leading to the permeabilization of cellular membranes and, subsequently, cell death [190]. The reduction of the pro-inflammatory response and promotion of apoptosis increase the frequency and severity of secondary bacterial infection in vivo [202].
The PB1-F2 protein of some IAV strains contains a cytotoxic sequence or “cytotoxic motif”, consisting of three amino acid residues (I68, L69 and V70) at the C-terminus, which can trigger cytotoxic death in immune and epithelial cells in lungs. This motif enhances immunopathological processes in lungs and accelerates the development of secondary bacterial infection [51,152].
3.1.2. PB1-F2 Pathogenic Markers Enhancing Secondary Bacterial Infection
At present, five specific amino acid markers found in the C-terminal region of PB1-F2 are recognized, which are associated with the induction of the host inflammatory response [154], as well as with complications after secondary bacterial infection [203]. The first marker, amino acid exchange at position 66 in the PB1-F2 sequence (N66S), correlates with pathogenicity, increased virulence [155], and early IFN response inhibition [148]. IFN suppression results in the increased susceptibility of the host to secondary bacterial infection. The other four markers in the C-terminal region—specifically L62, R75, R79, and L82—represent the PB1-F2 “proinflammatory domain” or “inflammatory motif” linked with significantly higher morbidity and mortality in mouse models of all pandemic IAVs from 1918, 1957, and 1968 [51,153]. These “inflammatory residues” enhance the development of secondary bacterial pneumonia, as they are responsible for increased lung destruction and significant pulmonary inflammation by inflammatory cells and proinflammatory cytokines present in the lungs. Interestingly, PB1-F2 protein without changes in these locations (i.e., motifs P62, H75, Q79, and S82) is referred to as “noninflammatory protein” and possesses antibacterial activity [153].
Another function of PB1-F2 that influences the inflammatory response is its ability to regulate the NLRP3-inflammasome in humans and mice, which consequently induces the secretion of the pyrogenic interleukin IL-1β [156,204,205]. NLRP3-inflammasome is a cytoplasmic multi-protein complex that is activated upon IAV infection. This complex mediates proteolysis of interleukins from the IL-1 family to their mature and fully active form [206]. After IAV is recognized by NOD-like receptors (NLRs), the NLRP3-inflammasome complex is up-regulated and, subsequently, mature IL-1β is secreted [157,207]. PB1-F2 protein has been associated with the activation of the NLRP3-inflammasome through various mechanisms, such as the induction of apoptosis, the inhibition of IFN I activation, the acidification of phagolysosomes, mitochondrial disruption, the activation of reactive oxygen species (ROS), and the formation of aggregates of the PB1-F2 C-terminal region [156,157,204,205,207,208]. At the same time, PB1-F2 can also inhibit the activation of the NLRP3-inflammasome by decreasing the mitochondrial membrane potential. PB1-F2 translocates into mitochondrial inner membrane space by the major outer mitochondrial membrane channel (Tom40). Accumulated PB1-F2 protein decreases the membrane potential and accelerates mitochondrial fragmentation. The resulting pathway appears to depend on the secondary structure of the PB1-F2 spliceosomes [158,205,209].
Overall, PB1-F2 is a critical virulence factor; its properties and functions differ according to its amino acid sequence, strain specificity, cell type, and host specificity [210]. Although the exact mechanism by which this protein influences the virulence or immunopathogenesis is not yet fully understood, it is clear that PB1-F2 is involved in the enhancement of pathological processes during IAV infection, which may lead to the development of secondary bacterial infection [153,156,202].
3.2. Characterization of Hemagglutinin
Hemagglutinin (HA) is the major surface glycoprotein of influenza virus, which is essential for the onset of viral infection. It is encoded by the fourth segment of IAV and, as one of the virulence factors, is the main target of the immune response [41,211,212,213]. The synthesis of HA takes place on the ribosomes of the rough endoplasmic reticulum as a precursor molecule HA0, which is post-translationally proteolytically cleaved into two glycopeptides, HA1 and HA2, which are linked by disulfide bonds [38,214]. Depending on the nature of the cleavage site, HA0 can be cleaved intracellularly or extracellularly. Multi-basic cleavage sites are cleaved intracellularly with subtilisin-like cellular proteases (furin, PC6), and monobasic cleavage sites are cleaved extracellularly with trypsin-like serine proteases (tryptase Clara, HAT-protease, TMPRSS2, and plasmin) [215,216,217]. HA forms on the viral surface homotrimers. The globular domain of HA trimer (formed by HA1) ensures initial contact with the target cell through binding to sialic acid receptors present on the cell surface. The stem domain, created mainly by HA2 [218], mediates the viral–endosomal membrane fusion after low-pH-induced HA structural rearrangement and plays an essential role in the release of influenza virus genome into the cytoplasm [219,220].
3.2.1. Changes of HA Cleavage during Viral and Bacterial Co-Infection
It has been shown that some bacteria influence the replication of the influenza virus and pathogenesis during secondary bacterial infection by promoting HA0 cleavage to its fusion-active form. This happens through the secretion of bacterial proteases or activation of cellular proteases [221]. The direct effect of proteases on HA cleavage and exacerbation of infection has been observed during co-infection with bacterial strains such as Staphylococcus aureus [222], Aerococcus viridans [223], Streptomyces griseus [224], and Stenotrophomonas maltophilia [225]. However, Callan et al. have described a reduction in viral infectivity after HA cleavage at an alternative amino acid sequence of the cleavage site with Pseudomonas aeruginosa protease [226]. Some bacteria in the respiratory tract produce proteins known as streptokinases and staphylokinases, which are capable of forming complexes that convert host enzymes into their active forms (e.g., plasmin, kallikrein, thrombin) and, thus, may indirectly increase the cleavage of the HA glycoprotein and spread of viral infection [223,227,228,229]. However, none of the aforementioned mechanisms are used during secondary bacterial infection with S. pneumoniae. As S. pneumoniae is able to bind plasminogen and host-derived activator onto its surface [230,231,232] and transport both proteins to the site of viral infection, it can possibly facilitate and increase the cleavage of viral HA into its active form [233]. In the case of viral–bacterial co-infection, the interaction between HA and bacteria leads to the promotion of viral infection. Viral HA, expressed on the surface of infected pneumocytes or on free extracellular virions, can bind to ligands on the polysaccharide capsule of Staphylococcus aureus [234], Streptococcus pyogenes [235], and Streptococcus agalactiae [236] and ameliorate bacterial internalization.
3.2.2. The Role of Hemagglutinin in the Autophagy
HA of IAV is one of the proteins involved in the regulation of autophagy. Wang et al. have observed autophagy induced by HA binding to heat shock protein 90AA1 (HSP90AA1) present on the cell surface [159]. This strategy is exploited by IAV to prolong the time during which the virus replicates [118]. The process of exacerbation of the disease due to pneumonia is multi-factorial and dependent on viral, bacterial, and host factors [82]. The ways in which the other HA properties, such as HA tropism and affinity to sialic acid, extent of HA glycosylation, and the optimum pH for HA structural rearrangement into its fusion-active conformation, influence the development of bacterial superinfection are poorly understood at present [160,237].
3.3. Characterization of Neuraminidase
Neuraminidase (NA) and hemagglutinin (HA) are the major surface proteins of influenza viruses. NA is a tetramer comprising four identical monomers. Each NA monomer consists of four structural domains, namely, the catalytic head, the stem, the transmembrane region, and the cytoplasmic tail [23,238]. Four catalytic head domains create the enzymatic site with exo-sialidase activity. The stalk domain contributes to the stability of the NA tetramer. The structure and length of the stem differ among viral strains. Its length, in particular, affects the ability of the virus to replicate [238,239,240]. The transmembrane domain anchors the NA tetramer to the viral envelope, and its signals are necessary for translocation from the endoplasmic reticulum. The fourth domain—the cytoplasmic tail—is thought to interact with matrix M1 viral protein and affects virion morphology and virulence [238,241,242,243]. Neuraminidase is an enzyme that cleaves the terminal α-glycosidic bond between N-acetyl-neuraminic acid (sialic acid) and the carbohydrate residues of glycopeptides or glycolipids on the cell surface. The NA of influenza viruses functions at multiple levels during IAV infection. NA activity enables the release of de novo synthesized budding viral particles from the infected cell surface and prevents virion aggregation. This cleavage mechanism of NA prevents not only the aggregation of emerging virions but also the rebinding of these virions to the dying host cell [23,238,244]. Viral NA also cleaves neuraminic acid residues from airway mucin, allowing the movement of the virus into target cells [23,238,245]. Another function is its role in enhancing HA-mediated membrane fusion [238,246].
3.3.1. The Role of the Viral and Bacterial Neuraminidases in Co-Infection
Many bacterial receptors are coated with carbohydrates covered by sialic acids. Therefore, most bacterial strains synthesize their own bacterial neuraminidases, which cleave sialic acids on the host cell surfaces and allow for their adhesion. S. pneumoniae expresses three types of neuraminidases: NanA, NanB, and NanC [247,248]. The most expressed and active is NanA, with a conserved catalytic site present in all strains. Although viral and pneumococcal neuraminidases have different quaternary structures (bacterial NanA is a monomer), their active sites are similar but have different substrate specificities [249,250]. The epithelium of the upper part of the respiratory tract is densely coated with α2,6-linked sialic acids, while α2,3-linked sialic acids prevail in the lower respiratory tract. Microbial NanA and viral NA can cleave sialic acids bound by both types of glycosidic bonds [249]. S. pneumoniae NanA facilitates colonization of the respiratory tract by cleaving sialic acid from mucin and reducing the viscosity of the mucus. Influenza infection increases the production of mucus, which serves as a source of nutrients for S. pneumoniae [6]. Therefore, after previous infection with influenza viruses, pneumococci can adhere to the pulmonary epithelium and invade host cells to a greater extent. This has been described especially for more virulent strains of the H3N2 sub-type, revealing relatively high viral neuraminidase activity. Several sources have suggested viral–bacterial neuraminidase interactions [18,77,251,252].
The NA activity of IAV alters the glycosylation of the host cell surface in the airway epithelium, thus affecting local and systemic immune responses and enhancing the development of bacterial infection [138,139]. Neuraminidases of IAV and S. pneumoniae can desialylate surface glycans on lung epithelial cells and expose sub-terminal galactosyl groups as ligands for soluble β-galactoside-binding proteins (i.e., galectins).
3.3.2. Cooperation of NA and Galectins during Co-Infection
Galectins play a key regulatory role in cell development and immune homeostasis. They are involved in cell activation, differentiation, and signaling. Galectins also serve as immune recognition receptors and effector factors in leukocyte recruitment and apoptosis and mediate host–pathogen interactions [253,254,255,256,257,258]. Yang et al. have recently identified three galectins with anti-influenza effects—Gal-1, Gal-2, and Gal-3—released after IAV infection from various cells in the respiratory tract [258]. Current studies have confirmed that Gal-1, Gal-3, and Gal-9 alleviate the overall course of influenza infection by blocking viral binding and strengthening cell immune responses [162,258,259,260,261,262].
It has been found that primary IAV infection and subsequent S. pneumoniae infection increase galectin expression and release, including Gal-1 and Gal-3, which bind strongly to glycans on the surface of both pathogens. Subsequently, galectins, including galectin-3, bound on the surface of S. pneumoniae interact with ligands and mediate the adhesion of S. pneumoniae to epithelial cells and, as has been shown in both in vitro and in vivo models, support S. pneumoniae invasion during co-infection [162,263,264,265,266,267]. This interplay between galectins and viral NA may determine the intensity of pneumococcal infection.
3.3.3. Impact of the Viral NA Activity on the Innate Immunity
Neuraminidases of the influenza virus, as well as from S. pneumoniae, are some of the few reported pathogen-encoded molecules that directly activate an important cytokine, transforming growth factor β (TGF-β). TGF-β is an important mediator of interactions between the infectious pathogen and its host and is constantly expressed in the body in the form of mRNA or present as a latent protein. Thus, it rapidly becomes accessible in the event of a primary infection. The latent TGF-β consists of an N-terminal latency-associated peptide (LAP), which is non-covalently linked to a C-terminal mature TGF-β1 molecule. The release of mature TGF-β from LAP by NA activates the TGF-β, which binds to cellular receptors and induces a biological response [163,268,269]. In the respiratory tract, the expression of the TGF-β1 isoform controls the differentiation, proliferation, and activation status of leukocytes. TGF-β is an immunosuppressive molecule, and influenza infection potentiates its immunosuppressive ability. Elevated TGF-β levels can induce apoptosis of both immune cells and lung epithelial cells, confirming the potential role of NA in the pathogenesis not only of viral infection, but also of bacterial co-infection [163,270,271,272]. TGF-β, a regulator of the adaptive immune response, reduces the number of activated cytotoxic T-cells and induces the production of IL-10 by regulatory T-cells, thus facilitating lung colonization by S. pneumoniae [164,273]. Although TGF-β is not an effector cytokine that can mediate bacterial clearance, Roberts et al. have shown that hosts with allergic airway disease, which induces TGF-β production, were protected against severe influenza and bacterial co-infection [273]. On the contrary, Li et al. have demonstrated that TGF-β directly activated by influenza virus NA promotes the expression of cellular adhesins, leading to decreased bacterial clearance and increased colonization. Therefore, the exact role of NA-activated TGF-β in co-infection remains unclear [164].
3.4. Characterization of NS1
Non-structural protein 1 (NS1) has many functions and is known as a major viral antagonist of the interferon response [274,275,276]. Thus, NS1 represents a very important virulence factor affecting the pathogenesis of influenza disease, as well as viral and bacterial co-infection [35,141,277].
NS1 is a product of alternative splicing of the NS gene, occurring as a homodimer. The monomer is composed of two functional domains, the N-terminal RNA-binding domain (RBD) and the C-terminal effector domain, which are joined by a short inter-domain linker region [35,141,278]. Each domain interacts with different cellular factors. The homodimer of NS1 can bind to various species of RNA—most importantly, to double-stranded RNA (dsRNA) by RBD at the N-terminus. This binding property influences multiple functions of cellular proteins critical in the development of secondary bacterial infection, consequently affecting the onset, course, and severity of the bacterial co-infection [35,140,277]. The most significant is the influence of NS1 on the type I IFN response as an antagonist of type I IFN signaling. NS1 also interacts with other IAV and host proteins, controlling the autophagy and apoptosis of lung epithelial cells [279,280,281,282].
3.4.1. NS1 Interaction with Interferon Signaling Pathways Enhances the Development of Secondary Bacterial Infection
After the IAV invades the host, it is recognized by a cellular pathogen sensor called retinoic acid-inducible gene-I (RIG-I). Subsequently, the RIG-I-mediated signaling pathway induces the expression of interferons [283]. However, NS1 is capable of inhibiting this pathway in several ways: first, by the direct interaction with E3 ubiquitin ligases TRIM25 [166] and Riplet [167], which prevents the activation of the RIG-I receptor; second, indirectly by interaction with host factors through its effector domain at the C-terminus, which leads to the inhibition of IRF3 phosphorylation [141,174] and subsequent inhibition of the type I IFN response mediated by IRF-3, as well as interferon-stimulated genes (ISGs) [141,175,284]. These steps result in the prevention of virus detection by host cells [166,276,285,286,287]. Next, NS1 can bind its RBD domain to RIG-I and several other proteins with dsRNA-binding ability and block their activation—for example, dsRNA-dependent serine/threonine–protein kinase R (PKR) [168,276,288].
PKR is a crucial protein responsible for the host antiviral response. It activates nuclear factor κB (NF-κB), thus contributing to type I IFN response [168,276]. During IAV infection, NF-κB regulates the expression of many cytokine and chemokine genes, including the antiviral cytokine IFNβ [289]. PKR activates IKκB, part of the IKK kinase complex, which phosphorylates the NF-κB inhibitor IκB, resulting in the activation of NF-κβ [290,291]. PKR also significantly slows down viral protein synthesis through phosphorylation of eukaryotic initiation factor-2 α-subunit protein (eIF-2α). Binding with NS1 prevents its activation, which blocks subsequent antiviral responses [168,276,292]. IAV viruses with defective or deleted NS1 (delNS1 mutants) are unable to block PKR activation [169]. They can, however, replicate in the absence of PKR, which proves the role of NS1 in counteracting the PKR-mediated antiviral response [170]. Furthermore, the RBD of NS1 can also compete with the RNA-binding capacity of oligoadenylate synthase (OAS), which is able to cleave viral RNA by the activation of RNase L. Such degraded viral RNA elements can be recognized by RIG-I receptors that activate IFN production [141,172,274]. All of these steps and functions weaken the immune response and prevent the detection of the virus in the host cells, suggesting the important role of NS1 in enhancing the development of secondary bacterial infection [33].
3.4.2. NS1 Motif Directly Involved in Co-Infection with S. pneumoniae
The structure and function of NS1 differ among types of influenza viruses, and even among sub-types, and may be used to predict the potential of a particular strain to enhance the development of bacterial co-infection [29,203]. It has been reported that influenza infection with A/Puerto Rico/8/34-H1N1 (PR8) virus impaired IFN-β production, which led to increased susceptibility to secondary bacterial infection [282]. At 7 dpi, when the organism is most susceptible to the development of secondary bacterial infection [6], minimal IFN-β production [43,282,285] and extensive inflammatory response with insufficient bacterial clearance were observed [9,31,293]. This environment provides suitable conditions allowing for the rapid spread of bacteria, thus causing tissue damage. In 2019, Shepardson et al. have identified HA and NS1 of the influenza virus as individual regulators of secondary bacterial infection severity and described a motif binding PDZ (PDZ-bm) present at the C-terminus of the NS1 protein of the PR8. To unveil its function, the PDZ-bm domain was deleted from the NS1 protein of PR8, and the resulting virus was not capable of lowering the expression of IFN-β. Thus, they identified the PDZ-bm sequence, which is directly involved in controlling the susceptibility to secondary bacterial infection through the regulation of the IFN-β response [282]. As there are significant differences among influenza virus strains in this region, it was suggested that NS1 proteins from different viruses have different impacts on the host susceptibility to secondary bacterial infection.
3.4.3. NS1 Manipulates Apoptosis
NS1 exhibits a complex role in the modulation of apoptosis, depending on various factors including the virus strain and the cell type. It is well-known that NS1 can, for example, directly bind to the linker region of PKR by its effector domain and block PKR-mediated apoptosis by preventing its conformational change and autophosphorylation of PKR. NS1 can also activate the host cell phosphatidylinositol 3-kinase (PI3K) pathway and, in this way, impair apoptosis [276,277]. The NS1 of H3N2 and H5N1 sub-types interact with heatshock protein Hsp90 and mediate the apoptosis of lymphocytes through the caspase cascade [181]. However, the interactions between NS1 and apoptotic pathways are still not well-understood and need to be further described in the future.
4. Conclusions
Influenza A virus infections in humans cause an acute respiratory disease with a spectrum of clinical symptoms. In many patients, the disease can be life-threatening and associated with various complications, depending on the comorbidities of the infected individual. The most frequently occurring complications are linked with bacterial co-infections or secondary superinfections, which enhance the pathological processes triggered by the IAV infection.
At present, many unanswered questions remain regarding the role of the host and its defense mechanisms in infection control, as well as about the effect of bacterial co-infection on the course of influenza infection and the host immune response. Additionally, many questions arise concerning the role of the primary viral infection and the function of viral proteins in this process [2]. Influenza infection damages lung tissue and promotes bacterial colonization through the alteration of the cytokine response. After the virus disrupts the epithelial barrier and induces the production of cytokines, thus reducing the number of Th17 cells in the lungs while also disrupting macrophage function and the production of suppressive cytokines released by regulatory T-cells, the primary influenza infection can lead to the development of a bacterial superinfection. Thus, the innate immune response does not function properly after being affected by viral proteins such as HA, NA, PB1-F2, and NS1. At least these IAV proteins are responsible for the disruption of the innate immunity and inflammatory response during viral and bacterial co-infection, as well as for the inability to trigger the antiviral response of the infected host. However, the synergy of the pathological impact of influenza–bacterial co-infections is a complex process, where not only viral but also bacterial and host factors play an important role. While indirect evidence of the roles of the various viral proteins in the pathogenesis of secondary bacterial infection has been supported by animal studies, exact direct clinical evidence in humans is, in general, lacking. Not only has such synergy has been observed during IAV pandemics or epidemics, but there are now data regarding similar complications in humans during SARS-CoV-2 infections. Understanding the mechanisms causing the devastating pneumonia through viral–bacterial co-infections can bring insight into the finding of targets for effective antiviral drugs or may help to improve prevention efforts through effective vaccines, consequently preventing the severe course of potentially dangerous respiratory infections.
Author Contributions
Conceptualization, K.T., M.M. and E.V.; Resources, M.M. and K.T.; Writing—original draft preparation, M.M. and K.T.; Writing—review and editing, K.B., E.V., F.K.; Funding acquisition, E.V. and K.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by research grants APVV-17–0445 (EV) from The Slovak Research and Development Agency of the Slovak Republic and grants VEGA No. 2/0048/19 (EV) and VEGA No. 2/0090/21 (KT) from The Scientific Grant Agency of the Ministry of Education, Science, Research and Sport of Slovak Republic and Slovak Academy of Sciences.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article.
Acknowledgments
The authors thank to Katarína Polčicová for critically reading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- The Top 10 Causes of Death. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 3 April 2022).
- Gavigan, P.; McCullers, J.A. Influenza: Annual seasonal severity. Curr. Opin. Pediatr. 2019, 1, 112–118. [Google Scholar] [CrossRef]
- Smith, A.M. Host-pathogen kinetics during influenza infection and coinfection: Insights from predictive modeling. Immunol. Rev. 2018, 285, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Mehta, D.; Petes, C.; Gee, K.; Basta, S. The Role of Virus Infection in Deregulating the Cytokine Response to Secondary Bacterial Infection. J. Interferon Cytokine Res. 2015, 35, 925–934. [Google Scholar] [CrossRef]
- Metersky, M.L.; Masterton, R.G.; Lode, H.; File, T.M., Jr.; Babinchak, T. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int. J. Infect. Dis. 2012, 16, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary Bacterial Infections Associated with Influenza Pandemics. Front. Microbiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Xie, J.; Zhao, J.; Cao, D.; Liang, Y.; Hou, X.; Wang, L.; Li, Z. Mechanisms of Severe Mortality-Associated Bacterial Co-infections Following Influenza Virus Infection. Front. Cell Infect. Microbiol. 2017, 7, 338. [Google Scholar] [CrossRef]
- Martin-Loeches, I.; van Someren Gréve, F.; Schultz, M.J. Bacterial pneumonia as an influenza complication. Curr. Opin. Infect. Dis. 2017, 30, 201–207. [Google Scholar] [CrossRef]
- Metzger, D.W.; Sun, K. Immune Dysfunction and Bacterial Co-Infections following Influenza. J. Immunol. 2013, 191, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.G.; Sanders, E.A.; Van Der Ende, A.; Van Loon, A.M.; Hoes, A.W.; Hak, E. Invasive pneumococcal and meningococcal disease: Association with influenza virus and respiratory syncytial virus activity? Epidemiol. Infect. 2008, 136, 1448–1454. [Google Scholar] [CrossRef]
- Weiser, J.N.; Ferreira, D.M.; Paton, J.C. Streptococcus pneumoniae: Transmission, colonization and invasion. Nat. Rev. Microbiol. 2018, 16, 355–367. [Google Scholar] [CrossRef]
- Chertow, D.S.; Memoli, M.J. Bacterial Coinfection in Influenza. JAMA 2013, 309, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Briestenská, K.; Mikušová, M.; Tomčíková, K.; Kostolanský, F.; Varečková, E. Quantification of bacteria by in vivo bioluminescence imaging in comparison with standard spread plate method and reverse transcription quantitative PCR (RT-qPCR). Arch. Microbiol. 2021, 203, 4737–4742. [Google Scholar] [CrossRef]
- Seki, M.; Kosai, K.; Yanagihara, K.; Higashiyama, Y.; Kurihara, S.; Izumikawa, K.; Miyazaki, Y.; Hirakata, Y.; Tashiro, T.; Kohno, S. Disease Severity in Patients with Simultaneous Influenza and Bacterial Pneumonia. Intern. Med. J. 2007, 46, 953–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- COVID-19 & Antibiotic Resistance. Available online: https://www.cdc.gov/drugresistance/covid19.html (accessed on 3 April 2022).
- Mirzaei, R.; Goodarzi, P.; Asadi, M.; Soltani, A.; Aljanabi, H.; Jeda, A.S.; Dashtbin, S.; Jalalifar, S.; Mohammadzadeh, R.; Teimoori, A.; et al. Bacterial co-infections with SARS-CoV-2. IUBMB Life 2020, 72, 2097–2111. [Google Scholar] [CrossRef] [PubMed]
- Sender, V.; Hentrich, K.; Henriques-Normark, B. Virus-Induced Changes of the Respiratory Tract Environment Promote Secondary Infections With Streptococcus pneumoniae. Front. Cell. Infect. Microbiol. 2021, 11, 643326. [Google Scholar] [CrossRef]
- McCullers, J.A. Insights into the Interaction between Influenza Virus and Pneumococcus. Clin. Microbiol. Rev. 2006, 19, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef]
- Fislová, T.; Gocník, M.; Sládková, T.; Durmanová, V.; Rajcáni, J.; Varecková, E.; Mucha, V.; Kostolanský, F. Multiorgan distribution of human influenza A virus strains observed in a mouse model. Arch. Virol. 2009, 154, 409–419. [Google Scholar] [CrossRef]
- Kuiken, T.; Riteau, B.; Fouchier, R.A.M.; Rimmelzwaan, G.F. Pathogenesis of influenza virus infections: The good, the bad and the ugly. Curr. Opin. Virol. 2012, 2, 276–286. [Google Scholar] [CrossRef]
- Peteranderl, C.; Herold, S.; Schmoldt, C. Human Influenza Virus Infections. Semin. Respir. Crit. Care Med. 2016, 37, 487–500. [Google Scholar] [CrossRef]
- Francis, M.E.; King, M.L.; Kelvin, A.A. Back to the Future for Influenza Preimmunity—Looking Back at Influenza Virus History to Infer the Outcome of Future Infections. Viruses 2019, 11, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, L.P.; Teixeira, M.M.; Garcia, C.C. The inflamatory response triggered by Influenza virus: A two edged sword. Inflamm. Res. 2017, 66, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- LeMessurier, K.S.; Tiwary, M.; Morin, N.P.; Samarasinghe, A.E. Respiratory Barrier as a Safeguard and Regulator of Defense Against Influenza A Virus and Streptococcus pneumoniae. Front. Immunol. 2020, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Shim, J.M.; Kim, J.; Tenson, T.; Min, J.Y.; Kainov, D.E. Influenza Virus Infection, Interferon Response, Viral Counter-Response, and Apoptosis. Viruses 2017, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Metcalf, J.P. The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J. Innate Immun. 2020, 12, 437–447. [Google Scholar] [CrossRef]
- Nogales, A.; Aydillo, T.; Ávila-Pérez, G.; Escalera, A.; Chiem, K.; Cadagan, R.; Dediego, M.L.; Li, F.; García-Sastre, A.; Martínez-Sobrido, L. Functional Characterization and Direct Comparison of Influenza A, B, C, and D NS1 Proteins in vitro and in vivo. Front. Microbiol. 2019, 10, 2862. [Google Scholar] [CrossRef]
- Chen, W.H.; Toapanta, F.R.; Shirey, K.A.; Zhang, L.; Giannelou, A.; Page, C.; Frieman, M.B.; Vogel, S.N.; Cross, A.S. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol. Lett. 2012, 141, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011, 186, 1666–1674. [Google Scholar] [CrossRef] [Green Version]
- Shahangian, A.; Chow, E.K.; Tian, X.; Kang, J.R.; Ghaffari, A.; Liu, S.Y.; Belperio, J.A.; Cheng, G.; Deng, J.C. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Investig. 2009, 119, 1910–1920. [Google Scholar] [CrossRef]
- Shepardson, K.M.; Larson, K.; Morton, R.V.; Prigge, J.R.; Schmidt, E.E.; Huber, V.C.; Rynda-Apple, A. Differential Type I Interferon Signaling Is a Master Regulator of Susceptibility to Postinfluenza Bacterial Superinfection. MBio 2016, 7, e00506-16. [Google Scholar] [CrossRef] [Green Version]
- Techasaensiri, B.; Techasaensiri, C.; Mejias, A.; McCracken, G.H., Jr.; Ramilo, O. Viral coinfections in children with invasive pneumococcal disease. Pediatr. Infect. Dis. J. 2010, 29, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Wang, L.; Li, S. Roles of the Non-Structural Proteins of Influenza A Virus. Pathogens 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martínez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chlanda, P.; Zimmerberg, J. Protein-lipid interactions critical to replication of the influenza A virus. FEBS Lett. 2016, 590, 1940–1954. [Google Scholar] [CrossRef] [Green Version]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [Green Version]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Li, S.; Sieben, C.; Ludwig, K.; Höfer, C.T.; Chiantia, S.; Herrmann, A.; Eghiaian, F.; Schaap, I.A. pH-Controlled two-step uncoating of influenza virus. Biophys. J. 2014, 106, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.L.; Palese, P. Orthomyxoviridae: The Viruses and Their Replication. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P., Eds.; Lippincott Williams & Wilkins, A Wolters Kluwer: Philadelphia, PA, USA, 2013; Volume 40, pp. 1151–1185. ISBN 978-1-45-110563-6. [Google Scholar]
- Engel, D. The influenza virus NS1 protein as a therapeutic target. Antivir. Res. 2013, 99, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, J.; Chen, Z. A second CRM1-dependent nuclear export signal in the influenza A virus NS2 protein contributes to the nuclear export of viral ribonucleoproteins. J. Virol. 2013, 87, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, D.; Fodor, E. Emerging Roles for the Influenza A Virus Nuclear Export Protein (NEP). PLoS Pathog. 2012, 8, e1003019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and viral determinants of influenza A virus species specificity. Nat. Rev. Microbiol. 2018, 17, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Boianelli, A.; Nguyen, V.K.; Ebensen, T.; Schulze, K.; Wilk, E.; Sharma, N.; Stegemann-Koniszewski, S.; Bruder, D.; Toapanta, F.R.; Guzmán, C.A.; et al. Modeling Influenza Virus Infection: A Roadmap for Influenza Research. Viruses 2015, 7, 5274–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.; Nogales, A.; Martínez-Sobrido, L. Influenza A Virus Studies in a Mouse Model of Infection. JoVE 2017, 127, 55898. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, S.; Kawaoka, Y. The pathogenesis of influenza virus infections: The contributions of virus and host factors. Curr. Opin. Immunol. 2011, 23, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Godlee, A.; Almond, M.H.; Dong, T. Pathogenesis of influenza: Virus–host interactions. Expert Rev. Anti Infect. Ther. 2011, 9, 573–575. [Google Scholar] [CrossRef]
- Kamal, R.P.; Alymova, I.V.; York, I.A. Evolution and Virulence of Influenza A Virus Protein PB1-F2. Int. J. Mol. Sci. 2017, 19, 96. [Google Scholar] [CrossRef] [Green Version]
- Košík, I.; Práznovská, M.; Košíková, M.; Bobišová, Z.; Hollý, J.; Varečková, E.; Kostolanský, F.; Russ, G. The Ubiquitination of the Influenza A Virus PB1-F2 Protein Is Crucial for Its Biological Function. PLoS ONE 2015, 10, e0118477. [Google Scholar] [CrossRef] [Green Version]
- Types of Influenza Viruses. Available online: https://www.cdc.gov/flu/about/viruses/types.htm (accessed on 3 April 2022).
- Bakaletz, L.O. Viral–bacterial co-infections in the respiratory tract. Curr. Opin. Microbiol. 2017, 35, 30–35. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumpitsch, C.; Koskinen, K.; Schöpf, V.; Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streptococcus Pneumoniae. Available online: https://www.cdc.gov/pneumococcal/clinicians/streptococcus-pneumoniae.html (accessed on 3 April 2022).
- Brooks, L.R.K.; Mias, G.I. Streptococcus pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front. Immunol. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, A.; Weiser, J.N.; Paton, J.C.; Andrew, P.W. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 2008, 6, 288–301. [Google Scholar] [CrossRef]
- Paton, J.C.; Trappetti, C. Streptococcus pneumoniae Capsular Polysaccharide. Microbiol. Spectr. 2019, 7, 33. [Google Scholar] [CrossRef]
- Chao, Y.; Marks, L.R.; Pettigrew, M.M.; Hakansson, A.P. Streptococcus pneumoniae biofilm formation and dispersion during colonisation and disease. Front. Cell. Infect. Microbiol. 2015, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Domenech, M.; García, E.; Moscoso, M. Biofilm formation in Streptococcus pneumoniae. Microb. Biotechnol. 2012, 5, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.R.; Reddinger, R.M.; Hakansson, A.P. High Levels of Genetic Recombination during Nasopharyngeal Carriage and Biofilm Formation in Streptococcus pneumoniae. MBio 2012, 3, e00200-12. [Google Scholar] [CrossRef] [Green Version]
- Shak, J.R.; Vidal, J.E.; Klugman, K.P. Influence of bacterial interactions on pneumococcal colonization of the nasopharynx. Trends Microbiol. 2013, 21, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Bogaert, D.; De Groot, R.; Hermans, P. Streptococcus pneumoniae colonisation: The key to pneumococcal disease. Lancet Infect. Dis. 2004, 4, 144–154. [Google Scholar] [CrossRef]
- Henriques-Normark, B.; Tuomanen, E.I. The Pneumococcus: Epidemiology, Microbiology, and Pathogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a010215. [Google Scholar] [CrossRef] [PubMed]
- Yau, B.; Hunt, N.; Mitchell, A.; Too, L. Blood–Brain Barrier Pathology and CNS Outcomes in Streptococcus pneumoniae Meningitis. Int. J. Mol. Sci. 2018, 19, 3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Egea, V.; Muñoz, P.; Valerio, M.; de Alarcón, A.; Lepe, J.A.; Miró, J.M.; Gálvez-Acebal, J.; García-Pavía, P.; Navas, E.; Goenaga, M.A.; et al. Characteristics and Outcome of Streptococcus pneumoniae Endocarditis in the XXI Century: A Systematic Review of 111 Cases (2000–2013). Medicine 2015, 94, e1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergenfelz, C.; Hakansson, A.P. Streptococcus pneumoniae Otitis Media Pathogenesis and How It Informs Our Understanding of Vaccine Strategies. Curr. Otorhinolaryngol. 2017, 5, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Peña, M.T.; Preciado, D.; Orestes, M.; Choi, S. Orbital complications of acute sinusitis: Changes in the post-pneumococcal vaccine era. JAMA Otolaryngol. Head Neck Surg. 2013, 139, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Tinkelman, D.G.; Silk, H.J. Clinical and Bacteriologic Features of Chronic Sinusitis in Children. Am. J. Dis. Child. 1989, 143, 938–941. [Google Scholar] [CrossRef]
- Kim, P.E.; Musher, D.M.; Glezen, W.P.; Rodriguez-Barradas, M.C.; Nahm, W.K.; Wright, C.E. Association of Invasive Pneumococcal Disease with Season, Atmospheric Conditions, Air Pollution, and the Isolation of Respiratory Viruses. Clin. Infect. Dis. 1996, 22, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Klugman, K.P.; Chien, Y.W.; Madhi, S.A. Pneumococcal pneumonia and influenza: A deadly combination. Vaccine 2009, 27, C9–C14. [Google Scholar] [CrossRef]
- Nair, H.; Brooks, W.A.; Katz, M.; Roca, A.; Berkley, J.A.; Madhi, S.A.; Simmerman, J.M.; Gordon, A.; Sato, M.; Howie, S.; et al. Global burden of respiratory infections due to seasonal influenza in young children: A systematic review and meta-analysis. Lancet 2011, 378, 1917–1930. [Google Scholar] [CrossRef] [Green Version]
- Rudd, J.M.; Ashar, H.K.; Chow, V.T.; Teluguakula, N. Lethal Synergism between Influenza and Streptococcus pneumoniae. J. Infect. Pulm. Dis. 2016, 2, 10-16966. [Google Scholar] [CrossRef]
- Pettigrew, M.M.; Marks, L.R.; Kong, Y.; Gent, J.F.; Roche-Hakansson, H.; Hakansson, A.P. Dynamic Changes in the Streptococcus pneumoniae Transcriptome during Transition from Biofilm Formation to Invasive Disease upon Influenza A Virus Infection. Infect. Immun. 2014, 82, 4607–4619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullers, J.A.; Bartmess, K.C. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J. Infect. Dis. 2003, 187, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, E.R.; Lenz, L.L. Inflammation as a Modulator of Host Susceptibility to Pulmonary Influenza, Pneumococcal, and Co-Infections. Front. Immunol. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Brealey, J.C.; Sly, P.D.; Young, P.R.; Chappell, K.J. Viral bacterial co-infection of the respiratory tract during early childhood. FEMS Microbiol. Lett. 2015, 362, fnv062. [Google Scholar] [CrossRef] [Green Version]
- Gounder, A.P.; Boon, A.C.M. Influenza Pathogenesis: The Effect of Host Factors on Severity of Disease. J. Immunol. 2019, 202, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Kash, J.C.; Taubenberger, J.K. The Role of Viral, Host, and Secondary Bacterial Factors in Influenza Pathogenesis. Am. J. Pathol. 2015, 185, 1528–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oggioni, M.R.; Trappetti, C.; Kadioglu, A.; Cassone, M.; Iannelli, F.; Ricci, S.; Andrew, P.W.; Pozzi, G. Switch from planktonic to sessile life: A major event in pneumococcal pathogenesis. Mol. Microbiol. 2006, 61, 1196–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taubenberger, J.K.; Kash, J.C. Insights on influenza pathogenesis from the grave. Virus Res. 2011, 162, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Pittet, L.A.; Hall-Stoodley, L.; Rutkowski, M.R.; Harmsen, A.G. Influenza Virus Infection Decreases Tracheal Mucociliary Velocity and Clearance of Streptococcus pneumoniae. Am. J. Respir. Cell Mol. 2010, 42, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Low, D. Reducing antibiotic use in influenza: Challenges and rewards. Clin. Microbiol. Infect. 2008, 14, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.R.; Davidson, B.A.; Knight, P.R.; Hakansson, A.P. Interkingdom Signaling Induces Streptococcus pneumoniae Biofilm Dispersion and Transition from Asymptomatic Colonization to Disease. MBio 2013, 4, e00438-13. [Google Scholar] [CrossRef] [Green Version]
- McNamee, L.A.; Harmsen, A.G. Both Influenza-Induced Neutrophil Dysfunction and Neutrophil-Independent Mechanisms Contribute to Increased Susceptibility to a Secondary Streptococcus pneumoniae Infection. Infect. Immun. 2006, 74, 6707–6721. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, M.N.; Standiford, T.J. Postinfluenza bacterial pneumonia: Host defenses gone awry. J. Interferon Cytokine Res. 2010, 30, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Ghoneim, H.E.; Thomas, P.G.; McCullers, J.A. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 2013, 191, 1250–1259. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Chen, C.J.; Mullarkey, C.E.; Hamilton, J.R.; Wong, C.K.; Leon, P.E.; Uccellini, M.B.; Chromikova, V.; Henry, C.; Hoffman, K.W.; et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat. Commun. 2017, 8, 846. [Google Scholar] [CrossRef]
- Nicol, M.Q.; Dutia, B.M. The role of macrophages in influenza A virus infection. Future Virol. 2014, 9, 847–862. [Google Scholar] [CrossRef]
- Wilden, J.J.; Jacob, J.C.; Ehrhardt, C.; Ludwig, S.; Boergeling, Y. Altered Signal Transduction in the Immune Response to Influenza Virus and S. pneumoniae or S. aureus Co-Infections. Int. J. Mol. Sci. 2021, 22, 5486. [Google Scholar] [CrossRef] [PubMed]
- LeVine, A.M.; Koeningsknecht, V.; Stark, J.M. Decreased pulmonary clearance of S. pneumoniae following influenza A infection in mice. J. Virol. Methods 2001, 94, 173–186. [Google Scholar] [CrossRef]
- McCullers, J.A.; Rehg, J.E. Lethal synergism between influenza virus and Streptococcus pneumoniae: Characterization of a mouse model and the role of platelet-activating factor receptor. J. Infect. Dis. 2002, 186, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; Adler, F.R.; Ribeiro, R.M.; Gutenkunst, R.N.; McAuley, J.L.; McCullers, J.A.; Perelson, A.S. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathog. 2013, 9, e1003238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.M.; McCullers, J.A. Secondary Bacterial Infections in Influenza Virus Infection Pathogenesis. In Influenza Pathogenesis and Control—Volume I. Current Topics in Microbiology and Immunology; Compans, R., Oldstone, M., Eds.; Springer: Cham, Switzerland, 2014; Volume 385, pp. 327–356. ISBN 978-3-319-11155-1. [Google Scholar]
- Robinson, K.M.; Kolls, J.K.; Alcorn, J.F. The immunology of influenza virus-associated bacterial pneumonia. Curr. Opin. Immunol. 2015, 34, 59–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didierlaurent, A.; Goulding, J.; Patel, S.; Snelgrove, R.; Low, L.; Bebien, M.; Lawrence, T.; van Rijt, L.S.; Lambrecht, B.N.; Sirard, J.C.; et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 2008, 205, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef]
- Li, W.; Moltedo, B.; Moran, T.M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 2008, 14, 558–564. [Google Scholar] [CrossRef]
- Branchett, W.J.; Lloyd, C.M. Regulatory cytokine function in the respiratory tract. Mucosal Immunol. 2019, 12, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Madan, R.; Karp, C.L.; Braciale, T.J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009, 15, 277–284. [Google Scholar] [CrossRef]
- Camp, J.V.; Jonsson, C.B. A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 2017, 8, 550. [Google Scholar] [CrossRef] [Green Version]
- Colamussi, M.L.; White, M.R.; Crouch, E.; Hartshorn, K.L. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood 1999, 93, 2395–2403. [Google Scholar] [CrossRef]
- Fernandez, M.V.; Miller, E.; Krammer, F.; Gopal, R.; Greenbaum, B.D.; Bhardwaj, N. Ion efflux and influenza infection trigger NLRP3 inflammasome signaling in human dendritic cells. J. Leukoc. Biol. 2016, 99, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, H.; Liu, Y.; Sia, S.F.; Peiris, J.S.M.; Lau, Y.L.; Tu, W. Avian influenza virus directly infects human natural killer cells and inhibits cell activity. Virol. Sin. 2017, 32, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.; Paust, S. Dynamic Natural Killer Cell and T Cell Responses to Influenza Infection. Front. Cell. Infect. Microbiol. 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, J.; Liu, Y.; Wen, L.; Huang, L.; Xiang, Z.; Lam, K.T.; Lv, A.; Mao, H.; Lau, Y.L.; et al. Uncompromised NK cell activation is essential for virus-specific CTL activity during acute influenza virus infection. Cell. Mol. Immunol. 2018, 15, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz-Cherry, S. Viral Interference: The Case of Influenza Viruses. J. Infect. Dis. 2015, 212, 1690–1691. [Google Scholar] [CrossRef] [Green Version]
- Small, C.L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef] [Green Version]
- Hinshaw, V.S.; Olsen, C.W.; Dybdahl-Sissoko, N.; Evans, D. Apoptosis: A mechanism of cell killing by influenza A and B viruses. J. Virol. 1994, 68, 3667–3673. [Google Scholar] [CrossRef] [Green Version]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [Green Version]
- Morris, S.J.; Price, G.E.; Barnett, J.M.; Hiscox, S.A.; Smith, H.; Sweet, C. Role of neuraminidase in influenza virus-induced apoptosis. J. Gen. Virol. 1999, 80, 137–146. [Google Scholar] [CrossRef]
- Zhou, A.; Zhang, W.; Dong, X.; Liu, M.; Chen, H.; Tang, B. The battle for autophagy between host and influenza A virus. Virulence 2021, 13, 46–59. [Google Scholar] [CrossRef]
- Atkin-Smith, G.K.; Duan, M.; Chen, W.; Poon, I.K.H. The induction and consequences of Influenza A virus-induced cell death. Cell Death Dis. 2018, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Yang, Y.; Wang, H.; Luo, J.; Huang, X.; You, J.; Wang, B.; Li, M. Role of Autophagy and Apoptosis in the Postinfluenza Bacterial Pneumonia. Biomed. Res. Int. 2016, 2016, 3801026. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, O.P.; Klenk, H.D. Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. J. Virol. 2013, 87, 13107–13114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhirnov, O.P.; Konakova, T.E.; Wolff, T.; Klenk, H.D. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 2002, 76, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.H.; Lee, D.C.; Leon, T.Y.; Lau, A.S. Role for autophagy in cellular response to influenza virus infection. Hong Kong Med. J. 2014, 20, 20–24. [Google Scholar]
- Thulasi Raman, S.N.; Zhou, Y. Networks of Host Factors that Interact with NS1 Protein of Influenza A Virus. Front. Microbiol. 2016, 7, 654. [Google Scholar] [CrossRef] [Green Version]
- Yordy, B.; Iwasaki, A. Autophagy in the control and pathogenesis of viral infection. Curr. Opin. Virol. 2011, 1, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Jiang, X.; Liu, D.; Fan, Z.; Hu, X.; Yan, J.; Wang, M.; Gao, G.F. Autophagy is involved in influenza A virus replication. Autophagy 2009, 5, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, W.; Yeung, A.C.; Chan, P.K. Apoptosis, cytokine and chemokine induction by non-structural 1 (NS1) proteins encoded by different influenza subtypes. Virol. J. 2011, 8, 554. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sun, Q.; Mi, R.; Zhang, H. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J. Genet. Genom. 2011, 38, 533–537. [Google Scholar] [CrossRef]
- Sun, Y.; Li, C.; Shu, Y.; Ju, X.; Zou, Z.; Wang, H.; Rao, S.; Guo, F.; Liu, H.; Nan, W.; et al. Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 2012, 5, ra16. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Zhao, J.; Ren, C.; Li, P.; Chen, H.; Jin, M.; Zhou, H. Autophagy Promotes Replication of Influenza A Virus In Vitro. J. Virol. 2019, 93, e01984-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannagé, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Rämer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, S.; Ludwig, S.; Pleschka, S.; Wolff, T. Apoptosis signaling in influenza virus propagation, innate host defense, and lung injury. J. Leukoc. Biol. 2012, 92, 75–82. [Google Scholar] [CrossRef]
- Iwai, A.; Shiozaki, T.; Miyazaki, T. Relevance of signaling molecules for apoptosis induction on influenza A virus replication. Biochem. Biophys. Res. Commun. 2013, 441, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Schultz-Cherry, S.; Dybdahl-Sissoko, N.; Neumann, G.; Kawaoka, Y.; Hinshaw, V.S. Influenza Virus Ns1 Protein Induces Apoptosis in Cultured Cells. J. Virol. 2001, 75, 7875–7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Yeganeh, B.; Ghavami, S.; Rahim, M.N.; Klonisch, T.; Halayko, A.J.; Coombs, K.M. Autophagy activation is required for influenza A virus-induced apoptosis and replication. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 364–378. [Google Scholar] [CrossRef]
- Zhang, R.; Chi, X.; Wang, S.; Qi, B.; Yu, X.; Chen, J.L. The regulation of autophagy by influenza A virus. Biomed. Res. Int. 2014, 2014, 498083. [Google Scholar] [CrossRef]
- Kosai, K.; Seki, M.; Tanaka, A.; Morinaga, Y.; Imamura, Y.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Yanagihara, K.; Tomono, K.; et al. Increase of apoptosis in a murine model for severe pneumococcal pneumonia during influenza A virus infection. Jpn. J. Infect. Dis. 2011, 64, 451–457. [Google Scholar] [CrossRef]
- Cauley, L.S.; Vella, A.T. Why is coinfection with influenza virus and bacteria so difficult to control? Discov. Med. 2015, 19, 33–40. [Google Scholar]
- Feng, C.; Zhang, L.; Nguyen, C.; Vogel, S.N.; Goldblum, S.E.; Blackwelder, W.C.; Cross, A.S. Neuraminidase reprograms lung tissue and potentiates lipopolysaccharide-induced acute lung injury in mice. J. Immunol. 2013, 191, 4828–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, S.J.; Roche, A.M.; Weiser, J.N. Influenza promotes pneumococcal growth during coinfection by providing host sialylated substrates as a nutrient source. Cell Host Microbe 2014, 16, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billharz, R.; Zeng, H.; Proll, S.C.; Korth, M.J.; Lederer, S.; Albrecht, R.; Goodman, A.G.; Rosenzweig, E.; Tumpey, T.M.; García-Sastre, A.; et al. The NS1 protein of the 1918 pandemic influenza virus blocks host interferon and lipid metabolism pathways. J Virol. 2009, 83, 10557–10570. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.X.; Wang, X.Q.; Liu, X.F. NS1: A Key Protein in the “Game” Between Influenza A Virus and Host in Innate Immunity. Front. Cell Infect. Microbiol. 2021, 11, 670177. [Google Scholar] [CrossRef]
- Halder, U.C.; Bagchi, P.; Chattopadhyay, S.; Dutta, D.; Chawla-Sarkar, M. Cell death regulation during influenza A virus infection by matrix (M1) protein: A model of viral control over the cellular survival pathway. Cell Death Dis. 2011, 2, e197. [Google Scholar] [CrossRef] [Green Version]
- Mayank, A.K.; Sharma, S.; Nailwal, H.; Lal, S.K. Nucleoprotein of influenza A virus negatively impacts antiapoptotic protein API5 to enhance E2F1-dependent apoptosis and virus replication. Cell Death Dis. 2015, 6, e2018. [Google Scholar] [CrossRef]
- Tripathi, S.; Batra, J.; Cao, W.; Sharma, K.; Patel, J.R.; Ranjan, P.; Kumar, A.; Katz, J.M.; Cox, N.J.; Lal, R.B.; et al. Influenza A virus nucleoprotein induces apoptosis in human airway epithelial cells: Implications of a novel interaction between nucleoprotein and host protein Clusterin. Cell Death Dis. 2013, 4, 562. [Google Scholar] [CrossRef] [Green Version]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef]
- Cheung, P.H.; Lee, T.T.; Chan, C.P.; Jin, D.Y. Influenza A virus PB1-F2 protein: An ambivalent innate immune modulator and virulence factor. J. Leukoc. Biol. 2020, 107, 763–771. [Google Scholar] [CrossRef]
- Laghlali, G.; Lawlor, K.E.; Tate, M.D. Die Another Way: Interplay between Influenza A Virus, Inflammation and Cell Death. Viruses 2020, 12, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conenello, G.M.; Tisoncik, J.R.; Rosenzweig, E.; Varga, Z.T.; Palese, P.; Katze, M.G. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 2011, 85, 652–662. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; García-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; Samarasinghe, A.; Vogel, P.; Green, A.M.; Weinlich, R.; McCullers, J.A. A novel cytotoxic sequence contributes to influenza A viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J. Virol. 2014, 88, 503–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alymova, I.V.; Green, A.M.; van de Velde, N.; McAuley, J.L.; Boyd, K.L.; Ghoneim, H.E.; McCullers, J.A. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 2011, 85, 12324–12333. [Google Scholar] [CrossRef] [Green Version]
- McAuley, J.; Deng, Y.M.; Gilbertson, B.; Mackenzie-Kludas, C.; Barr, I.; Brown, L. Rapid evolution of the PB1-F2 virulence protein expressed by human seasonal H3N2 influenza viruses reduces inflammatory responses to infection. Virol. J. 2017, 14, 162. [Google Scholar] [CrossRef] [Green Version]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar] [CrossRef]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [Green Version]
- Pinar, A.; Dowling, J.K.; Bitto, N.J.; Robertson, A.A.; Latz, E.; Stewart, C.R.; Drummond, G.R.; Cooper, M.A.; McAuley, J.L.; Tate, M.D.; et al. PB1-F2 Peptide Derived from Avian Influenza A Virus H7N9 Induces Inflammation via Activation of the NLRP3 Inflammasome. J. Biol. Chem. 2017, 292, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, T.; Lin, L.; Zhang, Y.; Peng, X.; Yan, Y.; Lei, J.; Zhou, J.; Hu, B. Influenza A Virus Induces Autophagy By Its Hemagglutinin Binding To Cell Surface Heat Shock Protein 90aa1. Front. Microbiol. 2020, 11, 566348. [Google Scholar] [CrossRef]
- Klonoski, J.M.; Watson, T.; Bickett, T.E.; Svendsen, J.M.; Gau, T.J.; Britt, A.; Nelson, J.T.; Schlenker, E.H.; Chaussee, M.S.; Rynda-Apple, A.; et al. Contributions of Influenza Virus Hemagglutinin and Host Immune Responses Toward the Severity of Influenza Virus: Streptococcus pyogenes Superinfections. Viral immunol. 2018, 31, 457–469. [Google Scholar] [CrossRef]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Amin, M.N.; Frieman, M.B.; Chen, W.H.; Cross, A.S.; Wang, L.X.; Vasta, G.R. Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding. Mol. Immunol. 2015, 65, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita-Lazar, M.; Banerjee, A.; Feng, C.; Vasta, G.R. Galectins regulate the inflammatory response in airway epithelial cells exposed to microbial neuraminidase by modulating the expression of SOCS1 and RIG1. Mol. Immunol. 2015, 68, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, D.R.; Bielefeldt-Ohmann, H. Transforming growth factor beta in infectious disease: Always there for the host and the pathogen. Trends Microbiol. 1999, 7, 232–236. [Google Scholar] [CrossRef]
- Li, N.; Ren, A.; Wang, X.; Fan, X.; Zhao, Y.; Gao, G.F.; Cleary, P.; Wang, B. Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 238–243. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Huang, C.T.; Chen, T.C.; Lin, C.Y.; Chiu, C.H.; Lin, Y.C.; Chang, C.S.; He, Y.C. IL-10 inhibits neuraminidase-activated TGF-β and facilitates Th1 phenotype during early phase of infection. Nat. Commun. 2015, 6, 6374. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Hatada, E.; Fukuda, R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 1992, 73, 3325–3329. [Google Scholar] [CrossRef]
- Hatada, E.; Saito, S.; Fukuda, R. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J. Virol. 1999, 73, 2425–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef] [Green Version]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A Site on the Influenza A Virus NS1 Protein Mediates Both Inhibition of PKR Activation and Temporal Regulation of Viral RNA Synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthe-tase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; García-Sastre, A. Activation of Interferon Regulatory Factor 3 Is Inhibited by the Influenza A Virus NS1 Protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Wei, X.; Guo, K.; Li, Y.; Lin, X.; Zou, Z.; Zhou, H.; Jin, M. The C-Terminal Effector Domain of Non-Structural Protein 1 of Influenza A Virus Blocks IFN-β Production by Targeting TNF Receptor-Associated Factor 3. Front. Immunol. 2017, 8, 779. [Google Scholar] [CrossRef]
- Feng, W.; Sun, X.; Shi, N.; Zhang, M.; Guan, Z.; Duan, M. Influenza a virus NS1 protein induced A20 contributes to viral replication by suppressing interferon-induced antiviral response. Biochem. Biophys. Res. Commun. 2017, 482, 1107–1113. [Google Scholar] [CrossRef]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Li, Y.; Liu, Q.; Anderson, D.H.; Babiuk, L.A.; Zhou, Y. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 2007, 81, 12730–12739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Majumdar, S.; Vipat, V.C.; Mishra, A.C.; Chakrabarti, A.K. Non Structural Protein of Avian Influenza A (H11N1) Virus Is a Weaker Suppressor of Immune Responses But Capable of Inducing Apoptosis in Host Cells. Virol. J. 2012, 9, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Yang, Y.; Zhou, X.; Yang, Z.; Liu, X.; Cao, Z.; Song, H.; He, Y.; Huang, P. The NS1 protein of influenza A virus interacts with heat shock protein Hsp90 in human alveolar basal epithelial cells: Implication for virus-induced apoptosis. Virol. J. 2011, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Chen, P.; Tong, G.; Ma, Z. The non-Structural (NS1) Protein of Influenza A Virus Associates With p53 and Inhibits p53-mediated Transcriptional Activity and Apoptosis. Biochem. Biophys. Res. Commun. 2010, 395, 141–145. [Google Scholar] [CrossRef]
- Yan, Y.; Du, Y.; Wang, G.; Li, K. Non-structural protein 1 of H3N2 influenza A virus induces nucleolar stress via interaction with nucleolin. Sci. Rep. 2017, 7, 17761. [Google Scholar] [CrossRef] [Green Version]
- Zhirnov, O.P.; Klenk, H.D. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis 2007, 12, 1419–1432. [Google Scholar] [CrossRef]
- Martin-Sancho, L.; Tripathi, S.; Rodriguez-Frandsen, A.; Pache, L.; Sanchez-Aparicio, M.; McGregor, M.J.; Haas, K.M.; Swaney, D.L.; Nguyen, T.T.; Mamede, J.I.; et al. Restriction factor compendium for influenza A virus reveals a mechanism for evasion of autophagy. Nat. Microbiol. 2021, 6, 1319–1333. [Google Scholar] [CrossRef]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Alymova, I.V.; York, I.A.; McCullers, J.A. Non-avian animal reservoirs present a source of influenza A PB1-F2 proteins with novel virulence-enhancing markers. PLoS ONE 2014, 9, e111603. [Google Scholar] [CrossRef] [PubMed]
- Alymova, I.V.; McCullers, J.A.; Kamal, R.P.; Vogel, P.; Green, A.M.; Gansebom, S.; York, I.A. Virulent PB1-F2 residues: Effects on fitness of H1N1 influenza A virus in mice and changes during evolution of human influenza A viruses. Sci. Rep. 2018, 8, 7474. [Google Scholar] [CrossRef]
- Bruns, K.; Studtrucker, N.; Sharma, A.; Fossen, T.; Mitzner, D.; Eissmann, A.; Tessmer, U.; Roder, R.; Henklein, P.; Wray, V.; et al. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. J. Biol. Chem. 2007, 282, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, C.; Al Bazzal, A.; Vidic, J.; Février, V.; Bourdieu, C.; Bouguyon, E.; Le Goffic, R.; Vautherot, J.F.; Bernard, J.; Moudjou, M.; et al. PB1-F2 influenza A virus protein adopts a beta-sheet conformation and forms amyloid fibers in membrane environments. J. Biol. Chem. 2010, 285, 13233–13243. [Google Scholar] [CrossRef] [Green Version]
- Henklein, P.; Bruns, K.; Nimtz, M.; Wray, V.; Tessmer, U.; Schubert, U. Influenza A virus protein PB1-F2: Synthesis and characterization of the biologically active full length protein and related peptides. J. Peptide Sci. 2005, 11, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Mazur, I.; Anhlan, D.; Mitzner, D.; Wixler, L.; Schubert, U.; Ludwig, S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell. Microbiol. 2008, 10, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, G.W.; Wang, C.H.; Huang, C.H.; Wang, Y.C.; Shih, S.R. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 2010, 84, 10051–10062. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Krejnusova, I.; Bystricka, M.; Polakova, K.; Russ, G. N-terminal region of the PB1-F2 protein is responsible for increased expression of influenza a viral protein PB1. Acta Virol. 2011, 55, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.K.; Pasricha, G. An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology 2013, 440, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.M.; Adler, F.R.; McAuley, J.L.; Gutenkunst, R.N.; Ribeiro, R.M.; McCullers, J.A.; Perelson, A.S. Effect of 1918 pb1-f2 expression on influenza a virus infection kinetics. PLoS Comput. Biol. 2011, 7, e1001081. [Google Scholar] [CrossRef] [Green Version]
- Coleman, J.R. The PB1-F2 protein of Influenza A virus: Increasing pathogenicity by disrupting alveolar macrophages. Virol. J. 2007, 4, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.H.; Bird, N.; Mackenzie-Kludas, C.; Mansell, A.; Kedzierska, K.; Brown, L.; McAuley, J. Induction of memory cytotoxic T cells to influenza A virus and subsequent viral clearance is not modulated by PB1-F2-dependent inflammasome activation. Immunol. Cell. Biol. 2016, 94, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The Influenza A Virus PB1-F2 Protein Targets the Inner Mitochondrial Membrane via a Predicted Basic Amphipathic Helix That Disrupts Mitochondrial Function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Chounan, R.; Higashi, Y.; Kurihara, N.; Kido, H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004, 578, 331–336. [Google Scholar] [CrossRef] [Green Version]
- Varga, Z.T.; Palese, P. The influenza A virus protein PB1-F2. Virulence 2011, 2, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks-Gorospe, J.N.; Hurtig, H.R.; Iverson, A.R.; Schuneman, M.J.; Webby, R.J.; McCullers, J.A.; Huber, V.C. Naturally occurring swine influenza A virus PB1-F2 phenotypes that contribute to superinfection with Gram-positive respiratory pathogens. J. Virol. 2012, 86, 9035–9043. [Google Scholar] [CrossRef] [Green Version]
- Tate, M.D.; Mansell, A. An update on the NLRP3 inflammasome and influenza: The road to redemption or perdition? Curr. Opin. Immunol. 2018, 54, 80–85. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3Inflammasome—A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Solbak, S.M.; Sharma, A.; Bruns, K.; Röder, R.; Mitzner, D.; Hahn, F.; Niebert, R.; Vedeler, A.; Henklein, P.; Henklein, P.; et al. Influenza A virus protein PB1-F2 from different strains shows distinct structural signatures. Biochim. Biophys. Acta 2013, 1834, 568–582. [Google Scholar] [CrossRef] [PubMed]
- Košík, I.; Hollý, J.; Russ, G. PB1-F2 expedition from the whole protein through the domain to aa residue function. Acta Virol. 2013, 57, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Staneková, Z.; Varečková, E. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol. J. 2010, 7, 351. [Google Scholar] [CrossRef] [Green Version]
- Tomčíková, K.; Varečková, E. Different mechanisms of the protection against influenza A infection mediated by broadly reactive HA2-specific antibodies. Acta Virol. 2019, 63, 347–365. [Google Scholar] [CrossRef]
- Varečková, E.; Mucha, V.; Kostolanský, F. HA2 glycopolypeptide of influenza A virus and antiviral immunity. Acta Virol. 2013, 57, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Steinhauer, D.A. Role of Hemagglutinin Cleavage for the Pathogenicity of Influenza Virus. Virology 1999, 258, 1–20. [Google Scholar] [CrossRef]
- Bertram, S.; Glowacka, I.; Steffen, I.; Kühl, A.; Pöhlmann, S. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 2010, 20, 298–310. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Freuer, C.; Sielaff, F.; Schmidt, S.; Eickmann, M.; Uhlendorff, J.; Steinmetzer, T.; Klenk, H.D.; Garten, W. Cleavage of Influenza Virus Hemagglutinin by Airway Proteases TMPRSS2 and HAT Differs in Subcellular Localization and Susceptibility to Protease Inhibitors. J. Virol. 2010, 84, 5605–5614. [Google Scholar] [CrossRef] [Green Version]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef]
- Varečková, E.; Mucha, V.; Wharton, S.A.; Kostolanský, F. Inhibition of fusion activity of influenza A haemagglutinin mediated by HA2-specific monoclonal antibodies. Arch. Virol. 2003, 148, 469–486. [Google Scholar] [CrossRef]
- Jakubcová, L.; Hollý, J.; Varečková, E. The role of fusion activity of influenza A viruses in their biological properties. Acta Virol. 2016, 60, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Jakubcová, L.; Vozárová, M.; Hollý, J.; Tomčíková, K.; Fogelová, M.; Polčicová, K.; Kostolanský, F.; Fodor, E.; Varečková, E. Biological properties of influenza A virus mutants with amino acid substitutions in the HA2 glycoprotein of the HA1/HA2 interaction region. J. Gen. Virol. 2019, 100, 1282–1292. [Google Scholar] [CrossRef]
- Böttcher-Friebertshäuser, E.; Klenk, H.D.; Garten, W. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog. Dis. 2013, 69, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Tashiro, M.; Ciborowski, P.; Klenk, H.D.; Pulverer, G.; Rott, R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature 1987, 325, 536–537. [Google Scholar] [CrossRef]
- Scheiblauer, H.; Reinacher, M.; Tashiro, M.; Rott, R. Interactions between Bacteria and Influenza A Virus in the Development of Influenza Pneumonia. J. Infect. Dis. 1992, 166, 783–791. [Google Scholar] [CrossRef]
- Klenk, H.; Rott, R.; Orlich, M. Further Studies on the Activation of Influenza Virus by Proteolytic Cleavage of the Haemagglutinin. J. Gen. Virol. 1977, 36, 151–161. [Google Scholar] [CrossRef]
- Mancini, D.A.; Mendonça, R.M.; Dias, A.L.; Mendonça, R.Z.; Pinto, J.R. Co-infection between influenza virus and flagellated bacteria. Rev. Inst. Med. Trop. Sao Paulo 2005, 47, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Callan, R.J.; Hartmann, F.A.; West, S.E.; Hinshaw, V.S. Cleavage of influenza A virus H1 hemagglutinin by swine respiratory bacterial proteases. J. Virol. 1997, 71, 7579–7585. [Google Scholar] [CrossRef] [Green Version]
- Berri, F.; Rimmelzwaan, G.F.; Hanss, M.; Albina, E.; Foucault-Grunenwald, M.-L.; Lê, V.B.; Vogelzang-van Trierum, S.E.; Gil, P.; Camerer, E.; Martinez, D.; et al. Plasminogen Controls Inflammation and Pathogenesis of Influenza Virus Infections via Fibrinolysis. PLoS Pathog. 2013, 9, e1003229. [Google Scholar] [CrossRef] [Green Version]
- Tse, L.V.; Marcano, V.C.; Huang, W.; Pocwierz, M.S.; Whittaker, G.R. Plasmin-Mediated Activation of Pandemic H1N1 Influenza Virus Hemagglutinin Is Independent of the Viral Neuraminidase. J. Virol. 2013, 87, 5161–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, L.V.; Whittaker, G.R. Modification of the hemagglutinin cleavage site allows indirect activation of avian influenza virus H9N2 by bacterial staphylokinase. Virology 2015, 482, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhard, T.; Kronvall, G.; Ullberg, M. Surface bound plasmin promotes migration of Streptococcus pneumoniae through reconstituted basement membranes. Microb. Pathog. 1999, 26, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Fulde, M.; Steinert, M.; Bergmann, S. Interaction of streptococcal plasminogen binding proteins with the host fibrinolytic system. Front. Cell. Infect. Microbiol. 2013, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Fulde, M.; Bernardo-García, N.; Rohde, M.; Nachtigall, N.; Frank, R.; Preissner, K.; Klett, J.; Morreale, A.; Chhatwal, G.S.; Hermoso, J.; et al. Pneumococcal phosphoglycerate kinase interacts with plasminogen and its tissue activator. J. Thromb. Haemost. 2014, 111, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Habets, M.N.; Hermans, P.W.; Diavatopoulos, D.A. Interactions between Streptococcus pneumoniae and influenza virus: A mutually beneficial relationship? Future Microbiol. 2012, 7, 609–624. [Google Scholar] [CrossRef]
- Passariello, C.; Nencioni, L.; Sgarbanti, R.; Ranieri, D.; Torrisi, M.R.; Ripa, S.; Garaci, E.; Palamara, A.T. Viral hemagglutinin is involved in promoting the internalisation of Staphylococcus aureus into human pneumocytes during influenza A H1N1 virus infection. Int. J. Med. Microbiol. 2011, 301, 97–104. [Google Scholar] [CrossRef]
- Okamoto, S.; Kawabata, S.; Nakagawa, I.; Okuno, Y.; Goto, T.; Sano, K.; Hamada, S. Influenza A Virus-Infected Hosts Boost an Invasive Type of Streptococcus pyogenes Infection in Mice. J. Virol. 2003, 77, 4104–4112. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, Y.; Ikeura, A.; Harada, Y.; Kuroda, K.; Hamayasu, H.; Suzuki, T.; Yamada, K.; Kawase, Y.; Suzuki, Y. Binding of influenza type A viruses to group B Streptococcus and haemagglutination by virus-bound bacteria. J. Electron. Microsc. 2000, 49, 765–773. [Google Scholar] [CrossRef]
- Short, K.R.; Habets, M.N.; Payne, J.; Reading, P.C.; Diavatopoulos, D.A.; Wijburg, O.L. Influenza A virus induced bacterial otitis media is independent of virus tropism for α2,6-linked sialic acid. Virol. J. 2013, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Castrucci, M.R.; Kawaoka, Y. Biologic importance of neuraminidase stalk length in influenza A virus. J. Virol. 1993, 67, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin-Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, 11, 346. [Google Scholar] [CrossRef] [Green Version]
- Barman, S.; Adhikary, L.; Chakrabarti, A.K.; Bernas, C.; Kawaoka, Y.; Nayak, D.P. Role of transmembrane domain and cytoplasmic tail amino acid sequences of influenza a virus neuraminidase in raft association and virus budding. J. Virol. 2004, 78, 5258–5269. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Leser, G.P.; Zhang, J.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997, 16, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Mitnaul, L.J.; Castrucci, M.R.; Murti, K.G.; Kawaoka, Y. The cytoplasmic tail of influenza A virus neuraminidase (NA) affects NA incorporation into virions, virion morphology, and virulence in mice but is not essential for virus replication. J. Virol. 1996, 70, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Wen, F.; Wan, X.F. Influenza Neuraminidase: Underrated Role in Receptor Binding. Trends. Microbiol. 2019, 27, 477–479. [Google Scholar] [CrossRef]
- Du, R.; Cui, Q.; Rong, L. Competitive Cooperation of Hemagglutinin and Neuraminidase during Influenza A Virus Entry. Viruses 2019, 11, 458. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Wurtzer, S.; Rameix-Welti, M.A.; Dwyer, D.; van der Werf, S.; Naffakh, N.; Clavel, F.; Labrosse, B. Enhancement of the influenza A hemagglutinin (HA)-mediated cell-cell fusion and virus entry by the viral neuraminidase (NA). PLoS ONE 2009, 4, e8495. [Google Scholar] [CrossRef] [Green Version]
- McCombs, J.E.; Kohler, J.J. Pneumococcal Neuraminidase Substrates Identified through Comparative Proteomics Enabled by Chemoselective Labeling. Bioconjug. Chem. 2016, 27, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Pettigrew, M.M.; Fennie, K.P.; York, M.P.; Daniels, J.; Ghaffar, F. Variation in the presence of neuraminidase genes among Streptococcus pneumoniae isolates with identical sequence types. Infect. Immun. 2006, 74, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Gut, H.; Xu, G.; Taylor, G.L.; Walsh, M.A. Structural basis for Streptococcus pneumoniae NanA inhibition by influenza antivirals zanamivir and oseltamivir carboxylate. J. Mol. Biol. 2011, 409, 496–503. [Google Scholar] [CrossRef]
- Walther, E.; Xu, Z.; Richter, M.; Kirchmair, J.; Grienke, U.; Rollinger, J.M.; Krumbholz, A.; Saluz, H.P.; Pfister, W.; Sauerbrei, A.; et al. Dual Acting Neuraminidase Inhibitors Open New Opportunities to Disrupt the Lethal Synergism between Streptococcus pneumoniae and Influenza Virus. Front. Microbiol. 2016, 7, 357. [Google Scholar] [CrossRef]
- Gubareva, L.V.; Kaiser, L.; Hayden, F.G. Influenza virus neuraminidase inhibitors. Lancet 2000, 355, 827–835. [Google Scholar] [CrossRef]
- Peltola, V.T.; Murti, K.G.; McCullers, J.A. influenza virus neuraminidase contributes to secondary bacterial pneumonia. J. Infect. Dis. 2005, 192, 249–257. [Google Scholar] [CrossRef]
- Nieminen, J.; St-Pierre, C.; Bhaumik, P.; Poirier, F.; Sato, S. Role of galectin-3 in leukocyte recruitment in a murine model of lung infection by Streptococcus pneumoniae. J. Immunol. 2008, 180, 2466–2473. [Google Scholar] [CrossRef] [Green Version]
- Rabinovich, G.A.; Toscano, M.A.; Jackson, S.S.; Vasta, G.R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 2007, 17, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Stowell, S.R.; Karmakar, S.; Arthur, C.M.; Ju, T.; Rodrigues, L.C.; Riul, T.B.; Dias-Baruffi, M.; Miner, J.; McEver, R.P.; Cummings, R.D. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol. Biol. Cell. 2009, 20, 1408–1418. [Google Scholar] [CrossRef] [Green Version]
- Vasta, G.R. Roles of galectins in infection. Nat. Rev. Microbiol. 2009, 7, 424–438. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.Y.; Rabinovich, G.A.; Liu, F.T. Galectins: Structure, function and therapeutic potential. Expert Rev. Mol. Med. 2008, 10, e17. [Google Scholar] [CrossRef]
- Yang, Z.S.; Lin, C.Y.; Huang, S.W.; Wang, W.H.; Urbina, A.N.; Tseng, S.P.; Lu, P.L.; Chen, Y.H.; Wang, S.F. Regulatory roles of galectins on influenza A virus and their potential as a therapeutic strategy. Biomed. Pharmacother. 2021, 139, 111713. [Google Scholar] [CrossRef]
- Bao, J.; Wang, X.; Liu, S.; Zou, Q.; Zheng, S.; Yu, F.; Chen, Y. Galectin-1 ameliorates influenza A H1N1pdm09 virus-induced acute lung injury. Front. Microbiol. 2020, 11, 1293. [Google Scholar] [CrossRef]
- Hattori, T.A.T.; Fujioka, Y.; Maruyama, J.; Nakayama, Y.; Ohba, Y.; Niki, T.; Miyazaki, T.; Hirashima, M.; Kida, H. Inhibition of influenza A virus infection by Galectin-9. Jpn. J. Vet. Res. 2013, 61, 5–18. [Google Scholar] [CrossRef]
- Katoh, S.; Ikeda, M.; Shimizu, H.; Mouri, K.; Obase, Y.; Kobashi, Y.; Fukushima, K.; Hirashima, M.; Oka, M. Increased levels of plasma galectin-9 in patients with influenza virus infection. Tohoku J. Exp. Med. 2014, 232, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Sundararajan, A.; Suryawanshi, A.; Kumar, N.; Veiga-Parga, T.; Kuchroo, V.K.; Thomas, P.G.; Sangster, M.Y.; Rouse, B.T. T cell immunoglobulin and mucin protein-3 (Tim-3)/galectin-9 interaction regulates influenza A virus-specific humoral and CD8 T-cell responses. Proc. Natl. Acad. Sci. USA 2011, 108, 19001–19006. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Wang, S.F.; Weng, I.C.; Hong, M.H.; Lo, T.H.; Jan, J.T.; Hsu, L.C.; Chen, H.Y.; Liu, F.T. Galectin-3 enhances avian H5N1 influenza A virus-induced pulmonary inflammation by promoting NLRP3 inflammasome activation. Am. J. Pathol. 2018, 188, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Li, S.W.; Yang, T.C.; Lai, C.C.; Huang, S.H.; Liao, J.M.; Wan, L.; Lin, Y.J.; Lin, C.W. Antiviral activity of aloe-emodin against influenza A virus via galectin-3 up-regulation. Eur. J. Pharmacol. 2014, 738, 125–132. [Google Scholar] [CrossRef]
- Lin, C.Y.; Yang, Z.S.; Wang, W.H.; Urbina, A.N.; Lin, Y.T.; Huang, J.C.; Liu, F.T.; Wang, S.F. The Antiviral Role of Galectins toward Influenza A Virus Infection—An Alternative Strategy for Influenza Therapy. Pharmaceuticals 2021, 14, 490. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Orwenyo, J.; Banerjee, A.; Vasta, G.R.; Wang, L.X. Design and synthesis of glycoprotein-based multivalent glyco-ligands for influenza hemagglutinin and human galectin-3. Bioorg. Med. Chem. 2013, 21, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.H.; Lin, C.Y.; Chang, M.R.; Urbina, A.N.; Assavalapsakul, W.; Thitithanyanont, A.; Chen, Y.H.; Liu, F.T.; Wang, S.F. The role of galectins in virus infection—A systemic literature review. J. Microbiol. Immunol. Infect. 2020, 53, 925–935. [Google Scholar] [CrossRef]
- Brydon, E.W.; Morris, S.J.; Sweet, C. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 2005, 29, 837–850. [Google Scholar] [CrossRef] [Green Version]
- Schultz-Cherry, S.; Hinshaw, V.S. Influenza virus neuraminidase activates latent transforming growth factor beta. J. Virol. 1996, 70, 8624–8629. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.M.; Turpin, E.A.; Moser, L.A.; O’Brien, K.B.; Cline, T.D.; Jones, J.C.; Tumpey, T.M.; Katz, J.M.; Kelley, L.A.; Gauldie, J.; et al. Transforming growth factor-β: Activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 2010, 6, e1001136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Huang, S.; Chen, J.; Zhang, S.; Chen, Z. NA proteins of influenza A viruses H1N1/2009, H5N1, and H9N2 show differential effects on infection initiation, virus release, and cell-cell fusion. PLoS ONE 2013, 8, e54334. [Google Scholar] [CrossRef] [Green Version]
- Letterio, J.J.; Roberts, A.B. Regulation of immune responses by TGF-beta. Annu. Rev. Immunol. 1998, 16, 137–161. [Google Scholar] [CrossRef] [Green Version]
- Roberts, S.; Salmon, S.L.; Steiner, D.J.; Williams, C.M.; Metzger, D.W.; Furuya, Y. Allergic Airway Disease Prevents Lethal Synergy of Influenza A Virus-Streptococcus pneumoniae Coinfection. MBio 2019, 10, e01335-19. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.C. Influenza Virus: A Master Tactician in Innate Immune Evasion and Novel Therapeutic Interventions. Front. Immunol. 2018, 9, 743. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Rahbar, R.; Chan, R.W.; Lee, S.M.; Chan, M.C.; Wang, B.X.; Baker, D.P.; Sun, B.; Peiris, J.S.; Nicholls, J.M.; et al. Influenza virus non-structural protein 1 (NS1) disrupts interferon signaling. PLoS ONE 2010, 5, e13927. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jeong, M.S.; Jang, S.B. Structure and Activities of the NS1 Influenza Protein and Progress in the Development of Small-Molecule Drugs. Int. J. Mol. Sci. 2021, 22, 4242. [Google Scholar] [CrossRef]
- Ayllon, J.; García-Sastre, A. The NS1 protein: A multitasking virulence factor. Curr. Top. Microbiol. Immunol. 2015, 386, 73–107. [Google Scholar] [CrossRef]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Sesma, A. The influenza virus NS1 protein: Inhibitor of innate and adaptive immunity. Infect. Disord. Drug Targets 2007, 7, 336–343. [Google Scholar] [CrossRef]
- Fernandez-Sesma, A.; Marukian, S.; Ebersole, B.J.; Kaminski, D.; Park, M.S.; Yuen, T.; Sealfon, S.C.; García-Sastre, A.; Moran, T.M. Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 2006, 80, 6295–6304. [Google Scholar] [CrossRef] [Green Version]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [Green Version]
- Shepardson, K.; Larson, K.; Cho, H.; Johns, L.L.; Malkoc, Z.; Stanek, K.; Wellhman, J.; Zaiser, S.; Daggs-Olson, J.; Moodie, T.; et al. A Novel Role for PDZ-Binding Motif of Influenza A Virus Nonstructural Protein 1 in Regulation of Host Susceptibility to Postinfluenza Bacterial Superinfections. Viral Immunol. 2019, 32, 131–143. [Google Scholar] [CrossRef]
- Hu, M.M.; Shu, H.B. Cytoplasmic Mechanisms of Recognition and Defense of Microbial Nucleic Acids. Annu. Rev. Cell Dev. Biol. 2018, 34, 357–379. [Google Scholar] [CrossRef]
- Sun, X.; Feng, W.; Guo, Y.; Wang, Q.; Dong, C.; Zhang, M.; Guan, Z.; Duan, M. MCPIP1 attenuates the innate immune response to influenza A virus by suppressing RIG-I expression in lung epithelial cells. J. Med. Virol. 2018, 90, 204–211. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, L.M.; Zeng, H.; Gomez, J.A.; Plowden, J.; Fujita, T.; Katz, J.M.; Donis, R.O.; Sambhara, S. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am. J. Respir. Cell Mol. 2007, 36, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Mibayashi, M.; Martínez-Sobrido, L.; Loo, Y.M.; Cárdenas, W.B.; Gale, M., Jr.; García-Sastre, A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [Green Version]
- Weber-Gerlach, M.; Weber, F. To conquer the host, influenza virus is packing it in: Interferon-antagonistic strategies beyond NS1. J. Virol. 2016, 90, 8389–8394. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Planz, O. Influenza viruses and the NF-kappaB signaling pathway—towards a novel concept of antiviral therapy. Biol. Chem. 2008, 389, 1307–1312. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Zamanian-Daryoush, M.; Mogensen, T.H.; DiDonato, J.A.; Williams, B.R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell. Biol. 2000, 20, 1278–1290. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; García-Sastre, A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar] [CrossRef] [Green Version]
- Rynda-Apple, A.; Robinson, K.M.; Alcorn, J.F. Influenza and Bacterial Superinfection: Illuminating the Immunologic Mechanisms of Disease. Infect Immun. 2015, 83, 3764–3770. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Illustration of influenza virus protein functions in the enhancement of S. pneumoniae lung infection. The mucosal layer in the lungs is damaged due to sialidase activity of viral NA, and ciliated epithelial cells in lungs, together with immune cells, are infected with IAV. During infection, IAV proteins PB1-F2, NS1, HA, NA, and M2 cause impairment of the immune response, the apoptosis and inflammation, leading to the destruction of tissue integrity. All of these viral protein functions can lead to enhanced susceptibility to secondary S. pneumoniae infection observed approximately 7 days after viral infection (7 dpi).
Figure 1.
Illustration of influenza virus protein functions in the enhancement of S. pneumoniae lung infection. The mucosal layer in the lungs is damaged due to sialidase activity of viral NA, and ciliated epithelial cells in lungs, together with immune cells, are infected with IAV. During infection, IAV proteins PB1-F2, NS1, HA, NA, and M2 cause impairment of the immune response, the apoptosis and inflammation, leading to the destruction of tissue integrity. All of these viral protein functions can lead to enhanced susceptibility to secondary S. pneumoniae infection observed approximately 7 days after viral infection (7 dpi).
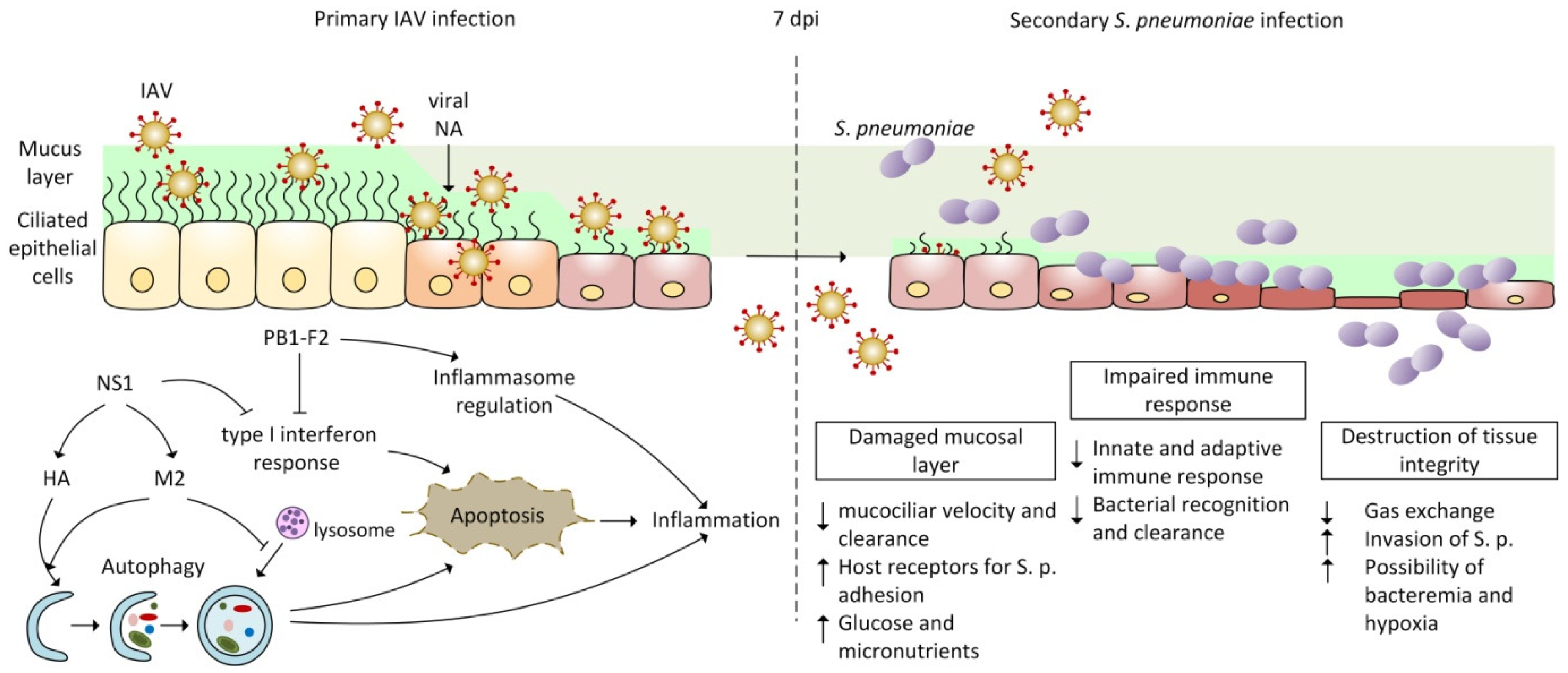

{kind=link}
Table 1.
Function of IAV proteins predisposing host to pneumococcal co-infection.
| IAV Protein | Function | Functional Domain | References |
|---|---|---|---|
| PB1-F2 | Inhibition of IFN response | N66S mutation | [148,149] |
| Apoptosis of epithelial and immune cells | - | [150,151] | |
| Cytotoxic death of epithelial and immune cells | Cytotoxic motif | [152] | |
| Induction of rapid inflammatory response | Inflammatory motif | [153,154] | |
| Regulation of NLRP3 inflammasome activity | - | [155,156,157,158] | |
| HA | Regulation of autophagosome formation | - | [119,127,159,160] |
| NA | Creation of environment for bacterial entrance | ||
| Alteration of glycosylation on cell surface | Catalytic domain | [138,139] | |
| Desialylation of surface glycans | [161,162] | ||
| Affection of innate immunity | |||
| Direct activation of TGF-β | Catalytic domain | [163,164,165] | |
| NS1 | Inhibition of IFN response in several ways | ||
| Blocking of RIG-I activation | RNA-binding domain | [166,167,168] | |
| Blocking of PKR activation | [168,169,170] | ||
| Blocking OAS function | [171,172] | ||
| Interaction with host factors | Effector domain PDZ-binding motif | [173,174,175,176] | |
| Manipulation of apoptosis in several ways | |||
| Binding to PKR linker domain | Effector domain | [120,132] | |
| Activation of PI3K pathway | SH3-binding motif aa 164–167 | [119,177,178,179] | |
| Interaction with Hsp90 | - | [180,181] | |
| Inhibition of p53 | aa 144–188 | [182,183,184] | |
| M2 | Induction of autophagosome formation | - | [119,124,129] |
| Inhibition of lysosomal degradation of autophagosomes | - | [129,185,186] | |
| NP | Induction of autophagosome formation | - | [128,129] |
(-) Multiple mechanisms or not defined.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mikušová, M.; Tomčíková, K.; Briestenská, K.; Kostolanský, F.; Varečková, E. The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection. Viruses 2022, 14, 1064. https://doi.org/10.3390/v14051064
AMA Style
Mikušová M, Tomčíková K, Briestenská K, Kostolanský F, Varečková E. The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection. Viruses. 2022; 14(5):1064. https://doi.org/10.3390/v14051064
Chicago/Turabian StyleMikušová, Miriam, Karolína Tomčíková, Katarína Briestenská, František Kostolanský, and Eva Varečková. 2022. "The Contribution of Viral Proteins to the Synergy of Influenza and Bacterial Co-Infection" Viruses 14, no. 5: 1064. https://doi.org/10.3390/v14051064
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.