

The Development of Pharmacophore Models for the Search of New Natural Inhibitors of SARS-CoV-2 Spike RBD–ACE2 Binding Interface


Abstract
1. Introduction

2. Results and Discussion
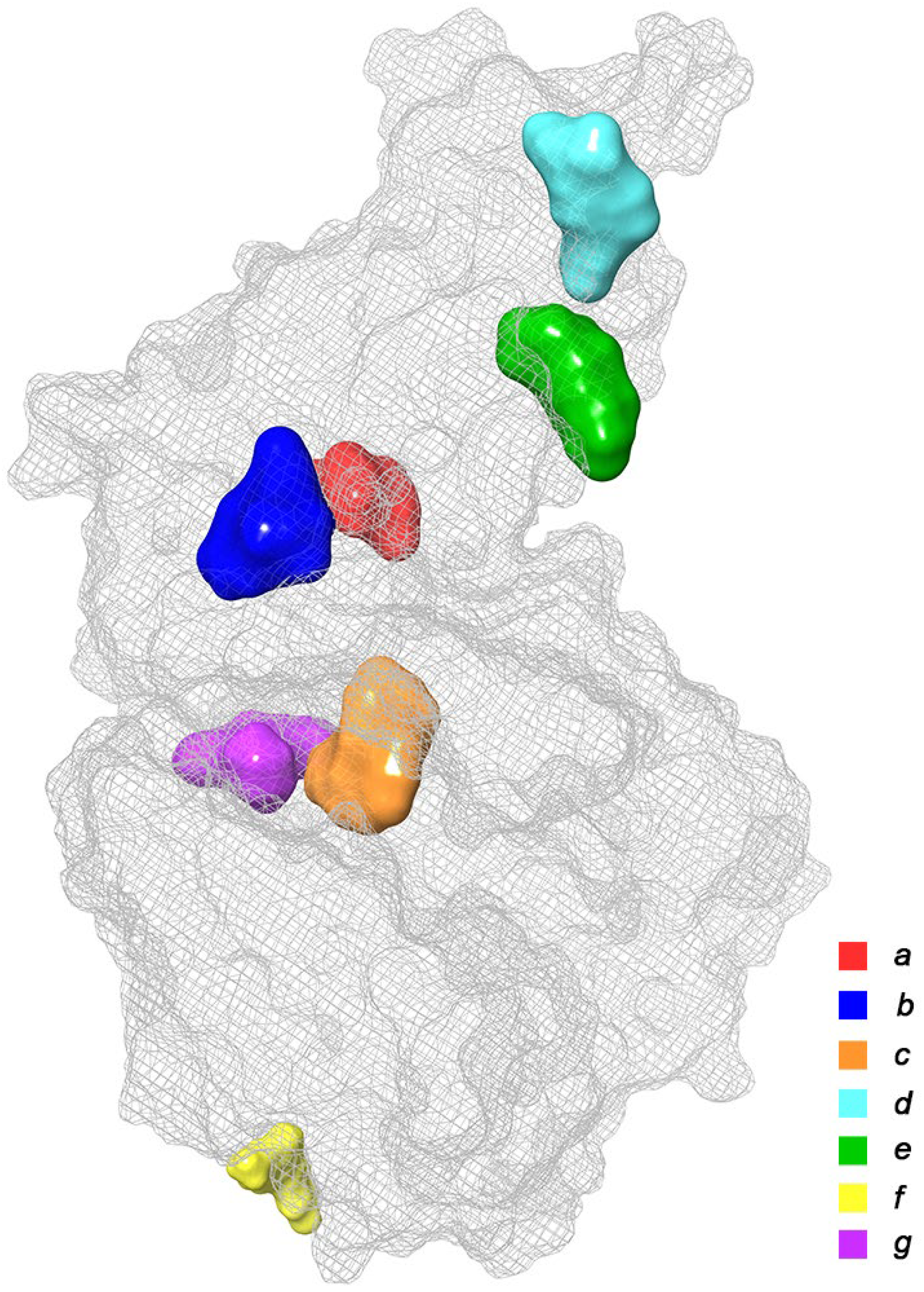
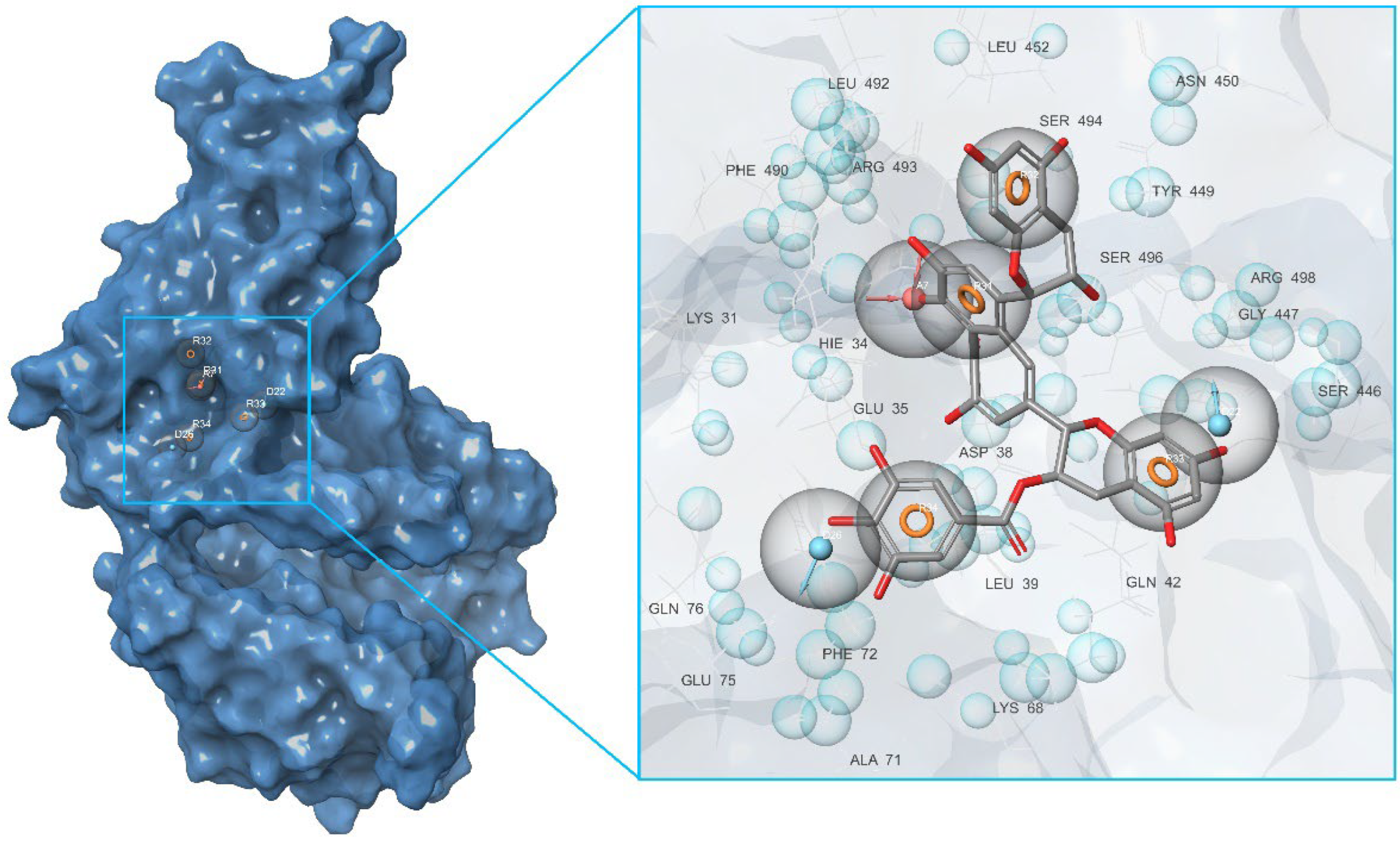
2.1. Initial Identification of Structural Similarity
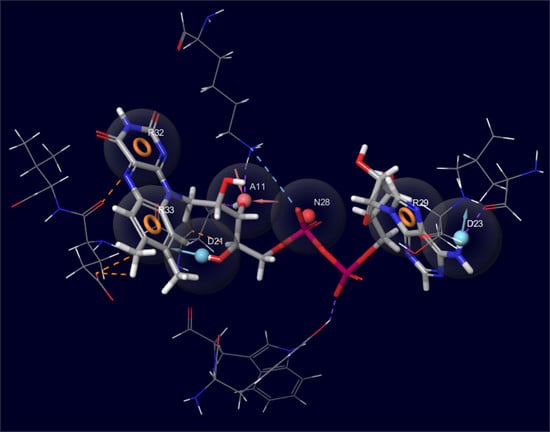
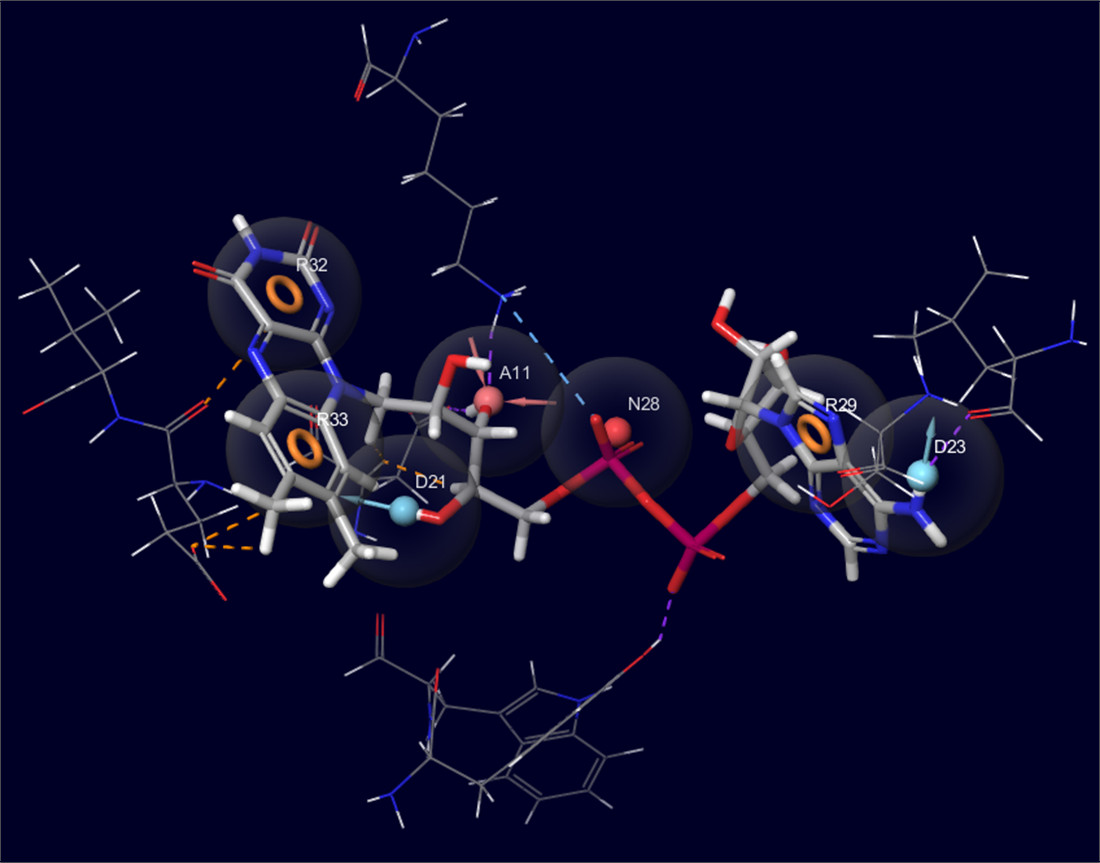
2.2. Development of Pharmacophore Models
2.3. Final Verification of Key Ligands
3. Materials and Methods
3.1. Preparation of Protein for Docking and Grid Generation
3.2. Preparation of Ligands
3.3. Development of Pharmacophore Models
- Hydrogen bond acceptor (A);
- Hydrogen bond donor (D);
- Aromatic ring (R);
- Positive ionizable (P);
- Negative ionizable (N);
- Hydrophobic center (H).
3.4. Molecular Docking Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Chen, Q.; Liu, K.; Wang, J.; Han, P.; Zhang, Y.; Hu, Y.; Meng, Y.; Pan, X.; Qiao, C.; et al. Broad host range of SARS-CoV-2 and the molecular basis for SARS-CoV-2 binding to cat ACE2. Cell Discov. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.-H.; Patel, N.; Haupt, R.; Zhou, H.; Weston, S.; Hammond, H.; Logue, J.; Portnoff, A.D.; Norton, J.; Guebre-Xabier, M.; et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVXCoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 2021, 12, 372. [Google Scholar] [CrossRef]
- Kang, Y.-F.; Sun, C.; Zhuang, Z.; Yuan, R.-Y.; Zheng, Q.; Li, J.-P.; Zhou, P.-P.; Chen, X.-C.; Liu, Z.; Zhang, X.; et al. Rapid development of SARS-CoV-2 spike protein receptor-binding domain self-assembled nanoparticle vaccine candidates. ACS Nano 2021, 15, 2738–2752. [Google Scholar] [CrossRef]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A. BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Abubakar, M.B.; Usman, D.; El-Saber Batiha, G.; Cruz-Martins, N.; Malami, I.; Ibrahim, K.G.; Abubakar, B.; Bello, M.B.; Muhammad, A.; Gan, S.H.; et al. Natural Products Modulating Angiotensin Converting Enzyme 2 (ACE2) as Potential COVID-19 Therapies. Front. Pharmacol. 2021, 12, 629935. [Google Scholar] [CrossRef]
- Ngo, H.X.; Garneau-Tsodikova, S. What are the drugs of the future? MedChemComm 2018, 9, 757–758. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.-W.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Piccoli, L.; Park, Y.-J.; Tortorici, M.A.; Czudnochowski, N.; Walls, A.C.; Beltramello, M.; Silacci-Fregni, C.; Pinto, D.; Rosen, L.E.; Bowen, J.E.; et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell 2020, 183, 1024–1042. [Google Scholar] [CrossRef]
- Panda, P.K.; Arul, M.N.; Patel, P.; Verma, S.K.; Luo, W.; Rubahn, H.-G.; Mishra, Y.K.; Suar, M.; Ahuja, R. Structure-based drug designing and immunoinformatics approach for SARS-CoV-2. Sci. Adv. 2020, 6, eabb8097. [Google Scholar] [CrossRef]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeño, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 Spikeinduced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef]
- Kim, C.; Ryu, D.-K.; Lee, J.; Kim, Y.-I.; Seo, J.-M.; Kim, Y.-G.; Jeong, J.-H.; Kim, M.; Kim, J.-I.; Kim, P.; et al. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat. Commun. 2021, 12, 288. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Cai, L.; Guo, X.; Cao, Y.; Ying, P.; Hong, L.; Zhang, Y.; Yi, G.; Fu, M. Determining available strategies for prevention and therapy: Exploring COVID-19 from the perspective of ACE2. Int. J. Mol. Med. 2021, 47, 43. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, F.; Comunale, M.A.; Mehta, A.; Sehgal, M.; Jain, P.; Cuconati, A.; Lin, H.; Block, T.M.; Chang, J.; et al. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob. Agents Chemother. 2015, 59, 206–216. [Google Scholar] [CrossRef]
- Chen, H.; Du, Q. Potential Natural Compounds for Preventing SARS-CoV-2 (2019-nCoV) Infection. Preprints 2020, 2020010358. [Google Scholar] [CrossRef]
- Teli, D.M.; Shah, M.B.; Chhabria, M.T. In silico Screening of Natural Compounds as Potential Inhibitors of SARS-CoV-2 Main Protease and Spike RBD: Targets for COVID-19. Front. Mol. Biosci. 2021, 7, 599079. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, V. Repurposing the natural compounds as potential therapeutic agents for COVID-19 based on the molecular docking study of the main protease and the receptor-binding domain of spike protein. J. Mol. Model. 2022, 28, 153. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Sharma, N.; Singh, B.; Sen, A.; Roy, A. Natural compounds from Clerodendrum spp. as possible therapeutic candidates against SARS-CoV-2: An in silico investigation. J. Biomol. Struct. Dyn. 2021, 39, 4774–4785. [Google Scholar] [CrossRef] [PubMed]
- Nedyalkova, M.; Vasighi, M.; Sappati, S.; Kumar, A.; Madurga, S.; Simeonov, V. Inhibition Ability of Natural Compounds on Receptor-Binding Domain of SARS-CoV2: An In Silico Approach. Pharmaceuticals 2021, 14, 1328. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Joshi, T.; Sharma, P.; Mathpal, S.; Pundir, H.; Bhatt, V.; Chandra, S. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4529–4536. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Pan, Y.C.; Wu, T.Y.; Yao, T.Y.; Wang, W.J.; Shen, W.J.; Ahmed, A.; Chan, S.T.; Tang, C.H.; Huang, W.C.; et al. Potential natural products that target the SARS-CoV-2 spike protein identified by structure-based virtual screening, isothermal titration calorimetry and lentivirus particles pseudotyped (Vpp) infection assay. J. Tradit. Complement. Med. 2022, 12, 73–89. [Google Scholar] [CrossRef]
- Semenov, V.A.; Krivdin, L.B. Combined Computational NMR and Molecular Docking Scrutiny of Potential Natural SARS-CoV-2 Mpro Inhibitors. J. Phys. Chem. B 2022, 126, 2173–2187. [Google Scholar] [CrossRef]
- Lei, S.; Chen, X.; Wu, J.; Duan, X.; Men, K. Small molecules in the treatment of COVID-19. Signal Transduct. Target Ther. 2022, 7, 387. [Google Scholar] [CrossRef]
- Chapman, R.L.; Andurkar, S.V. A review of natural products, their effects on SARS-CoV-2 and their utility as lead compounds in the discovery of drugs for the treatment of COVID-19. Med. Chem. Res. 2022, 31, 40–51. [Google Scholar] [CrossRef]
- Kim, C.-H. Anti-SARS-CoV-2 Natural Products as Potentially Therapeutic Agents. Front. Pharmacol. 2021, 12, 590509. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, N.; Yang, L.; Song, X.-Q. Bioactive natural products in COVID-19 therapy. Front. Pharmacol. 2022, 13, 926507. [Google Scholar] [CrossRef]
- Muhseen, Z.T.; Kadhim, S.; Yahiya, Y.I.; Alatawi, E.A.; Aba Alkhayl, F.F.; Almatroudi, A. Insights into the Binding of Receptor-Binding Domain (RBD) of SARS-CoV-2 Wild Type and B.1.620 Variant with hACE2 Using Molecular Docking and Simulation Approaches. Biology 2021, 10, 1310. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef]
- Delgado, J.M.; Duro, N.; Rogers, D.M.; Tkatchenko, A.; Pandit, S.A.; Varma, S. Molecular basis for higher affinity of SARS-CoV-2 spike RBD for human ACE2 receptor. Proteins 2021, 89, 1134–1144. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef]
- Khan, A.; Gui, J.; Ahmad, W.; Haq, I.; Shahid, M.; Khan, A.A.; Shah, A.; Khan, A.; Ali, L.; Anwar, Z.; et al. The SARS-CoV-2 B.1.618 variant slightly alters the spike RBD-ACE2 binding affinity and is an antibody escaping variant: A computational structural perspective. RSC Adv. 2021, 11, 30132–30147. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Culletta, G.; Gulotta, M.R.; Perricone, U.; Zappalà, M.; Almerico, A.M.; Tutone, M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. [Google Scholar] [CrossRef]
- Sharanya, C.S.; Arun, K.G.; Abhithaj, J.; Sabu, A.; Haridas, M. Drug repurposing for COVID-19 from FDA approved and experiment stage drugs by in silico methods with SARS CoV-2 spike protein. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Kouznetsova, V.L.; Zhang, A.; Miller, M.A.; Tatineni, M.; Greenberg, J.P.; Tsigelny, I.F. Potential SARS-CoV-2 Spike Protein-ACE2 Interface Inhibitors: Repurposing FDA-approved Drugs. J. Explor. Res. Pharmacol. 2022, 7, 17–29. [Google Scholar] [CrossRef]
- Elekofehinti, O.O.; Iwaloye, O.; Molehin, O.R.; Famusiwa, C.D. Identification of lead compounds from large natural product library targeting 3C-like protease of SARS-CoV-2 using E-pharmacophore modelling, QSAR and molecular dynamics simulation. Silico Pharmacol. 2021, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Cruz, A.; Chen, L.; Ghattas, M.; Ji, Y.; Huang, K.; Ayoub, S., Jr.; Luchko, T.; McKay, D.J.; Kurtzman, T.; et al. An online repository of solvation thermodynamic and structural maps of SARS-CoV-2 targets. J. Comput. Aided Mol. Des. 2020, 12, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.-L.; Qiu, X.-D.; Wu, S.; Liu, Y.-T.; Zhao, T.; Sun, Z.-H.; Li, Z.-R.; Shan, G.-Z. Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology. Molecules 2021, 26, 57. [Google Scholar] [CrossRef] [PubMed]
- Gangadevi, S.; Badavath, V.N.; Thakur, A.; Yin, N.; De Jonghe, S.; Acevedo, O.; Jochmans, D.; Leyssen, P.; Wang, K.; Neyts, J.; et al. Kobophenol A Inhibits Binding of Host ACE2 Receptor with Spike RBD Domain of SARS-CoV-2, a Lead Compound for Blocking COVID-19. J. Phys. Chem. Lett. 2021, 12, 1793–1802. [Google Scholar] [CrossRef]
- Sethi, A.; Sanam, S.; Munagalasetty, S.; Jayanthi, S.; Alvala, M. Understanding the role of galectin inhibitors as potential candidates for SARS-CoV-2 spike protein: In silico studies. RSC Adv. 2020, 10, 29873–29884. [Google Scholar] [CrossRef]
- Xiong, J.; Xiang, Y.; Huang, Z.; Liu, X.; Wang, M.; Ge, G.; Chen, H.; Xu, J.; Zheng, M.; Chen, L.; et al. Structure-Based Virtual Screening and Identification of Potential Inhibitors of SARS-CoV-2 S-RBD and ACE2 Interaction. Front. Chem. 2021, 9, 40702. [Google Scholar] [CrossRef]
- Utomo, R.Y.; Ikawati, M.; Meiyanto, E. Revealing the Potency of Citrus and Galangal Constituents to Halt SARS-CoV-2 Infection. Preprints 2020, 2020030214. [Google Scholar] [CrossRef]
- Day, C.J.; Bailly, B.; Guillon, P.; Dirr, L.; Jen, F.E.-C.; Spillings, B.L.; Mak, J.; von Itzstein, M.; Haselhorst, T.; Jennings, M.P.; et al. Multidisciplinary approaches identify compounds that bind to human ACE2 or SARS CoV-2 spike protein as candidates to block SARS-CoV-2–ACE2 receptor interactions. mBio 2021, 12, e03681–e20. [Google Scholar] [CrossRef]
- Shoemark, D.K.; Colenso, C.K.; Toelzer, C.; Gupta, K.; Sessions, R.B.; Davidson, A.D.; Berger, I.; Schaffitzel, C.; Spencer, J.; Mulholland, A.J.; et al. Molecular Simulations suggest Vitamins, Retinoids and Steroids as Ligands of the Free Fatty Acid Pocket of the SARS-CoV-2 Spike Protein. Angew. Chem. Int. Ed. Engl. 2021, 60, 7098–7110. [Google Scholar] [CrossRef]
- Ahmad, S.; Abbasi, H.W.; Shahid, S.; Gul, S.; Abbasi, S.W. Molecular docking, simulation and MM-PBSA studies of nigella sativa compounds: A computational quest to identify potential natural antiviral for COVID-19 treatment. J. Biomol. Struct. Dyn. 2021, 12, 4225–4233. [Google Scholar] [CrossRef]
- Abdelli, I.; Hassani, F.; Bekkel Brikci, S.; Ghalem, S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. J. Biomol. Struct. Dyn. 2021, 9, 3263–3276. [Google Scholar] [CrossRef]
- Shahbazi, B.; Mafakher, L.; Teimoori-Toolabi, L. Different compounds against Angiotensin-Converting Enzyme 2 (ACE2) receptor potentially containing the infectivity of SARS-CoV-2: An in silico study. J. Mol. Model. 2022, 28, 82. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 5, 766–788. [Google Scholar] [CrossRef]
- Ogedjo, M.; Onoka, I.; Sahini, M.; Shadrack, D.M. Accommodating receptor flexibility and free energy calculation to reduce false positive binders in the discovery of natural products blockers of SARS-COV-2 spike RBD-ACE2 interface. Biochem. Biophys. Rep. 2021, 27, 101024. [Google Scholar] [CrossRef]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchianò, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 Receptor Interaction by Natural and Semi-synthetic Steroidal Agents Acting on Functional Pockets on the Receptor Binding Domain. Front. Chem. 2020, 8, 572885. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Khamitov, E.M.; Gureev, M.A.; Yarovaya, O.I.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakov, D.N.; Maksyutov, R.A.; Salakhutdinov, N.F. Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses 2022, 14, 119. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Biagioli, M.; Marchianò, S.; Roselli, R.; Di Giorgio, C.; Bellini, R.; Bordoni, M.; Gidari, A.; Sabbatini, S.; Francisci, D.; Fiorillo, B.; et al. Discovery of a AHR pelargonidin agonist that counter-regulates Ace2 expression and attenuates ACE2-SARS-CoV-2 interaction. Biochem. Pharmacol. 2021, 188, 114564. [Google Scholar] [CrossRef]
- Ngwa, W.; Kumar, R.; Thompson, D.; Lyerly, W.; Moore, R.; Reid, T.-E.; Lowe, H.; Toyang, N. Potential of Flavonoid-Inspired Phytomedicines against COVID-19. Molecules 2020, 25, 2707. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.-T.; Miao, J.; Wang, L.; Fu, H.; Li, Q.; Wen, W.-B.; Zhang, Z.-Y.; Song, R.-W.; Liu, X.-G.; et al. Network pharmacology studies on the effect of Chai-Ling decoction in coronavirus disease 2019. Trad. Med. Res. 2020, 5, 145–159. [Google Scholar] [CrossRef]
- Liu, W.; Zheng, W.; Cheng, L.; Li, M.; Huang, J.; Bao, S.; Xu, Q.; Ma, Z. Citrus fruits are rich in flavonoids for immunoregulation and potential targeting ACE2. Nat. Prod. Bioprospect. 2022, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Güler, H.I.; Kara, Y. Targeting CoV-2 Spike RBD: ACE-II Complex with Phenolic Compounds from Cistus (Cistus L.) Bee Pollen for COVID-19 Treatment by Molecular Docking Study. J. Apither. Nat. 2020, 3, 10–23. [Google Scholar] [CrossRef]
- Thuy, B.T.P.; My, T.T.A.; Hai, N.T.T.; Hieu, L.T.; Hoa, T.T.; Loan, H.T.P.; Triet, N.T.; Anh, T.T.V.; Quy, P.T.; Tat, P.V.; et al. Investigation into SARS-CoV-2 Resistance of Compounds in Garlic Essential Oil. ACS Omega 2020, 5, 8312–8320. [Google Scholar] [CrossRef] [PubMed]
- Poochi, S.P.; Easwaran, M.; Balasubramanian, B.; Anbuselvam, M.; Meyyazhagan, A.; Park, S.; Bhotla, H.K.; Anbuselvam, J.; Arumugam, V.A.; Keshavarao, S.; et al. Employing bioactive compounds derived from Ipomoea obscura (L.) to evaluate potential inhibitor for SARS-CoV-2 main protease and ACE2 protein. Food Front. 2020, 1, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Sajib, A. Repurposing of Approved Drugs with Potential to Interact with SARS-CoV-2 Receptor. Preprints 2020, 2020040369. [Google Scholar] [CrossRef]
- Yadav, R.; Hasan, S.; Mahato, S.; Celik, I.; Mary, Y.S.; Kumar, A.; Dhamija, P.; Sharma, A.; Choudhary, N.; Chaudhary, P.K.; et al. Molecular docking, DFT analysis, and dynamics simulation of natural bioactive compounds targeting ACE2 and TMPRSS2 dual binding sites of spike protein of SARS CoV-2. J. Mol. Liq. 2021, 342, 116942. [Google Scholar] [CrossRef]
- Bharadwaj, H.; Kashid, E.; Kumar, S.P.; Tutika, S.; Kaushik, S. A Comparative Docking Analysis of Remdesivir and Dexamethasone Efficacy on Patients with COVID-19 and Myasthenia Gravis. ECS Trans. 2022, 107, 1409. [Google Scholar] [CrossRef]
- Petit, L.; Vernès, L.; Cadoret, J.P. Docking and in silico toxicity assessment of Arthrospira compounds as potential antiviral agents against SARS-CoV-2. J. Appl. Phycol. 2021, 33, 1579–1602. [Google Scholar] [CrossRef]
- Farshi, P.; Kaya, E.C.; Hashempour-Baltork, F.; Khosravi-Darani, K. The effect of plant metabolites on coronaviruses: A comprehensive review focusing on their IC50 values and molecular docking scores. Mini Rev. Med. Chem. 2021, 22, 457–483. [Google Scholar] [CrossRef]
- Ohishi, T.; Hishiki, T.; Baig, M.S.; Rajpoot, S.; Saqib, U.; Takasaki, T.; Hara, Y. Epigallocatechin gallate (EGCG) attenuates severe acute respiratory coronavirus disease 2 (SARS-CoV-2) infection by blocking the interaction of SARS-CoV-2 spike protein receptor-binding domain to human angiotensin-converting enzyme 2. PLoS ONE 2022, 17, e0271112. [Google Scholar] [CrossRef]
- Henss, L.; Auste, A.; Schürmann, C.; Schmidt, C.; von Rhein, C.; Mühlebach, M.D.; Schnierle, B.S. The green tea catechin epigallocatechin gallate inhibits SARS-CoV-2 infection. J. Gen. Virol. 2021, 102, 001574. [Google Scholar] [CrossRef]
- Singh, A.; Dhar, R. A large-scale computational screen identifies strong potential inhibitors for disrupting SARS-CoV-2 S-protein and human ACE2 interaction. J. Biomol. Struct. Dyn. 2021, 1–14. [Google Scholar] [CrossRef]
- Zia, A.; Akhtar, S.; Ali Khan, M. Docking of Velpatasvir to the SARS-CoV-2 Viral Spike Protein-Human ACE2 Complex: Repurposing for COVID-19. Preprints 2021. [Google Scholar] [CrossRef]
- Chen, C.C.; Zhuang, Z.J.; Wu, C.W.; Tan, Y.L.; Huang, C.H.; Hsu, C.Y.; Tsai, E.M.; Hsieh, T.H. Venetoclax Decreases the Expression of the Spike Protein through Amino Acids Q493 and S494 in SARS-CoV-2. Cells 2022, 11, 1924. [Google Scholar] [CrossRef]
- Fürstenau, M.; Langerbeins, P.; De Silva, N.; Fink, A.M.; Robrecht, S.; von Tresckow, J.; Simon, F.; Hohloch, K.; Droogendijk, J.; van der Klift, M.; et al. COVID-19 among fit patients with CLL treated with venetoclax-based combinations. Leukemia 2020, 34, 2225–2229. [Google Scholar] [CrossRef]
- Maffucci, I.; Contini, A. In Silico Drug Repurposing for SARS-CoV-2 Main Proteinase and Spike Proteins. J. Proteome Res. 2020, 19, 4637–4648. [Google Scholar] [CrossRef]
- Guedes, I.A.; Costa, L.S.C.; Dos Santos, K.B.; Karl, A.L.M.; Rocha, G.K.; Teixeira, I.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custódio, F.L.; et al. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep. 2021, 11, 5543. [Google Scholar] [CrossRef]
- Júnior, M.; de Sousa Junior, R.; Nze, G.; Giozza, W.; Júnior, L. Evaluation of Peppermint Leaf Flavonoids as SARS-CoV-2 Spike Receptor-Binding Domain Attachment Inhibitors to the Human ACE2 Receptor: A Molecular Docking Study. Open J. Biophys. 2022, 12, 132–152. [Google Scholar] [CrossRef]
- Vivar-Sierra, A.; Araiza-Macías, M.J.; Hernández-Contreras, J.P.; Vergara-Castañeda, A.; Ramírez-Vélez, G.; Pinto-Almazán, R.; Salazar, J.R.; Loza-Mejía, M.A. In Silico Study of Polyunsaturated Fatty Acids as Potential SARS-CoV-2 Spike Protein Closed Conformation Stabilizers: Epidemiological and Computational Approaches. Molecules 2021, 26, 711. [Google Scholar] [CrossRef]
- Elfiky, A.A. Natural products may interfere with SARS-CoV-2 attachment to the host cell. J. Biomol. Struct. Dyn. 2021, 39, 3194–3203. [Google Scholar] [CrossRef]
- Kumar, S.; Kashyap, P.; Chowdhury, S.; Kumar, S.; Panwar, A.; Kumar, A. Identification of phytochemicals as potential therapeutic agents that binds to Nsp15 protein target of coronavirus (SARS-CoV-2) that are capable of inhibiting virus replication. Phytomedicine 2021, 85, 153317. [Google Scholar] [CrossRef]
- Srivastava, R.; Tripathi, S.; Unni, S.; Hussain, A.; Haque, S.; Dasgupta, N.; Singh, V.; Mishra, B.N. Silybin B and Cianidanol Inhibit Mpro and Spike Protein of SARS-CoV-2: Evidence from in silico Molecular Docking Studies. Curr. Pharm. Des. 2021, 27, 3476–3489. [Google Scholar] [CrossRef]
- Uma Reddy, B.; Routhu, N.K.; Kumar, A. Multifaceted roles of plant derived small molecule inhibitors on replication cycle of SARS-CoV-2. Microb. Pathog. 2022, 168, 105512. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- How Is the IFD Score Calculated and What Is Its Units? Available online: https://www.schrodinger.com/kb/307 (accessed on 11 November 2022).
- Schrödinger Release 2018-1: Induced Fit Docking Protocol; Schrödinger, LLC: New York, NY, USA, 2018.
- Messi, B.B.; Ndjoko-Ioset, K.; Hertlein-Amslinger, B.; Lannang, A.M.; Nkengfack, A.E.; Wolfender, J.-L.; Hostettmann, K.; Bringmann, G. Preussianone, a New Flavanone-Chromone Biflavonoid from Garcinia preussii Engl. Molecules 2012, 17, 6114–6125. [Google Scholar] [CrossRef]
- Stermitz, F.R.; Tawara-Matsuda, J.; Lorenz, P.; Mueller, P.; Zenewicz, L.; Lewis, K. 5′-Methoxyhydnocarpin-D and pheophorbide A: Berberis species components that potentiate berberine growth inhibition of resistant Staphylococcus aureus. J. Nat. Prod. 2000, 63, 1146–1149. [Google Scholar] [CrossRef]
- Han, J.; Liu, Y.; Wang, R.; Yang, J.; Ling, V.; Borchers, C.H. Metabolic profiling of bile acids in human and mouse blood by LC-MS/MS in combination with phospholipid-depletion solid-phase extraction. Anal. Chem. 2015, 87, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Erlund, I. Review of the flavonoids quercetin, hesperetin and naringenin. Dietary sources, bioactivities, bioavailability and epidemiology. Nutr. Res. 2004, 24, 851–874. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-1: Maestro; Schrödinger, LLC: New York, NY, USA, 2018; Available online: https://www.schrodinger.com/freemaestro (accessed on 11 November 2022).
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Source | Pharmacological Function(s) | Binding Energy Score, kcal/mol a | References |
|---|---|---|---|---|
| binding domain: a | ||||
| 7-Methyl-guanosine-5′-triphosphate-5′-guanosine | synthetic | a biomarker of some types of cancer | −9.1 | [40] |
| 8-Bromo-adenosine-5′-monophosphate | synthetic | inhibition of inosine monophosphate dehydrogenase in Escherichia coli | −8.1 | [40] |
| Acalabrutinib | synthetic | inhibition of mantle cell lymphoma and chronic lymphocytic leukemia | −7.2 | [66] |
| Acitretin | synthetic | treatment of severe psoriasis and other skin disorders in adults | −9.6 | [67] |
| Adenosine-2′-5′-diphosphate | agonist activity at P2Y1 receptor in turkey erythrocyte membranes | −8.6 | [40] | |
| Alpinumisoflavone | Erythrina lysistemon | antischistosomal activity | −10.7 | [56] |
| Cladribine | synthetic | a medication used to treat hairy cell leukemia and B-cell chronic lymphocytic leukemia | −7.9 | [40] |
| Clofarabine | synthetic | treating relapsed or refractory acute lymphoblastic leukaemia | −7.2 | [40] |
| Curcumin | genus Curcuma | antiinflammatory, antitumor activity | −9.0 | [48] |
| Demethylzeylasteral | Tripterygium wilfordii Hook F. | androgen receptor in human LNCAP cells | ND | [44] |
| Dexamethasone | synthetic | anti-inflammatory and immunosuppressant effects; treating arthritis, severe allergies, asthma, and certain types of cancer | −6.5 | [68] |
| Dieckol | Eisenia bicyclis | antithrombotic and profibrinolytic activities | −8.1 | [69] |
| Dimethylcurcumin | synthetic | antiandrogen activity | −11.2 | [48,70] |
| Dithymoquinone | Nigella sativa | therapeutic of inflamation | −8.6 | [51] |
| Epigallocatechin-3-gallate | Camellia sinensis | antioxidant effects, cancer chemoprevention, improving cardiovascular health, enhancing weight loss | ND | [49,71,72] |
| Ergocalciferol (Vitamin D2) | Fish oil | a dietary supplement to prevent and treat vitamin D deficiency | −14.8 | [73] |
| Evans Blue | synthetic | a negative allosteric modulator of the AMPA and kainate receptors and an inhibitor of vesicular glutamate transporters | ND | [49] |
| Fludarabine | synthetic | a chemotherapy medication used in the treatment of leukemia and lymphoma | −7.0 | [40] |
| Glycyrrhizin | Glycyrrhiza radix | emulsifier and gel-forming agent in foodstuffs and cosmetics | −9.0 | [20] |
| Hesperidin | Citrus aurantium | inhibitor of the TRPM3 channels | −9.5 | [48] |
| Indacaterol | synthetic | an ultra-long-acting beta-adrenoceptor agonist used for the treatment of chronic obstructive pulmonary disease in patients with asthma | −8.1 | [53] |
| Kobophenol A | Caragana chamlagu | inhibitor of acetylcholinesterase | −11.1 | [45] |
| Levodopa | Mucuna pruriens | an amino acid precursor of dopamine with antiparkinsonian properties | −6.1 | [67] |
| Luteolin | Reseda luteola | a principal yellow dye compound | −7.8 | [53] |
| Parvisoflavone B | Erythrina schliebenii | antitubercular and cytotoxic activity | −10.7 | [56] |
| Rutin | Fagopyrum esculantum | antioxidant and cytoprotective properties | −7.9 | [21] |
| Taraxerol | Taraxacum officinale | antiinflammatory activity | −7.5 | [23] |
| Tazarotene | synthetic | treatment of plaque psoriasis and acne and a therapeutic for photoaged and photodamaged skin | −6.1 | [67] |
| Tretinoin | a natural derivative of vitamin A | treatment of acne and follicular keratosis and the curing of acute promyelocytic leukemia | −6.0 | [67] |
| Ursodeoxycholic acid | genus Ursus | treatment of several diseases of the liver or bile ducts | −7.0 | [53] |
| Velpatasvir | synthetic | the NS5A inhibitor used in the treatment of hepatitis C infection | −11.1 | [74] |
| Venetoclax | synthetic | a medication used to treat adults with chronic lymphocytic leukemia, small lymphocytic lymphoma, and acute myeloid leukemia | ND | [49,75,76,77] |
| Vitamin B12 | Propionibacterium shermanii | hematopoiesis, neural metabolism, DNA and RNA production | −7.6 | [22] |
| Vitamin K2 | Mycobacterium tuberculosis | a common form of vitamin K, primarily necessary for the body to carry out vital processes, cleaning blood vessels, and blood clotting | −9.5 | [78] |
| binding domain: b | ||||
| Acetoside | Olea europaea | antioxidant, anti-inflammatory activity | −8.5 | [21] |
| Amentoflavone | Ginkgo biloba | inhibitor of CYP3A4 and CYP2C9, which are enzymes responsible for the metabolism of some drugs in the body; it is also an inhibitor of human cathepsin B | −8.5 | [26] |
| Arbidol | synthetic | a broadspectrum respiratory antiviral drug | −7.7 | [58] |
| Celastrol | Tripterygium wilfordii | antitumor action, inhibitor of inflammatory and human prostate cancer activities | −8.3 | [26] |
| Dioscin | Ophiopogon intermedius | antitumor, antimicrobial, anti-infammatory, antioxidative, and tissue-protective activities | −8.9 | [26] |
| Epimedin C | Herba epimedii | treatment of cardiovascular disease and bone loss | −8.1 | [26] |
| Epitheaflavin monogallate | Camellia sinensis | antitoxicant, antioxidant, and antiinflammatory activity | −7.5 | [21] |
| Saikosaponin | Bupleurum chinense | treatment of hepatitis in Chinese herbal medicine | −9.1 | [26] |
| Solanine | Solanum nigrum | fungicide, antimicrobial and pesticide properties | −9.5 | [21] |
| binding domain: c | ||||
| Anabsinthin | Artemisia absinthium L. | inhibition of the human immunodeficiency virus 1 (HIV1) protease, treating acute bacillary dysentery | −12.5 | [25] |
| Atazanavir | synthetic | the inhibitor of the HIV protease; selectively inhibits the virus-specific processing of viral Gag-Pol proteins in the HIV-infected cells, preventing the infection of other cells | −12.4 | [62] |
| Baicalin | Scutellaria baicalensis | antioxidant, anti-inflammatory, and anti-apoptosis properties | −8.5 | [20] |
| β-Sitosterol | Solanum trilobatum | reduction of benign prostatic hyperplasia and blood cholesterol levels | −10.9 | [62] |
| Caflanone | Cannabis sativa | selective activity against the human coronavirus (COVID-19) disease; vasorelaxant activity against phenylephrine-induced contraction in rat aorta | −7.9 | [61] |
| Chloroquine | genus Cinchona | a medication used to prevent and treat malaria | −6.5 | [52] |
| Demethylzeylasteral | Tripterygium wilfordii | antitumor effects in a variety of cancers, inhibits the proliferation, migration, and invasion of gastric cancer cells | ND | [44] |
| Epitheaflavin monogallate | Camellia sinensis | cancer-fighting chemical when combined with cisplatin against ovarian cancer cells | −7.5 | [21] |
| Ertapenem | synthetic | a carbapenem antibiotic medication used for the treatment of infections of the abdomen, the lungs, the upper part of the female reproductive system, and the diabetic foot | −8.8 | [41] |
| Flavin adenine dinucleotide | cow milk | a cofactor for cytochrome-b5 reductase, the enzyme that maintains hemoglobin in its functional reduced state | −8.6 | [41] |
| Indacaterol | synthetic | an ultra-long-acting beta-adrenoceptor agonist licensed for the treatment of chronic obstructive pulmonary disease | −8.1 | [41,53] |
| Kaempferol | Lycopodiella inundata | a multipotential neuroprotective action through the modulation of several proinflammatory signaling pathways | −10.4 | [62] |
| Ledipasvir | synthetic | a direct acting antiviral medication used as part of combination therapy to treat chronic hepatitis C and exhibiting many pharmacological activities | −9.1 | [41] |
| Naringenin | genus Citrus | inhibition of some drug-metabolizing cytochrome P450 enzymes including CYP3A4 and CYP1A2 | −6.4 | [79] |
| Nicotianamine | Glycine max | potent inhibitor of the angiotensin-converting enzyme ACE2 | −5.1 | [20] |
| Raltegravir | synthetic | a potent CYP3A inhibitor decreasing the amount of human immunodeficiency virus in human blood | −9.1 | [41] |
| Stigmasterol | Ophiopogon japonicus | maintaining the structure and physiology of cell membranes | −9.8 | [62] |
| binding domain: d | ||||
| Chrysin | Scutellaria baicalensis | antivirus and antiinflammatory properties | −6.5 | [53] |
| Glycyrrhizin | Glycyrrhiza radix | antihepatotoxic activity | −9.0 | [20] |
| Linoleic acid | Carthamus tinctorius | one of two essential fatty acids for humans, who must obtain it through their diet | −6.8 | [80] |
| Myricetin 3-(4″-galloylrhamnoside) | Limonium species | an excellent source of phytosterols and flavonoids | −8.3 | [24] |
| Myricetin 3-rhamnoside | Newtonia buchananii | active against B. cereus, E. coli, and S. aureus | −8.5 | [24] |
| Pelargonidin | genus Geranium | a type of plant pigment producing a characteristic orange color, which is used in food and industrial dyes | −7.7 | [59] |
| binding domain: e | ||||
| Betulinic acid | Betula pubescens | a naturally occurring pentacyclic triterpenoid providing antiretroviral, antimalarial, and anti-inflammatory properties, as well as a more recently discovered potential as an anticancer agent | −8.1 | [56] |
| Canrenone | active metabolite of spironolactone | an antimineralocorticoid and active metabolite of spironolactone used in the treatment of primary hyperaldosteronism | −7.9 | [56] |
| Glycyrrhizin | Glycyrrhiza radix | a component of licorice, causes apparent mineralocorticoid excess through the inhibition of the enzyme 11-β-hydroxysteroid dehydrogenase | −9.0 | [20] |
| Oleanolic acid | Olea europaea, Rosa woodsii | exhibiting antitumor and antiviral properties together with weak anti-HIV and weak anti-HCV activities in vitro | −8.2 | [25,56] |
| Potassium canrenoate | synthetic | an aldosterone antagonist of the spirolactone group, metabolizing to active canrenone | −6.9 | [56] |
| binding domain: f | ||||
| Hesperetin | Citrus aurantium | inhibitor of the Mpro of SARS-coronaviruses | −9.1 | [20] |
| Scutellarin | Erigeron breviscapus | antiplatelet and anticoagulation properties | −14.9 | [20] |
| binding domain: g | ||||
| 2-vinyl-4H-1,3-dithiine | Allium sativum | affecting the vascular smooth muscle cells isolated from spontaneous hypertensive rats | −14.0 | [64] |
| Abemaciclib | synthetic | a medication for the treatment of advanced or metastatic breast cancers | −9.9 | [41,67] |
| Allyl disulfid | Allium sativum | providing antioxidative, antiviral, neuroprotective, antiparasitic, anticancer, and antihyperlipidemic activities | −15.3 | [64] |
| Allyl methyl trisulfide | Allium chinense, Mansoa alliacea | used as flavoring agent and tumor inhibitor | −14.4 | [64] |
| Allyl propyl trisulfide | Azadirachta indica | used in food additives and flavors | −14.0 | [64] |
| Caffeic acid phenethyl ester | Propolis | antimitogenic, anticarcinogenic, anti-inflammatory, and immunomodulatory properties in vitro | −6.5 | [81,82] |
| Chrysin | Passiflora caerulea | an ingredient in dietary supplements | −7.1 | [53] |
| Cianidanol | Salix atrocinerea, Visnea mocanera | an antioxidant flavonoid, occurring especially in woody plants | −9.5 | [83] |
| Diallyl tetrasulfid | synthetic | shown to selectively kill cancerous cells in the prostate and breast, leaving healthy cells unharmed; providing also antioxidant, anti-inflammatory, and anti-apoptotic effects; and a promising treatment for cardiac arrhythmias | −14.5 | [64] |
| Flavin adenine dinucleotide | cow milk | a redox-active coenzyme associated with various proteins, which is involved with several enzymatic reactions in metabolism | −9.9 | [41] |
| Pinocembrin | Turnera diffusa | antioxidant, a drug to treat cerebral ischemia, intracerebral hemorrhage, neurodegenerative diseases, cardiovascular diseases, and atherosclerosis | −7.8 | [63] |
| Ponatinib | synthetic | treatment of chronic myeloid leukemia and chromosome-positive acute lymphoblastic leukemia, a multi-targeted tyrosine-kinase inhibitor | −9.9 | [41] |
| Saquinavir | synthetic | an antiretroviral drug used to treat or prevent HIV/AIDS | −11.7 | [41,62,67] |
| Siponimod | synthetic | a selective sphingosine-1-phosphate receptor modulator for oral use for multiple sclerosis | −9.9 | [41,67] |
| Ursodeoxycholic acid | genus Ursus | used as therapy in primary biliary cholangitis; for intrahepatic cholestasis of pregnancy; has been suggested to be an adequate treatment of bile reflux gastritis | −8.7 | [45,53,65,84] |
| Binding Domain | Peculiarities of Domain | Residues |
|---|---|---|
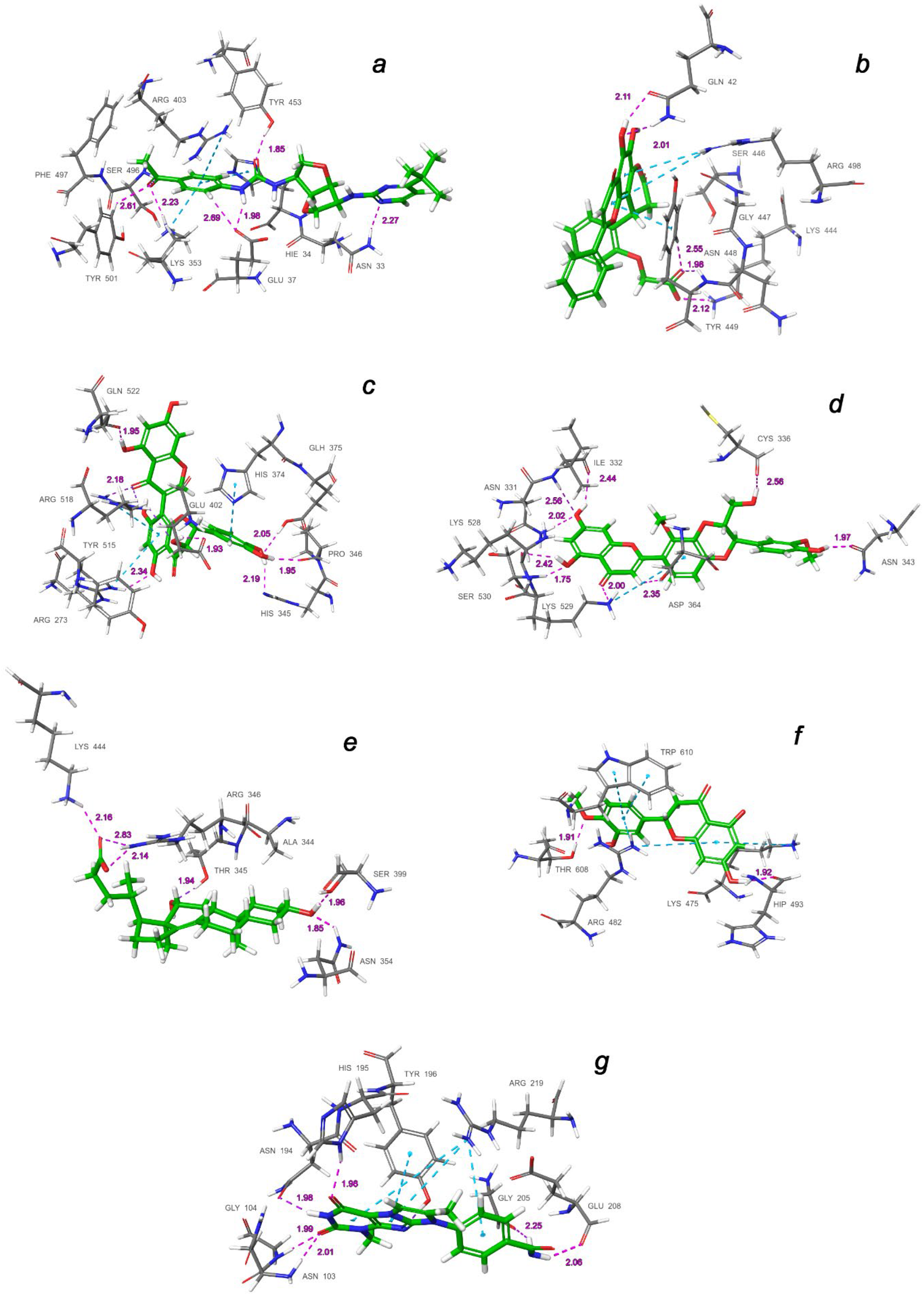
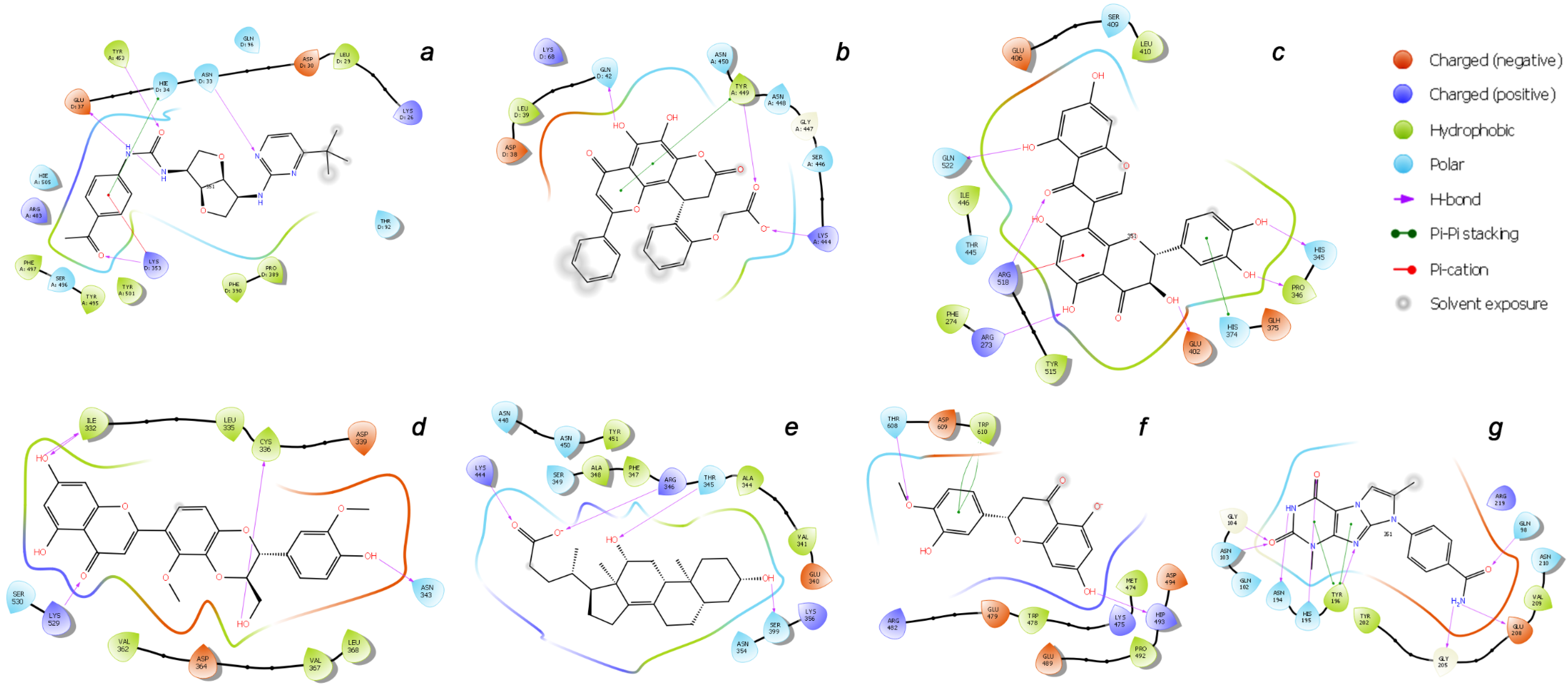
| a | Central contact area RBD with ACE2 | RBD: Glu406, Arg403, Ser496, His505 ACE2: His34, Asp30, Lys353, Thr27 |
| b | Hydrophobic pocket beside the interaction interface of RBD-ACE2 | RBD: Tyr449, Leu452, Ala352 |
| c | Catalytic cleft of ACE2 | ACE2: Thr371, Glu406, Arg273, His345, Asn149 |
| d | Bent FA hydrophobic tube of RBD | RBD: Leu368, Leu387, Phe388, Phe342, Ile434, Phe377, Phe338, Tyr365, Ala372 |
| e | β-sheet in the core of the RBD | RBD: Lys440, Ser438, Arg346, Asp442, Val445, Tyr451 |
| f | Deepening pocket at the ACE2 surface | ACE2: Arg482, Glu495 |
| g | Hydrophobic pocket alongside the cleft of ACE2 | ACE2: Ser511, Tyr196, Gln102, Glu208, Pro565, Trp 566, Ala 396, Gln 98, Leu91 |
| Library Size, Cmpds. | Hypothesis | Structure of Hypothesis | Key Ligand | Matched Ligand Sites | Phase Screen Score | Top Ligand (IFD) |
|---|---|---|---|---|---|---|
| binding domain: a | ||||||
| 2438 | a1 | ADDDDNR | CNP0260198 | DDNR | 1.418 | CNP0332318 |
| CNP0141274 | ADDR | 1.292 | ||||
| a2 | AADDDNR | CNP0363429 | ADDR | 1.606 | ||
| CNP0123143 | ADDR | 1.498 | ||||
| CNP0332318 | AADD | 1.477 | ||||
| a3 | AAADR | CNP0224071 | AAADR | 2.743 | ||
| AAARR | CNP0274243 | AAARR | 2.704 | |||
| AAARR | CNP0322514 | AAARR | 2.701 | |||
| a4 | AAADDDNNRR | CNP0305586 | AADR | 1.320 | ||
| CNP0429890 | ADRR | 1.291 | ||||
| binding domain: b | ||||||
| 3632 | b1 | ADDRRRR | CNP0129813 | ADRR | 1.811 | CNP0401960 |
| b2 | AAHNNNR | CNP0129813 | AHN | 1.516 | ||
| CNP0401960 | AHNR | 1.412 | ||||
| b3 | ADHR | CNP0131499 | ADHR | 1.898 | ||
| AADHR | CNP0146455 | AADHR | 1.860 | |||
| AADHR | CNP0403928 | AADHR | 1.808 | |||
| b4 | ADDNRRRR | CNP0128506 | ADNR | 1.393 | ||
| binding domain: c | ||||||
| 3657 | c1 | ADDDRRR | CNP0277806 | DDDRR | 1.665 | CNP0277806 |
| CNP0302437 | DDDRR | 1.646 | ||||
| CNP0318431 | DDRR | 1.622 | ||||
| CNP0129813 | ADRR | 1.620 | ||||
| c2 | AAADDRR | CNP0409641 | AARR | 1.604 | ||
| CNP0271209 | AAADRR | 1.555 | ||||
| CNP0406372 | AADR | 1.547 | ||||
| CNP0131497 | AAAR | 1.512 | ||||
| c3 | AAARR | CNP0437810 | AAARR | 2.781 | ||
| c4 | AAADDDRRR | CNP0168889 | AADDRR | 1.409 | ||
| CNP0153057 | AADDRR | 1.394 | ||||
| CNP0310325 | ADDDRR | 1.393 | ||||
| binding domain: d | ||||||
| 4953 | d1 | DDRRR | CNP0182350 | DDRR | 1.608 | CNP0380471 |
| CNP0191402 | DDRR | 1.508 | ||||
| d2 | AADDHNR | CNP0429546 | AAHNR | 1.453 | ||
| d3 | ADDRRR | CNP0318928 | ADDRRR | 2.630 | ||
| CNP0140035 | ADDRRR | 2.586 | ||||
| CNP0380471 | ADDRRR | 2.517 | ||||
| d4 | AADDDHNRRR | CNP0204419 | AAHNR | 1.327 | ||
| CNP0348217 | AADR | 1.220 | ||||
| binding domain: e | ||||||
| 4847 | e1 | HHN | CNP0161706 | HHN | 1.594 | CNP0340958 |
| e2 | AADNRRR | CNP0340958 | AADN | 1.348 | ||
| e3 | AHHHN | CNP0287935 | AHHHN | 2.688 | ||
| e4 | AADHNRRR | CNP0360609 | AAHH | 1.533 | ||
| CNP0364398 | AAHH | 1.504 | ||||
| CNP0329427 | AAHH | 1.475 | ||||
| binding domain: f | ||||||
| 4431 | f1 | ADRR | CNP0393256 | ADRR | 2.692 | CNP0393256 |
| f2 | ADDDRRR | CNP0104690 | ADRR | 1.540 | ||
| CNP0148806 | ADRR | 1.505 | ||||
| CNP0122888 | ADDRR | 1.499 | ||||
| f3 | AAADRR | CNP0393256 | AAADRR | 2.981 | ||
| f4 | AADDDRRRR | CNP0302437 | ADRRR | 1.481 | ||
| CNP0347670 | ADRR | 1.472 | ||||
| binding domain: g | ||||||
| 620 | g1 | ADDNRRR | CNP0342552 | ADRR | 1.413 | CNP0125042 |
| CNP0202472 | ADRR | 1.175 | ||||
| CNP0176937 | ADRR | 1.170 | ||||
| g2 | ADDDDDD | CNP0391500 | ADDD | 1.398 | ||
| CNP0005103 | ADDD | 1.389 | ||||
| CNP0176937 | ADDD | 1.352 | ||||
| g3 | AADRR | CNP0125042 | AARR | 2.126 | ||
| g4 | AADDDDNRR | CNP0071844 | ADRR | 1.298 | ||
| Binding Domain | Ligand | Binding Energy, kcal/mol | IFD Score, kcal/mol | Type of Interactions of Residues |
|---|---|---|---|---|
| a | CNP0332318 | −6.71 | −1673.0 | H-bond: Tyr453(A), Ser496(A), Tyr501(A), Asn33(D), Glu37(D), Lys353(D) π-alkyl: Arg403(A), Lys353(D) t-stacking: His34(D) |
| b | CNP0401960 | −7.93 | −1672.3 | H-bond: Lys444(A), Tyr449(A), Gln42(D) π-alkyl: Arg498(A) π-stacking: Tyr449(A) |
| c | CNP0277806 | −9.98 | −1677.2 | H-bond: Arg273(D), His345(D), Pro346(D), Gln375 (D), Glu402(D), Arg518(D), Gln522(D) π-alkyl: Arg273(D), Arg518(D) π-stacking: His374(D) |
| d | CNP0380471 | −8.68 | −1673.6 | H-bond: Asn331(A), Ile332(A), Cys336(A), Asn343(A), Asp364(A), Lys528(A), Lys529(A), Ser530(A) π-alkyl: Lys529(A) |
| e | CNP0340958 | −6.21 | −1671.4 | H-bond: Thr345(A), Arg346(A), Asn354(A), Ser399(A), Lys444(A) |
| f | CNP0393256 | −6.56 | −1671.8 | H-bond: His493(D), Thr608(D) π-alkyl: Lys475(D), Arg482(D) t-stacking: Trp610(D) |
| g | CNP0125042 | −9.16 | −1676.5 | H-bond: Gln98(D), Asn103(D), Gly104(D), Asn194(D), His195(D), Tyr196(D), Gly205(D), Glu208(D) π-alkyl: Arg219(D) t-stacking: Tyr196(D) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenov, V.A.; Krivdin, L.B. The Development of Pharmacophore Models for the Search of New Natural Inhibitors of SARS-CoV-2 Spike RBD–ACE2 Binding Interface. Molecules 2022, 27, 8938. https://doi.org/10.3390/molecules27248938
Semenov VA, Krivdin LB. The Development of Pharmacophore Models for the Search of New Natural Inhibitors of SARS-CoV-2 Spike RBD–ACE2 Binding Interface. Molecules. 2022; 27(24):8938. https://doi.org/10.3390/molecules27248938
Chicago/Turabian StyleSemenov, Valentin A., and Leonid B. Krivdin. 2022. "The Development of Pharmacophore Models for the Search of New Natural Inhibitors of SARS-CoV-2 Spike RBD–ACE2 Binding Interface" Molecules 27, no. 24: 8938. https://doi.org/10.3390/molecules27248938
APA StyleSemenov, V. A., & Krivdin, L. B. (2022). The Development of Pharmacophore Models for the Search of New Natural Inhibitors of SARS-CoV-2 Spike RBD–ACE2 Binding Interface. Molecules, 27(24), 8938. https://doi.org/10.3390/molecules27248938