


Characterization of Continuous Transcriptional Heterogeneity in High-Risk Blastemal-Type Wilms’ Tumors Using Unsupervised Machine Learning

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
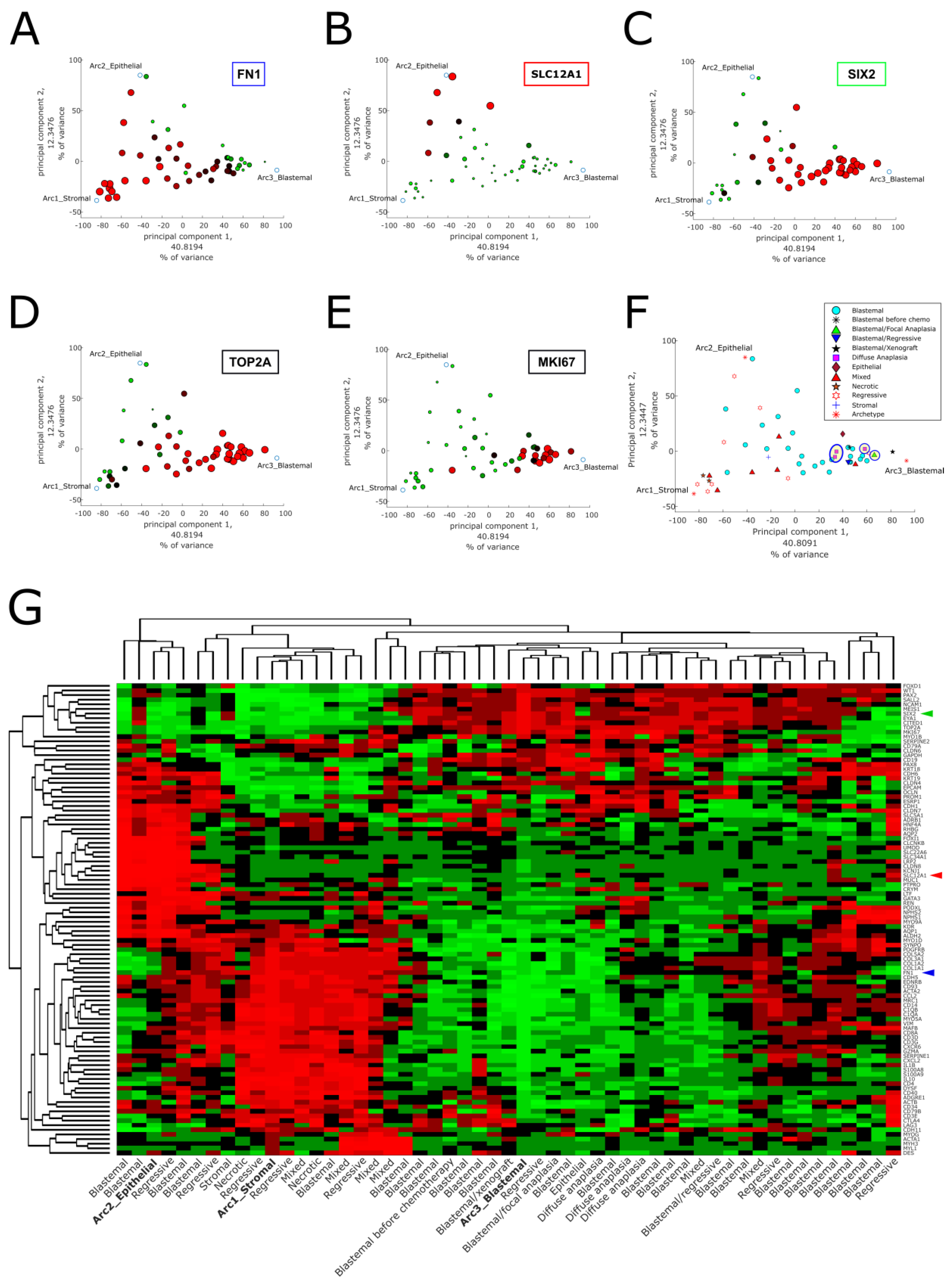
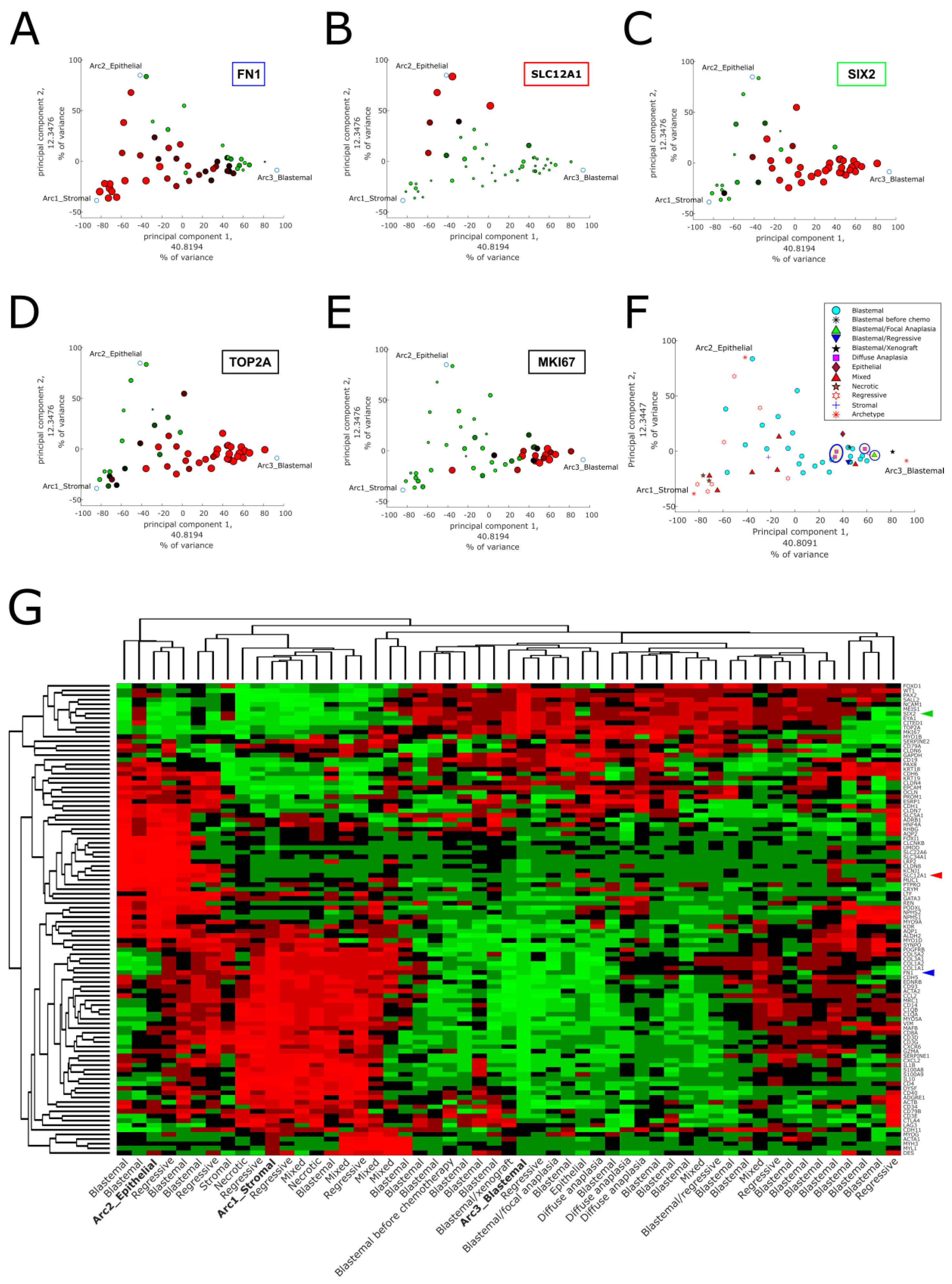
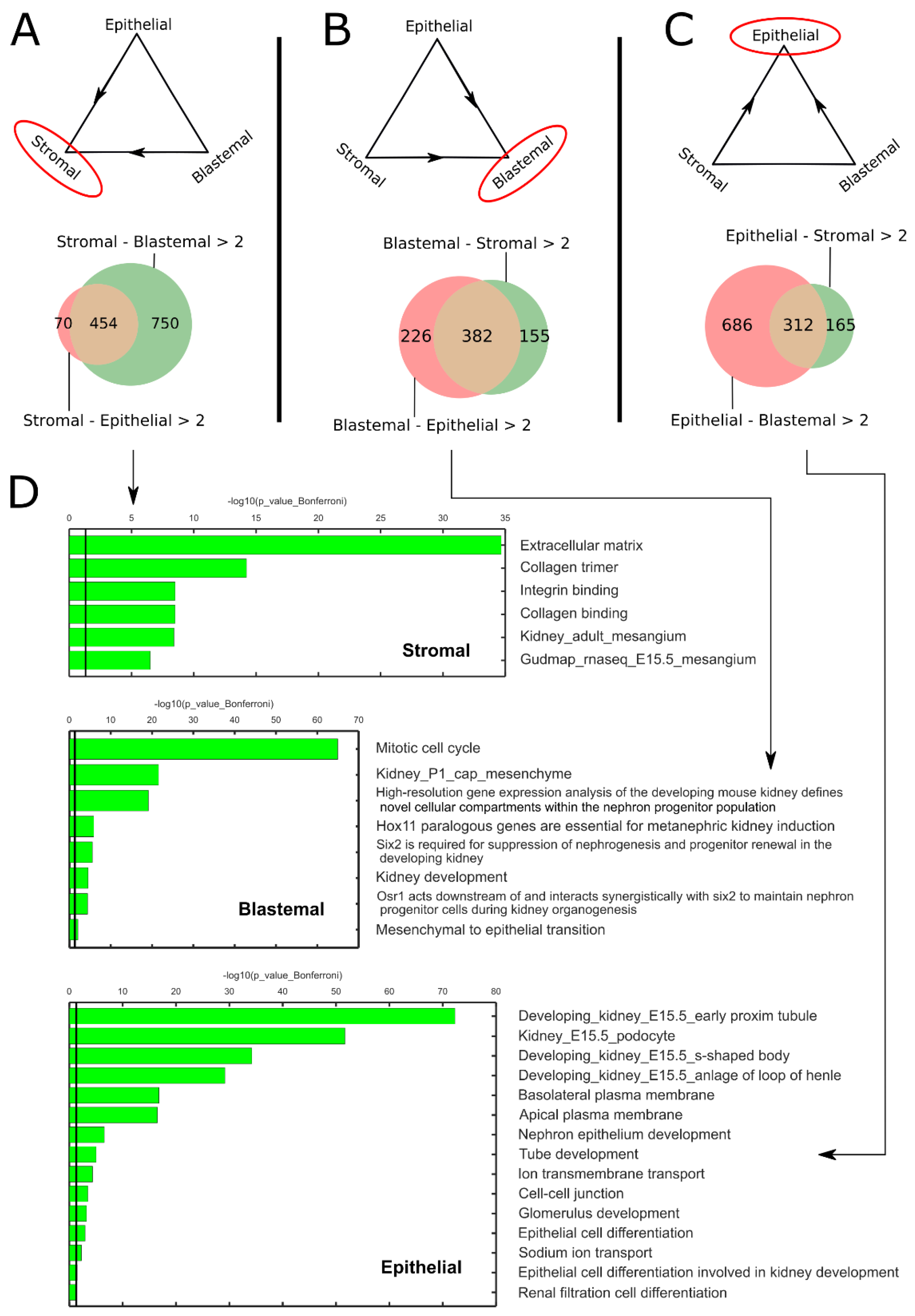
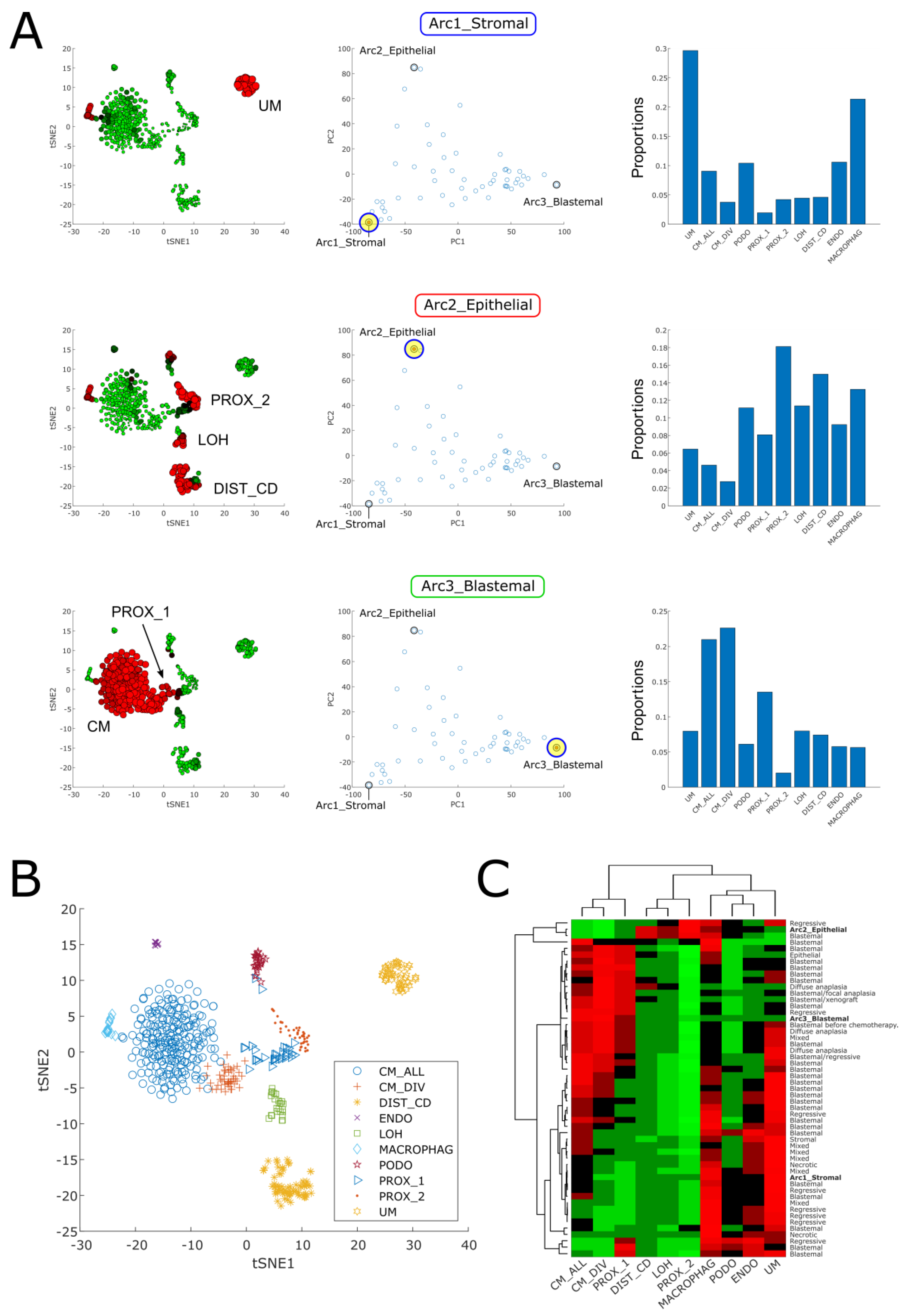
2.1. Pareto Task Inference Shows That Blastemal Type, Post-Operative Chemotherapy Wilms’ Tumors Fill a Triangle-Shaped Continuum in Latent Space That Is Bounded by Archetypes with Stromal, Epithelial, and Blastemal Characteristics
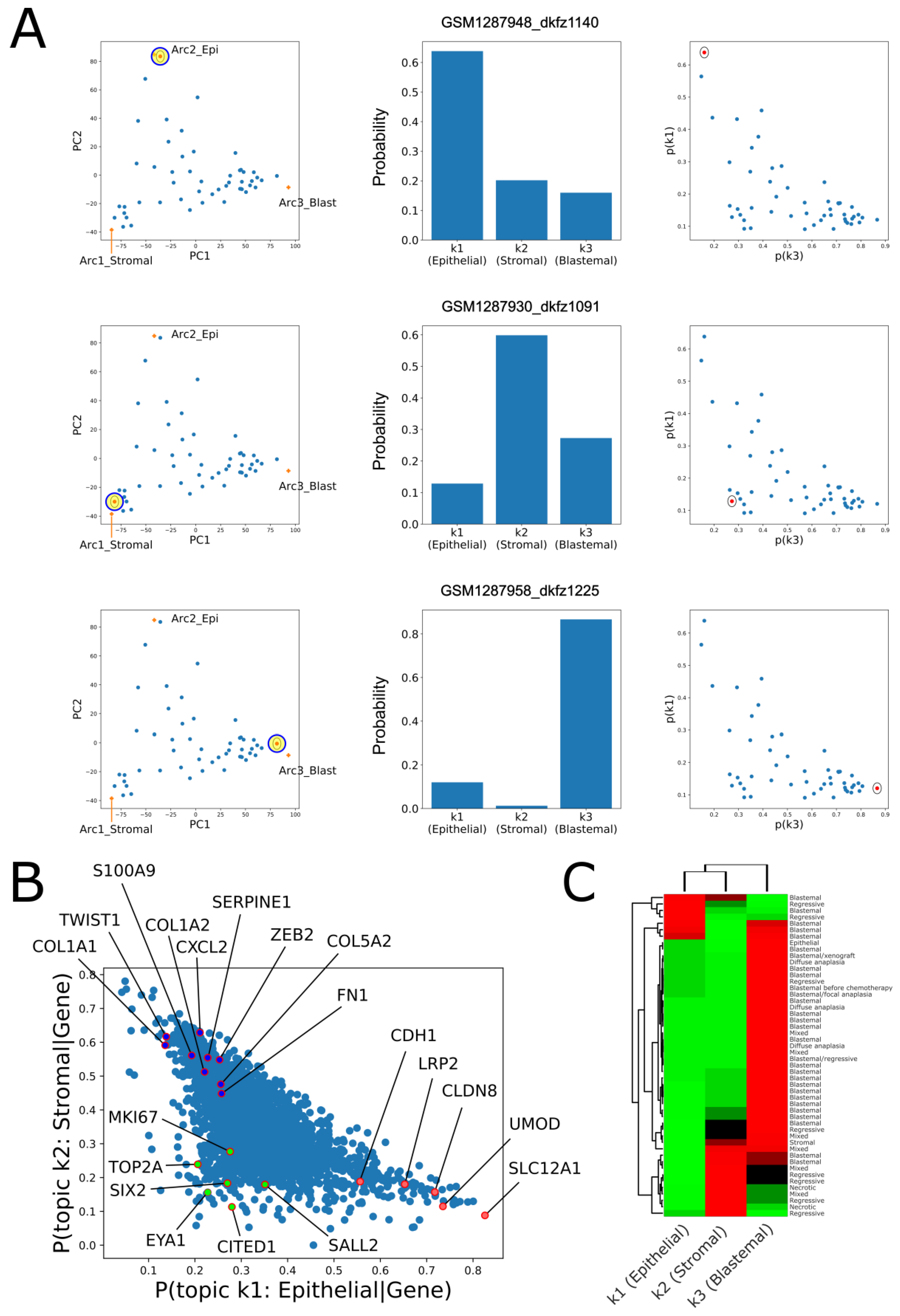
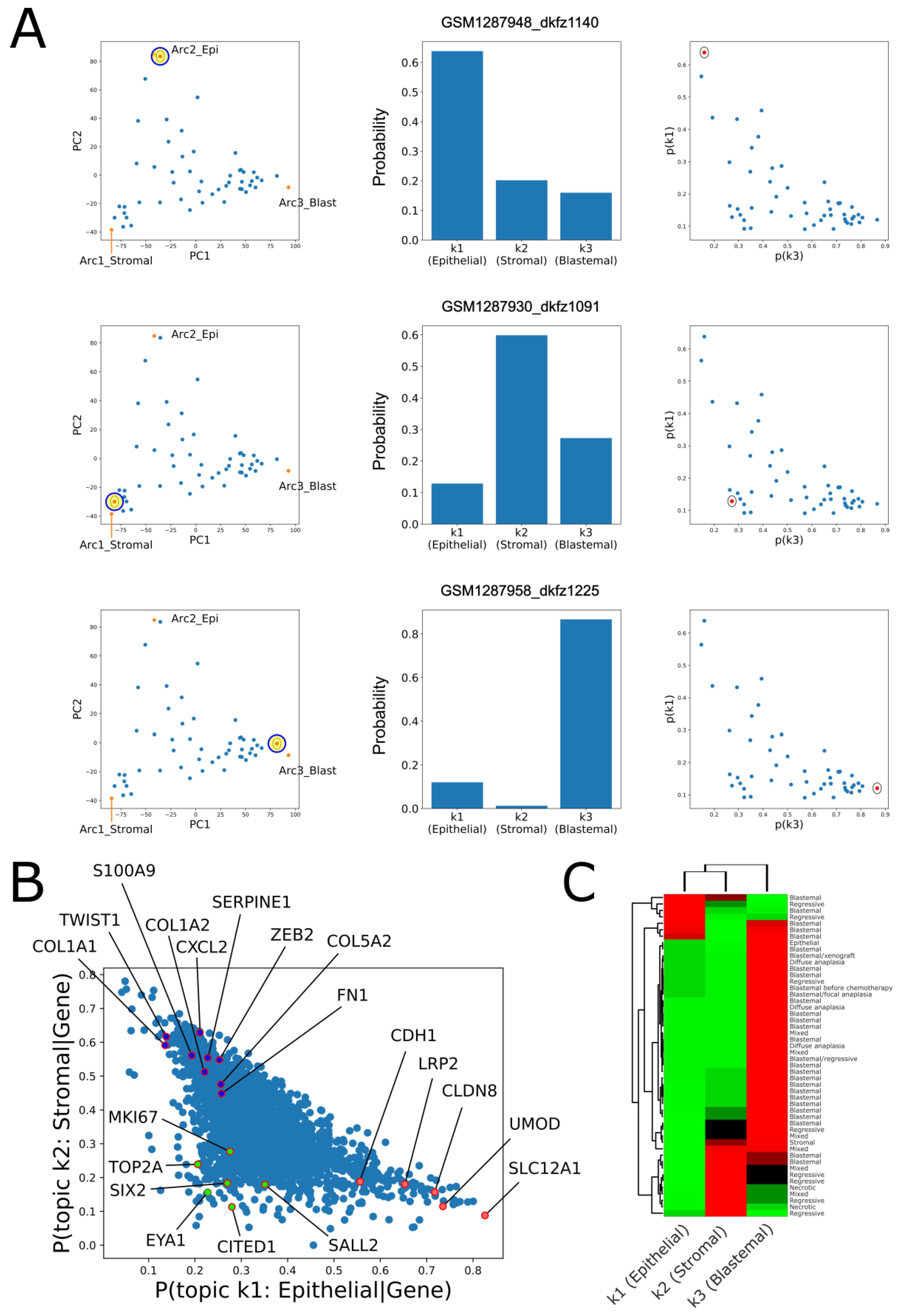
2.2. Topic Modeling Shows That Each Tumor Can Be Represented as a Unique Mixture of Three Hidden Topics with Blastemal, Stromal, and Epithelial Characteristics
2.3. Cellular Deconvolution Indicates That Each Tumor Is Composed of a Unique Mixture of Cell Populations Resembling Those of the Fetal Kidney
3. Discussion
4. Materials and Methods
4.1. Gene Expression Datasets
4.2. Data Preprocessing
4.3. Data Visualization and Clustering
4.4. Archetype Analysis
4.5. Topic Modeling
4.6. Cellular Deconvolution
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Servaes, S.E.; Hoffer, F.A.; Smith, E.A.; Khanna, G. Imaging of Wilms tumor: An update. Pediatr. Radiol. 2019, 49, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, P.; Pritchard-Jones, K.; Charlton, J. The yin and yang of kidney development and Wilms’ tumors. Genes Dev. 2015, 29, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Dome, J.S.; Fernandez, C.V.; Mullen, E.A.; Kalapurakal, J.A.; Geller, J.I.; Huff, V.; Gratias, E.J.; Dix, D.B.; Ehrlich, P.F.; Khanna, G.; et al. Children’s Oncology Group’s 2013 blueprint for research: Renal tumors. Pediatr. Blood Cancer 2013, 60, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Vujanić, G.M.; Sandstedt, B. The pathology of Wilms’ tumour (nephroblastoma): The International Society of Paediatric Oncology approach. J. Clin. Pathol. 2010, 63, 102–109. [Google Scholar] [CrossRef]
- Popov, S.D.; Sebire, N.J.; Vujanic, G.M. Wilms’ Tumour—Histology and Differential Diagnosis. In Wilms Tumor; van den Heuvel-Eibrink, M.M., Ed.; Codon Publications: Brisbane, Australia, 2016. [Google Scholar] [CrossRef]
- Trink, A.; Kanter, I.; Pode-Shakked, N.; Urbach, A.; Dekel, B.; Kalisky, T. Geometry of Gene Expression Space of Wilms’ Tumors From Human Patients. Neoplasia 2018, 20, 871–881. [Google Scholar] [CrossRef]
- Gadd, S.; Huff, V.; Huang, C.-C.; Ruteshouser, E.C.; Dome, J.S.; Grundy, P.E.; Breslow, N.; Jennings, L.; Green, D.M.; Beckwith, J.B.; et al. Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: A Children’s Oncology Group Study. Neoplasia 2012, 14, 742–756. [Google Scholar] [CrossRef]
- Gadd, S.; Sredni, S.T.; Huang, C.-C.; Perlman, E.J. Rhabdoid tumor: Gene expression clues to pathogenesis and potential therapeutic targets. Lab. Investig. 2010, 90, 724–738. [Google Scholar] [CrossRef]
- Huang, C.-C.; Gadd, S.; Breslow, N.; Cutcliffe, C.; Sredni, S.T.; Helenowski, I.B.; Dome, J.S.; Grundy, P.E.; Green, D.M.; Fritsch, M.K.; et al. Predicting relapse in favorable histology wilms tumor using gene expression analysis: A report from the renal tumor committee of the children’s oncology group. Clin. Cancer Res. 2009, 15, 1770–1778. [Google Scholar] [CrossRef]
- Sredni, S.T.; Gadd, S.; Huang, C.-C.; Breslow, N.; Grundy, P.; Green, D.M.; Dome, J.S.; Shamberger, R.C.; Beckwith, J.B.; Perlman, E.J.; et al. Subsets of very low risk Wilms tumor show distinctive gene expression, histologic, and clinical features. Clin. Cancer Res. 2009, 15, 6800–6809. [Google Scholar] [CrossRef] [Green Version]
- Wegert, J.; Ishaque, N.; Vardapour, R.; Geörg, C.; Gu, Z.; Bieg, M.; Ziegler, B.; Bausenwein, S.; Nourkami, N.; Ludwig, N.; et al. Mutations in the SIX1/2 Pathway and the DROSHA/DGCR8 miRNA Microprocessor Complex Underlie High-Risk Blastemal Type Wilms Tumors. Cancer Cell 2015, 27, 298–311. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [PubMed]
- Oyelade, J.; Isewon, I.; Oladipupo, F.; Aromolaran, O.; Uwoghiren, E.; Ameh, F.; Achas, M.; Adebiyi, E. Clustering Algorithms: Their Application to Gene Expression Data. Bioinform. Biol Insights 2016, 10, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; Hsiao, C.J.; Stephens, M. Visualizing the structure of RNA-seq expression data using grade of membership models. PLOS Genet. 2017, 13, e1006599. [Google Scholar] [CrossRef]
- Jew, B.; Alvarez, M.; Rahmani, E.; Miao, Z.; Ko, A.; Garske, K.M.; Sul, J.H.; Pietiläinen, K.H.; Pajukanta, P.; Halperin, E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020, 11, 1971. [Google Scholar] [CrossRef]
- Frishberg, A.; Peshes-Yaloz, N.; Cohn, O.; Rosentul, D.; Steuerman, Y.; Valadarsky, L.; Yankovitz, G.; Mandelboim, M.; Iraqi, F.A.; Amit, I.; et al. Cell composition analysis of bulk genomics using single cell data. Nat Methods 2019, 16, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Hart, Y.; Sheftel, H.; Hausser, J.; Szekely, P.; Ben-Moshe, N.B.; Korem, Y.; Tendler, A.; Mayo, A.E.; Alon, U. Inferring biological tasks using Pareto analysis of high-dimensional data. Nat. Methods 2015, 12, 233–235. [Google Scholar] [CrossRef]
- Korem, Y.; Szekely, P.; Hart, Y.; Sheftel, H.; Hausser, J.; Mayo, A.; Rothenberg, M.E.; Kalisky, T.; Alon, U. Geometry of the Gene Expression Space of Individual Cells. PLOS Comput. Biol. 2015, 11, e1004224. [Google Scholar] [CrossRef]
- Morup, M.; Hansen, L.K. Archetypal analysis for machine learning and data mining. Neurocomputing 2012, 80, 54–63. [Google Scholar] [CrossRef]
- Chen, J.; Xu, H.; Aronow, B.J.; Jegga, A.G. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinform. 2007, 8, 392. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.J.; Chen, X.; Pinto, E.M.; Williams, J.S.; Clay, M.R.; Pounds, S.B.; Cao, X.; Shi, L.; Lin, T.; Neale, G.; et al. Forty-five patient-derived xenografts capture the clinical and biological heterogeneity of Wilms tumor. Nat. Commun. 2019, 10, 5806. [Google Scholar] [CrossRef]
- Carbonetto, P.; Sarkar, A.; Wang, Z.; Stephens, M. Non-negative matrix factorization algorithms greatly improve topic model fits. arXiv 2022, arXiv:2105.13440. [Google Scholar]
- Wineberg, Y.; Bar-Lev, T.H.; Futorian, A.; Ben-Haim, N.; Armon, L.; Ickowicz, D.; Oriel, S.; Bucris, E.; Yehuda, Y.; Pode-Shakked, N.; et al. Single-Cell RNA Sequencing Reveals mRNA Splice Isoform Switching during Kidney Development. J. Am. Soc. Nephrol. 2020, 31, 2278–2291. [Google Scholar] [CrossRef] [PubMed]
- Bioucas-Dias, J.M. A variable splitting augmented Lagrangian approach to linear spectral unmixing. In Proceedings of the 2009 First Workshop on Hyperspectral Image and Signal Processing, Grenoble, France, 26–28 August 2009; pp. 1–4. [Google Scholar] [CrossRef]
- Blei, D.M.; Ng, A.Y.; Jordan, M.I. Latent Dirichlet Allocation. J. Mach. Learn. Res. 2003, 3, 993–1022. [Google Scholar]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trink, Y.; Urbach, A.; Dekel, B.; Hohenstein, P.; Goldberger, J.; Kalisky, T. Characterization of Continuous Transcriptional Heterogeneity in High-Risk Blastemal-Type Wilms’ Tumors Using Unsupervised Machine Learning. Int. J. Mol. Sci. 2023, 24, 3532. https://doi.org/10.3390/ijms24043532
Trink Y, Urbach A, Dekel B, Hohenstein P, Goldberger J, Kalisky T. Characterization of Continuous Transcriptional Heterogeneity in High-Risk Blastemal-Type Wilms’ Tumors Using Unsupervised Machine Learning. International Journal of Molecular Sciences. 2023; 24(4):3532. https://doi.org/10.3390/ijms24043532
Chicago/Turabian StyleTrink, Yaron, Achia Urbach, Benjamin Dekel, Peter Hohenstein, Jacob Goldberger, and Tomer Kalisky. 2023. "Characterization of Continuous Transcriptional Heterogeneity in High-Risk Blastemal-Type Wilms’ Tumors Using Unsupervised Machine Learning" International Journal of Molecular Sciences 24, no. 4: 3532. https://doi.org/10.3390/ijms24043532
APA StyleTrink, Y., Urbach, A., Dekel, B., Hohenstein, P., Goldberger, J., & Kalisky, T. (2023). Characterization of Continuous Transcriptional Heterogeneity in High-Risk Blastemal-Type Wilms’ Tumors Using Unsupervised Machine Learning. International Journal of Molecular Sciences, 24(4), 3532. https://doi.org/10.3390/ijms24043532