

Assessment of AI-Based Protein Structure Prediction for the NLRP3 Target
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Model Generation
2.2. Simulation Details
3. Results
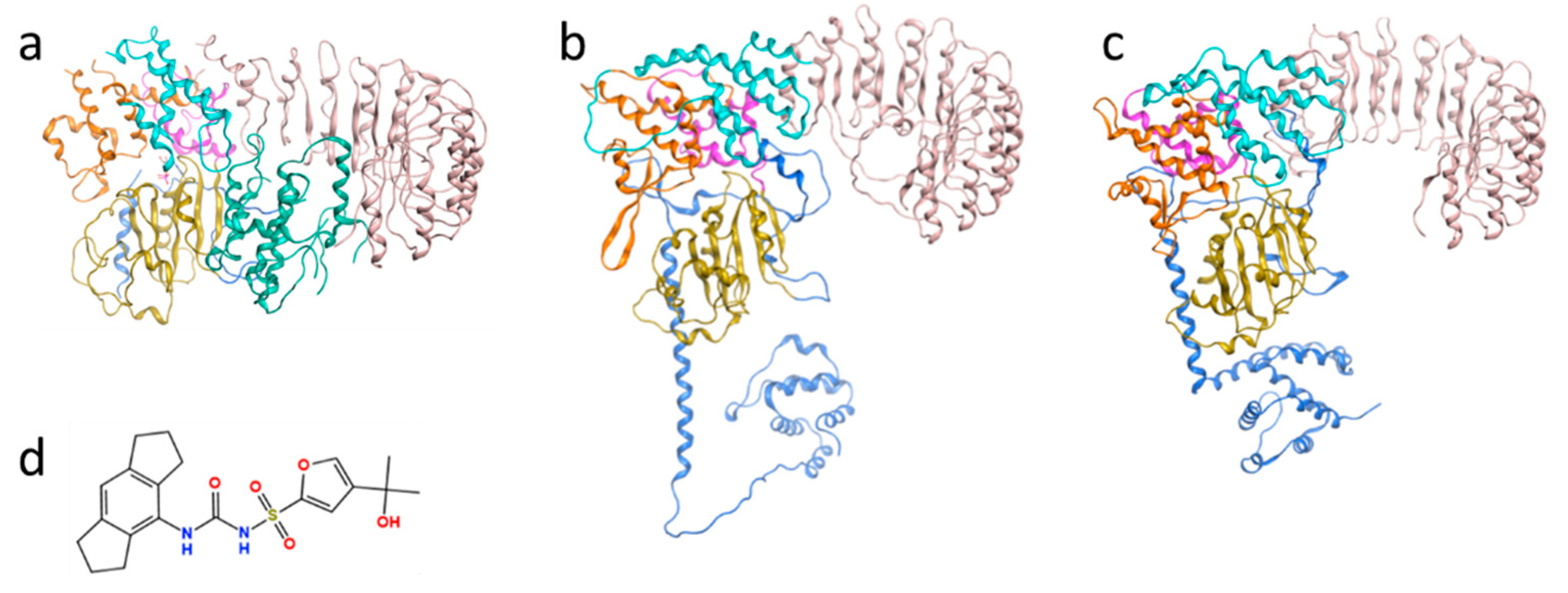
3.1. Evaluation of the AI-Predicted Models of NLRP3
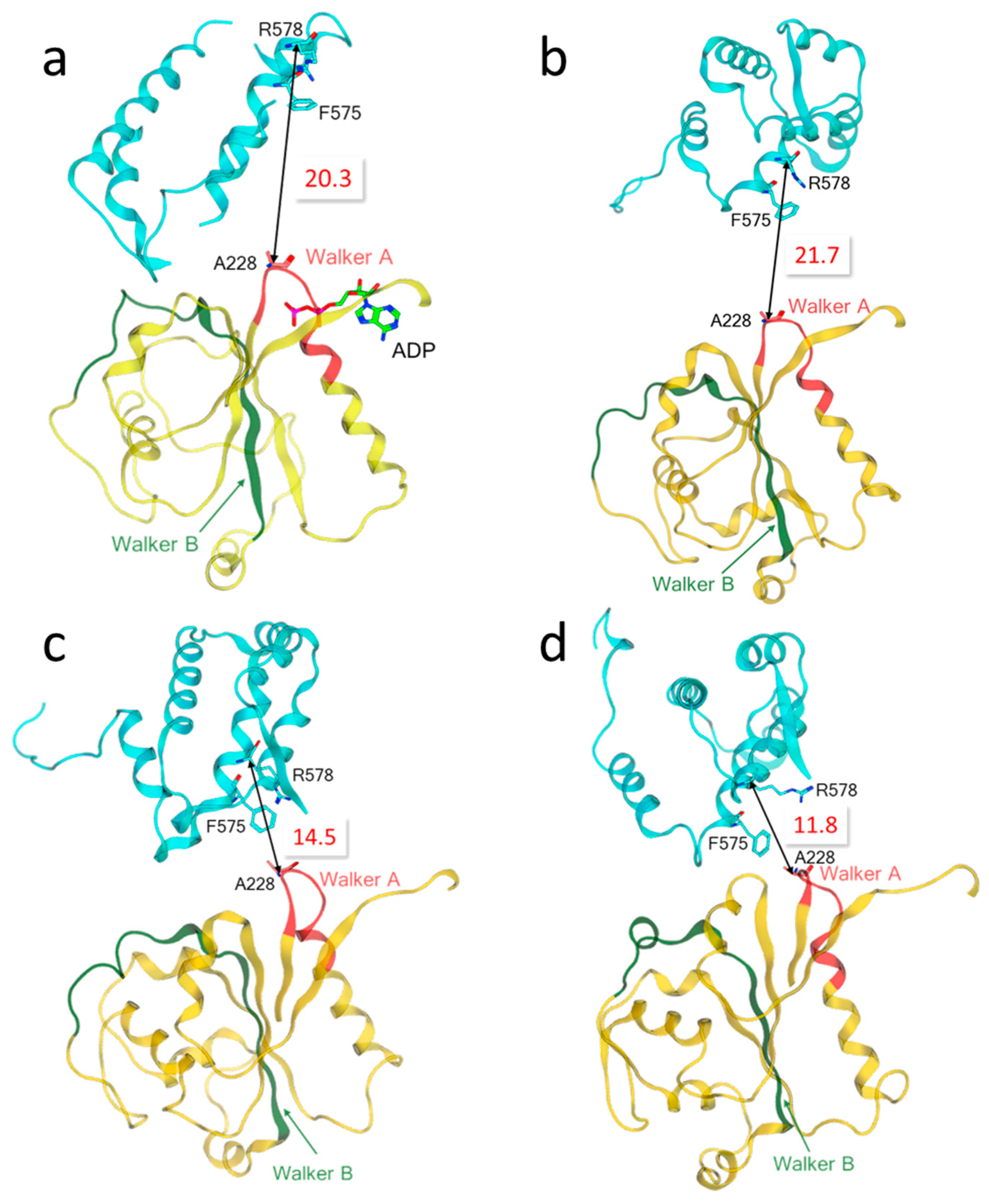
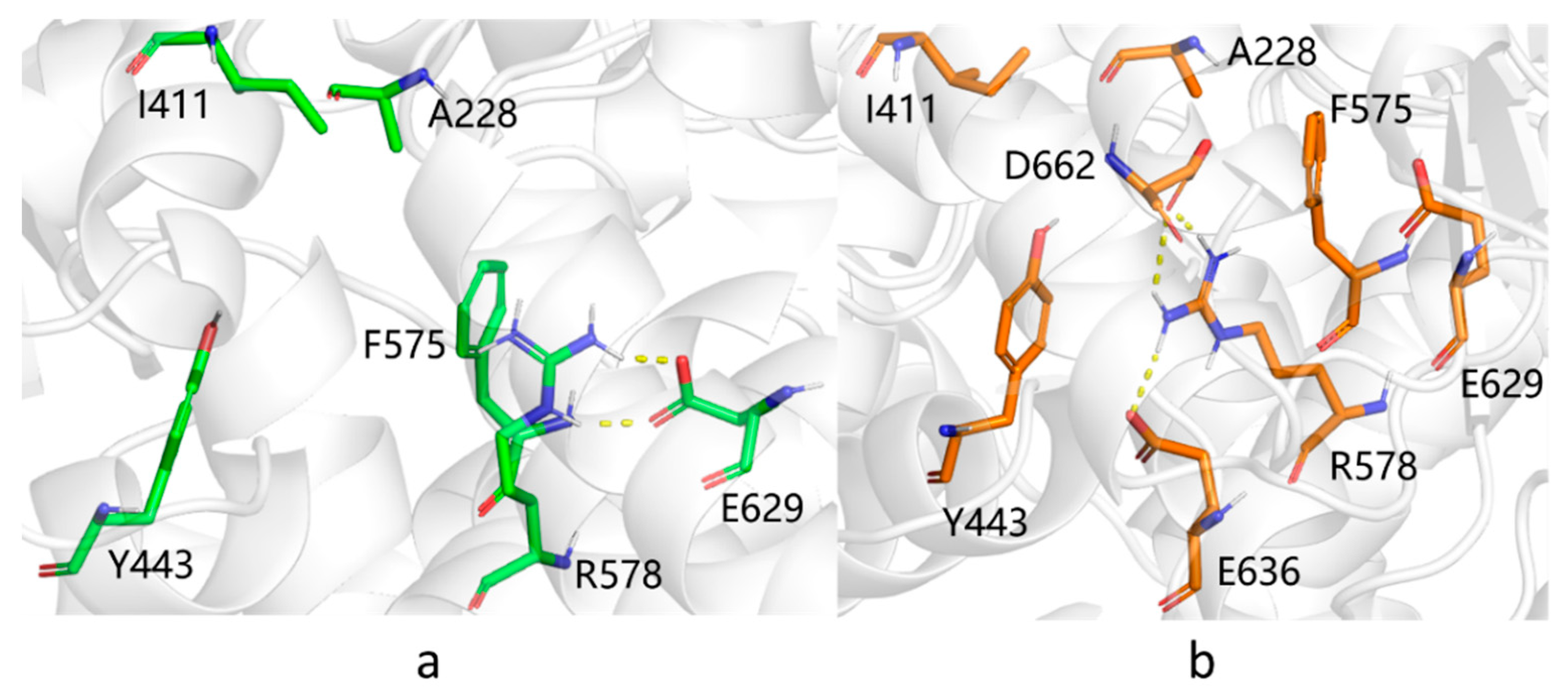
3.2. Characterization of the Binding Pocket of Compound MCC950
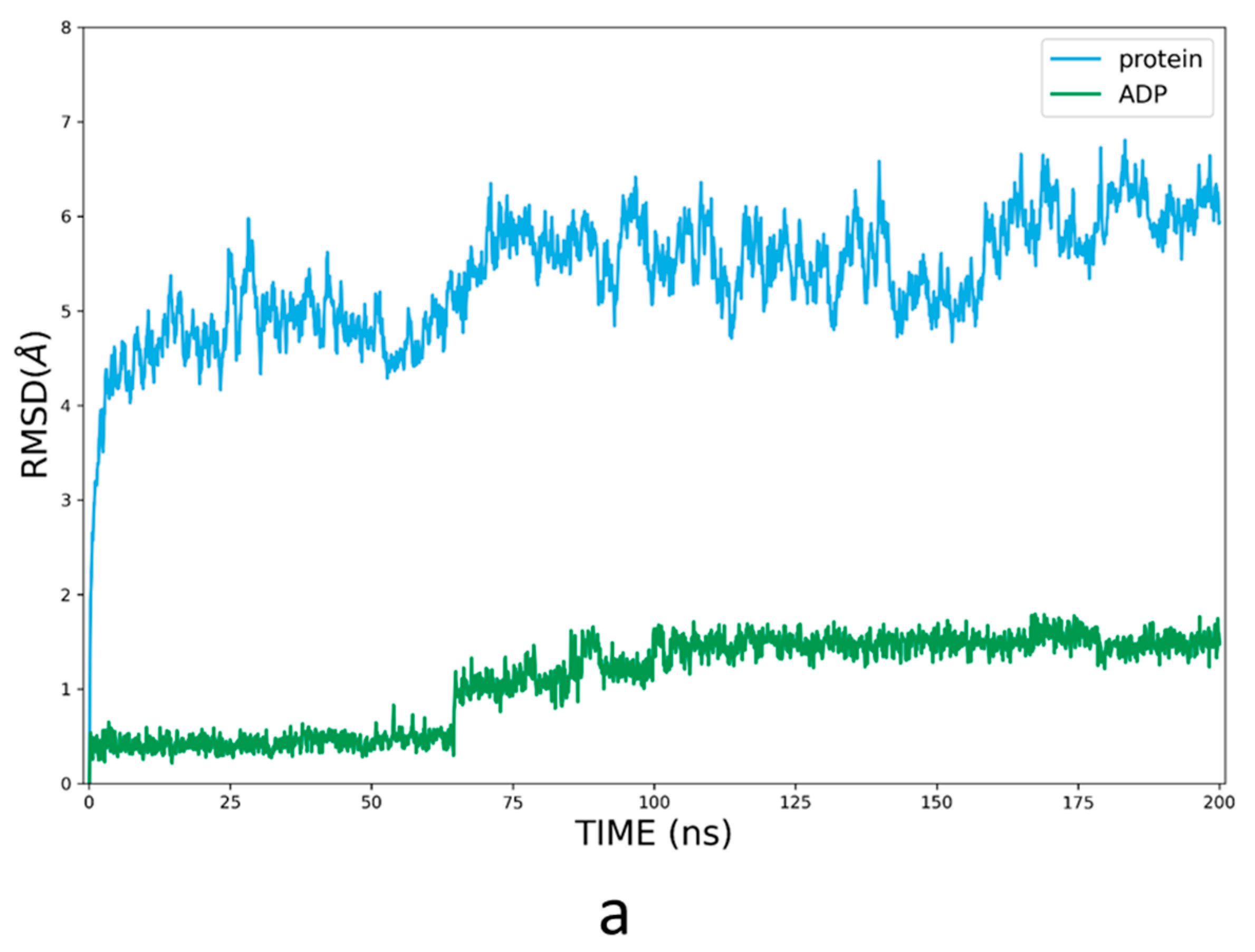
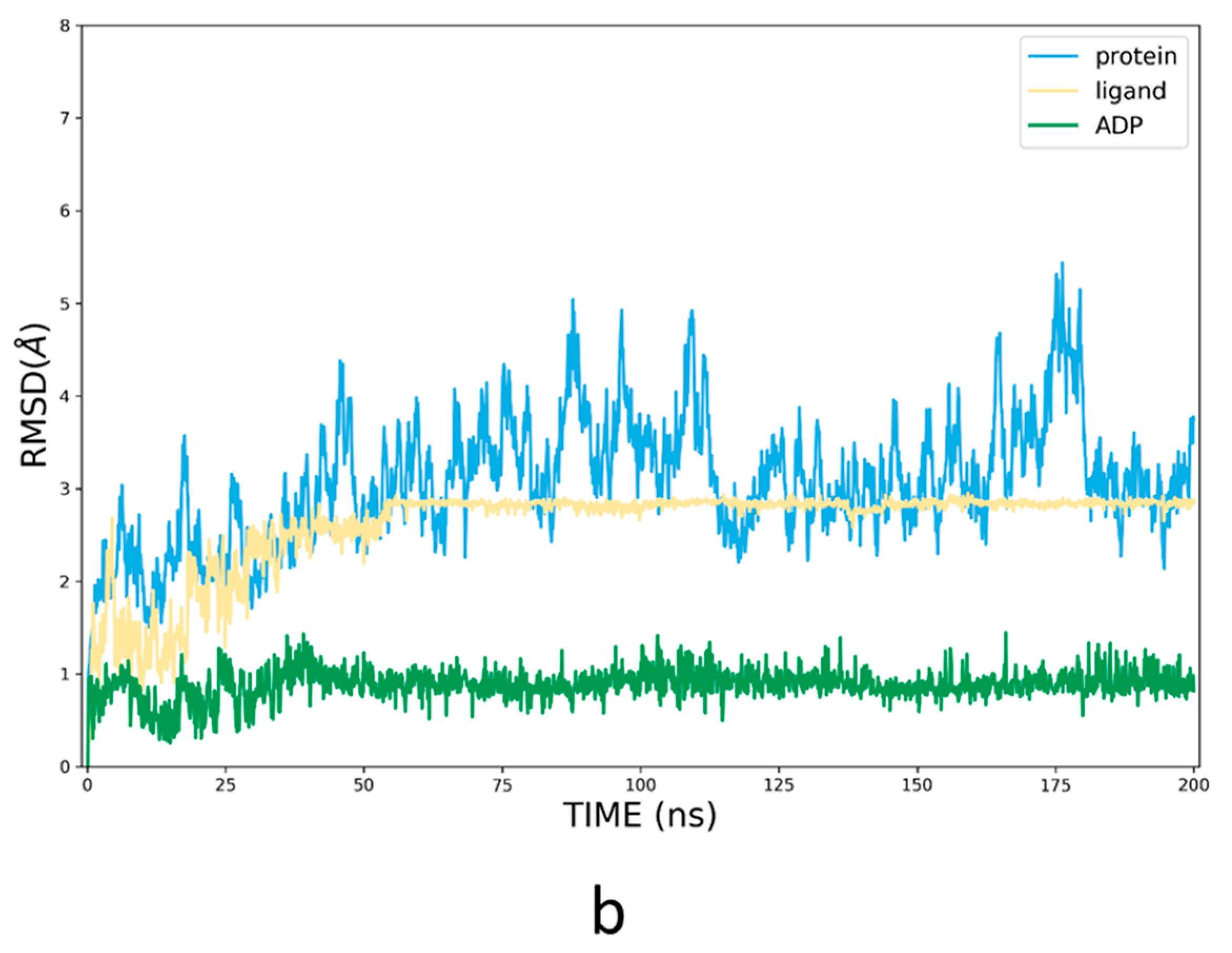
3.3. Refinement of the Docking Pose Using Molecular Dynamics Simulation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marrone, T.J.; Briggs, A.J.M.; McCammon, J.A. Structure-Based Drug Design:Computational Advances. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Schaeffer, R.D.; Kinch, L.; Kryshtafovych, A.; Grishin, N.V. Assessment of domain interactions in the fourteenth round of the Critical Assessment of Structure Prediction. Proteins Struct. Funct. Bioinform. 2021, 89, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Sharif, H.; Wang, L.; Wang, W.L.; Magupalli, V.G.; Andreeva, L.; Qiao, Q.; Hauenstein, A.; Wu, Z.; Núñez, G.; Mao, Y.; et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019, 570, 338–343. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Hochheiser, I.V.; Pilsl, M.; Hagelueken, G.; Moecking, J.; Michael, M.; Brinkschulte, R.; Latz, E.; Engel, C.; Geyer, M. Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 2022, 604, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Dekker, C.; Mattes, H.; Wright, M.; Boettcher, A.; Hinniger, A.; Hughes, N.; Kapps-Fouthier, S.; Eder, J.; Erbel, P.; Stiefl, N.; et al. Crystal Structure of NLRP3 NACHT Domain with an Inhibitor Defines Mechanism of Inflammasome Inhibition. J. Mol. Biol. 2021, 433, 167309. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2022; Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 19 August 2022).
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2019, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; de Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.; Wijekoon, C.P.; Liao, K.-C.; Muruve, D.A. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life 2013, 65, 851–862. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 |
|---|---|---|---|---|---|
| AlphaFold | |||||
| NBD | 83.3 | 85.3 | 86.8 | 83.5 | 85.7 |
| HD1 | 82.8 | 84.9 | 85.4 | 82.1 | 84.3 |
| WHD | 70.2 | 71.5 | 74.2 | 71.0 | 71.2 |
| HD2 | 76.8 | 78.6 | 79.4 | 77.2 | 79.5 |
| LRR | 84.6 | 85.0 | 86.4 | 85.3 | 84.7 |
| Whole | 79.1 | 80.3 | 81.4 | 79.1 | 79.8 |
| RoseTTAFold | |||||
| NBD | 83.4 | 81.6 | 82.5 | 80.7 | 79.5 |
| HD1 | 83.5 | 82.3 | 81.3 | 80.7 | 80.3 |
| WHD | 80.0 | 78.4 | 76.4 | 78.7 | 77.3 |
| HD2 | 75.2 | 73.1 | 70.2 | 73.0 | 71.1 |
| LRR | 82.3 | 82.9 | 83.1 | 81.9 | 81.8 |
| Whole | 79.7 | 79.4 | 77.9 | 77.6 | 76.8 |
| Domain | Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | No. of Aligned Atoms |
|---|---|---|---|---|---|---|
| AlphaFold | ||||||
| NBD | 4.4 (3.7) | 4.4 (3.7) | 4.4 (3.7) | 4.4 (3.7) | 4.4 (3.7) | 1178 (154) |
| HD1 | 2.7 (1.8) | 2.6 (1.7) | 3.0 (2.2) | 2.6 (1.6) | 2.7 (1.8) | 505 (62) |
| WHD | 6.0 (5.3) | 6.2 (5.5) | 6.1 (5.4) | 5.9 (5.2) | 6.1 (5.4) | 780 (98) |
| HD2 | 18.5 (18.3) | 18.6 (18.4) | 18.5 (18.3) | 18.5 (18.3) | 18.4 (18.3) | 587 (75) |
| LRR | 12.1 (11.8) | 12.5 (12.1) | 13.5 (13.1) | 11.4 (11.2) | 12.8 (12.5) | 2713 (359) |
| Full-length | 14.7 (14.4) | 14.8 (14.4) | 14.6 (14.2) | 10.5 (10.2) | 14.6 (14.2) | 6192 (798) |
| RoseTTAFold | ||||||
| NBD | 4.2 (3.5) | 4.3 (3.6) | 4.3 (3.6) | 4.2 (3.6) | 4.1 (3.5) | Same as AlphaFold |
| HD1 | 2.4 (1.4) | 2.6 (1.5) | 2.5 (1.5) | 2.5 (1.5) | 2.5 (1.4) | |
| WHD | 3.2 (2.3) | 3.2 (2.3) | 3.3 (2.4) | 3.2 (2.3) | 3.2 (2.4) | |
| HD2 | 16.3 (15.8) | 17.6 (17.2) | 17.6 (17.6) | 16.4 (16.0) | 16.5 (16.0) | |
| LRR | 7.9 (7.4) | 8.2 (7.7) | 8.2 (7.7) | 8.2 (7.7) | 8.1 (7.5) | |
| Full-length | 10.7 (10.2) | 10.5 (9.9) | 10.7 (10.2) | 10.5 (9.9) | 10.5 (9.9) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, J.; Lei, J.; Yu, J.; Cui, W.; Satz, A.L.; Zhou, Y.; Feng, H.; Deng, J.; Su, W.; Kuai, L. Assessment of AI-Based Protein Structure Prediction for the NLRP3 Target. Molecules 2022, 27, 5797. https://doi.org/10.3390/molecules27185797
Yin J, Lei J, Yu J, Cui W, Satz AL, Zhou Y, Feng H, Deng J, Su W, Kuai L. Assessment of AI-Based Protein Structure Prediction for the NLRP3 Target. Molecules. 2022; 27(18):5797. https://doi.org/10.3390/molecules27185797
Chicago/Turabian StyleYin, Jian, Junkun Lei, Jialin Yu, Weiren Cui, Alexander L. Satz, Yifan Zhou, Hua Feng, Jason Deng, Wenji Su, and Letian Kuai. 2022. "Assessment of AI-Based Protein Structure Prediction for the NLRP3 Target" Molecules 27, no. 18: 5797. https://doi.org/10.3390/molecules27185797
APA StyleYin, J., Lei, J., Yu, J., Cui, W., Satz, A. L., Zhou, Y., Feng, H., Deng, J., Su, W., & Kuai, L. (2022). Assessment of AI-Based Protein Structure Prediction for the NLRP3 Target. Molecules, 27(18), 5797. https://doi.org/10.3390/molecules27185797