
3D In Vitro Models: Novel Insights into Idiopathic Pulmonary Fibrosis Pathophysiology and Drug Screening


Abstract
:1. Introduction
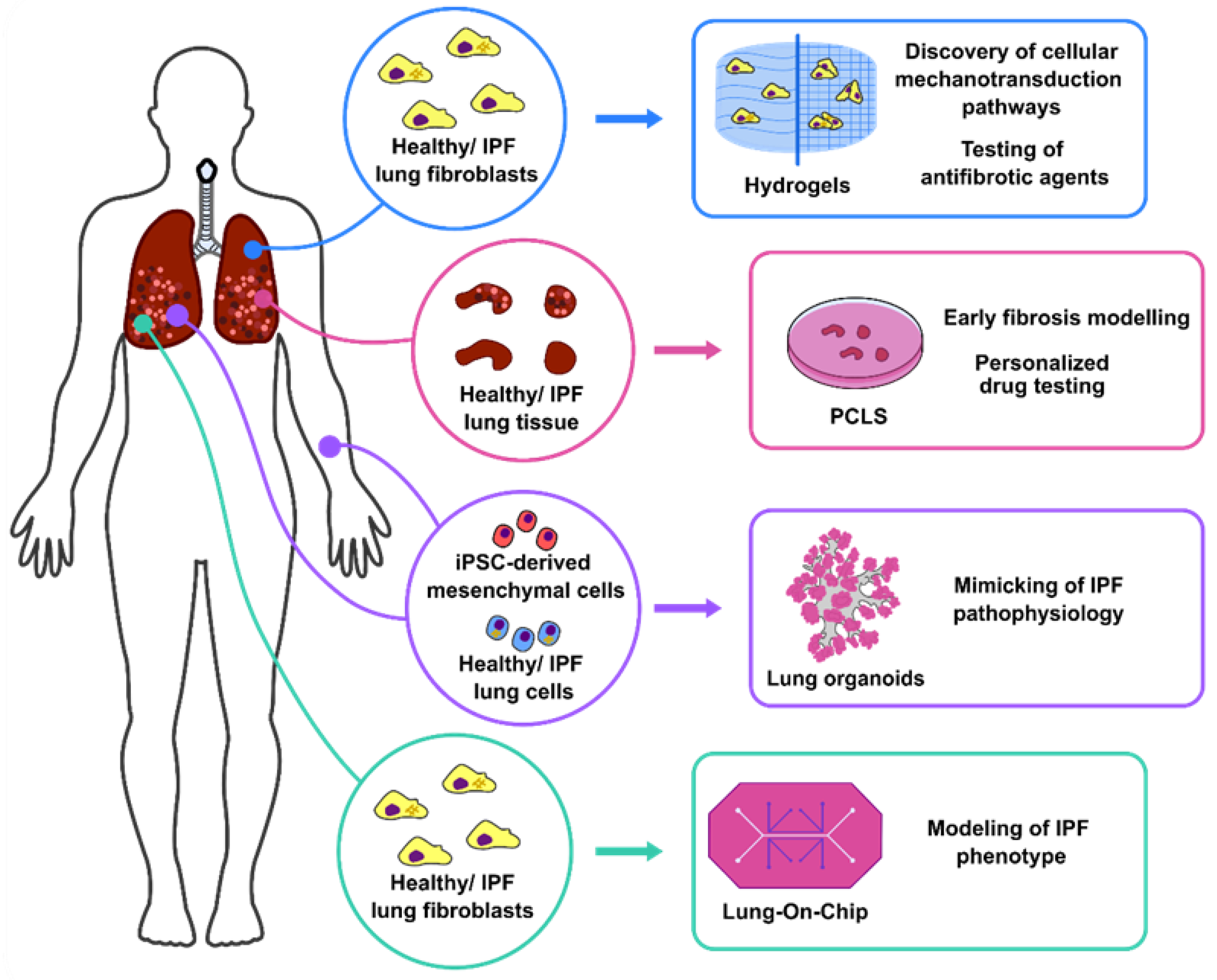
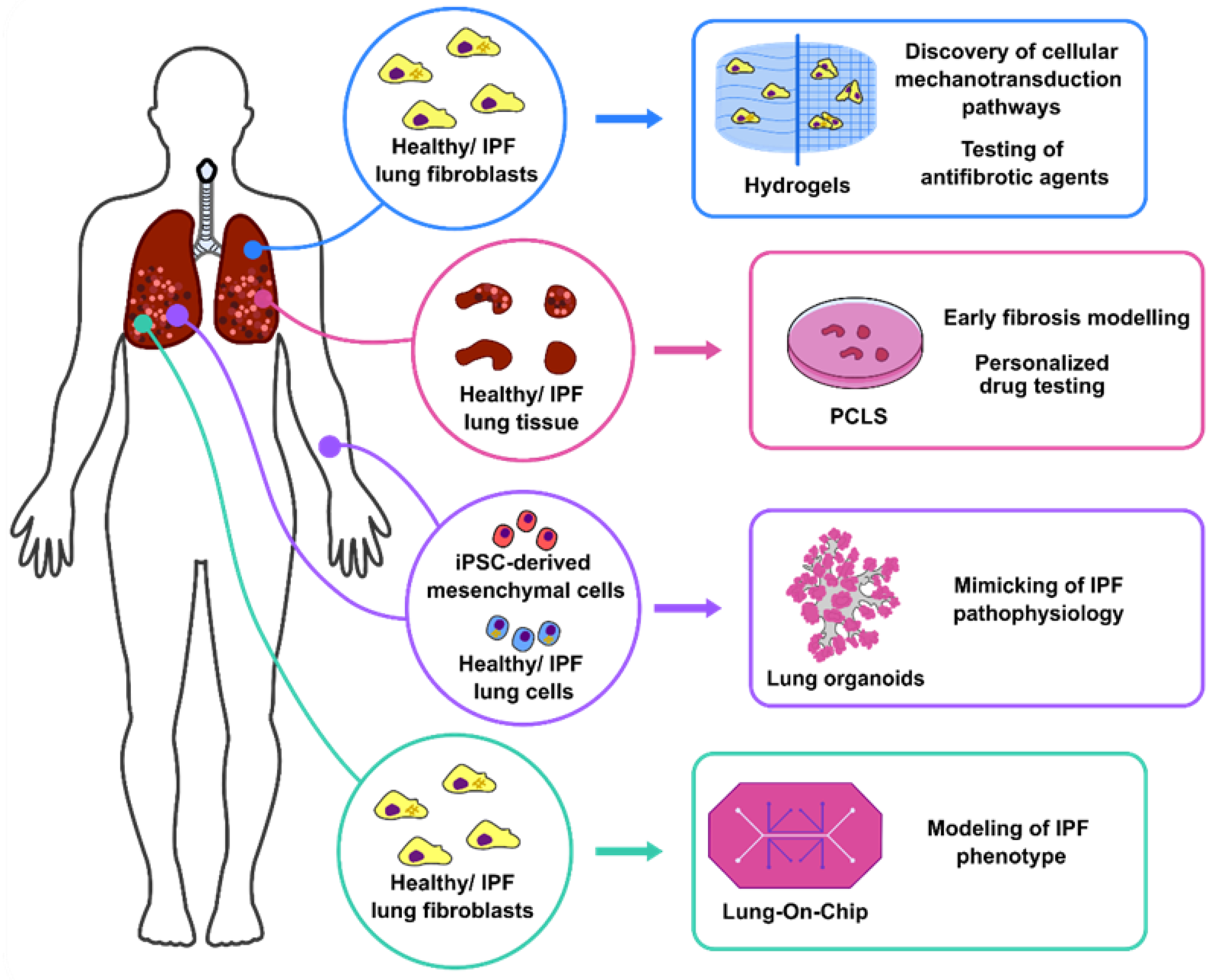
2. 3D Lung Culture Systems
2.1. Hydrogels: Modelling of Human Lung Fibroblast Migration and Differentiation
2.2. PCLS: Uncovering Antifibrotic/Regenerative Pathways to Treat IPF
2.3. Lung Organoids: Mimicking of Pulmonary Fibrosis Pathophysiology for Personalised Medicine
2.4. Lung in a Chip: Development of Complex Ex Vivo Systems for Pulmonary Fibrosis Modelling

{kind=link}
| 3D Culture System | Species | Source | Cellular Composition | Applicability/ Main Finding | Reference |
|---|---|---|---|---|---|
| PA hydrogels | Human | Healthy donors | Lung fibroblasts Distal lung epithelial cells, vascular and mesenchymal compartment | Cellular mechanotransduction | Tse et al. [21] |
| Asano et al. [22] | |||||
| IPF patients | Chen et al. [30] | ||||
| Mouse | Saline/Bleomycin-treated mice | PGE2-COX2 axis modulation in stiff matrixes | Liu et al. [28] | ||
| Collagen-rich hydrogels | Human | Healthy donors/ IPF patients | FAK/Akt signalling pathway activation promoting collagen deposition | Giménez et al. [24] | |
| Eicosanoid biosynthetic enzymes upregulation in fibrotic conditions | Berhan et al. [29] | ||||
| PCLS | Mouse | Saline/ Bleomycin-treated lung tissue | Drug testing: CSP7 | Marudamuthu et al. [33] | |
| Drug testing: senolytic drugs | Lehmann et al. [36] | ||||
| Drug testing: αvβ integrins | Tsoyi et al. [39] | ||||
| Human | Healthy donors/ IPF lung tissue | Disease modelling: early fibrosis | Alsafadi et al. [34] | ||
| Drug testing: metformin | Kheirollahi et al. [37] | ||||
| Drug testing: SDC2 ligands | Decaris et al. [40] | ||||
| Human /Mouse | Healthy lung tissue/Bleomycin-treated lung tissue | Disease modelling: early fibrosis | Lehmann et al. [35] | ||
| Lung organoids | Human | CF patients broncho-alveolar lavage | Airway organoids: basal, ciliated, secretory, and club cells | Disease modelling: CF pathophysiology | Sachs et al. [42] |
| CF patient nasal brushing | Airway organoids: basal, ciliated, and secretory cells | Personalised medicine: discovery of therapeutic targets and more effective CFTR modulators | Sette et al. [43]. | ||
| Healthy donors | Distal alveolar organoids: AECII, AECI, iPSC derived-mesenchymal cells | Disease modelling: epithelial-mesenchymal interactions during fibrosis | Wilkinson et al. [50] | ||
| Healthy/ IPF lung tissue | Pulmospheres: AECII, endothelial cells, macrophages, mesenchymal cells | Disease modelling: myofibroblast activation | Surolia et al. [51] | ||
| Healthy donors | ESC-derived lung organoids–epithelial cells and mesenchymal cells | Disease modelling: mutation in HPS1, HPS2 and HPS4 genes to induce fibrosis | Strikoudis et al. [52] | ||
| Mouse/ Human | HA- and TLR4-deficient mice/ IPF patients | Alveolospheres: AECI, AECII, fibroblasts | Disease modelling: AECII regeneration after fibrosis | Liang et al. [44] | |
| Lung-on-chip | Human | CF patients | CF bronchial epithelial cells and pulmonary microvascular endothelial cells | Disease modelling: CF pathology | Plebani et al. [58] |
| Healthy Donors/ IPF patients | SAEC, endothelial cells, fibroblast | Disease modelling: IPF phenotype | Mejías et al. [60] |
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hyldgaard, C.; Hilberg, O.; Bendstrup, E. How Does Comorbidity Influence Survival in Idiopathic Pulmonary Fibrosis? Respir. Med. 2014, 108, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, A.L.; Swigris, J.J. Idiopathic Pulmonary Fibrosis: Diagnosis and Epidemiology. Clin. Chest Med. 2012, 33, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.A.; Wijsenbeek, M.; Bondue, B.; Bouros, D.; Bresser, P.; Robalo Cordeiro, C.; Hilberg, O.; Magnusson, J.; Manali, E.D.; Morais, A.; et al. Idiopathic Pulmonary Fibrosis: Best Practice in Monitoring and Managing a Relentless Fibrotic Disease. Respiration 2020, 99, 73–82. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2014, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal Models of Fibrotic Lung Disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Maher, T.M.; Strek, M.E. Antifibrotic Therapy for Idiopathic Pulmonary Fibrosis: Time to Treat. Respir. Res. 2019, 20, 205. [Google Scholar] [CrossRef] [Green Version]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of Action of Nintedanib in the Treatment of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef]
- Meyer, K.C.; Decker, C.A. Role of Pirfenidone in the Management of Pulmonary Fibrosis. Ther. Clin. Risk Manag. 2017, 13, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Sgalla, G.; Cocconcelli, E.; Tonelli, R.; Richeldi, L. Novel Drug Targets for Idiopathic Pulmonary Fibrosis. Expert Rev. Respir. Med. 2016, 10, 393–405. [Google Scholar] [CrossRef]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front. Med. 2017, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Hilster, R.H.J.; Sharma, P.K.; Jonker, M.R.; White, E.S.; Gercama, E.A.; Roobeek, M.; Timens, W.; Harmsen, M.C.; Hylkema, M.N.; Burgess, J.K. Human Lung Extracellular Matrix Hydrogels Resemble the Stiffness and Viscoelasticity of Native Lung Tissue. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L698–L704. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, H.N.; Uhl, F.E.; Pineda, R.H.; Bailey, K.E.; Rojas, M.; Wagner, D.E.; Königshoff, M. Applications and Approaches for Three-Dimensional Precision-Cut Lung Slices. Disease Modeling and Drug Discovery. Am. J. Respir. Cell Mol. Biol. 2020, 62, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; An, G.H.; Kim, J.-Y.; Rasaei, R.; Kim, W.J.; Jin, X.; Woo, D.-H.; Han, C.; Yang, S.-R.; Kim, J.-H.; et al. Human Pluripotent Stem Cell-Derived Alveolar Organoids for Modeling Pulmonary Fibrosis and Drug Testing. Cell Death Discov. 2021, 7, 48. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, Characterization, and Applications: A Review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Betts, C.; Cunoosamy, D.M.; Åberg, P.M.; Hornberg, J.J.; Sivars, K.B.; Cohen, T.S. Use of Precision Cut Lung Slices as a Translational Model for the Study of Lung Biology. Respir. Res. 2019, 20, 162. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Miller, A.J.; Spence, J.R. In Vitro Models to Study Human Lung Development, Disease and Homeostasis. Physiology 2017, 32, 246–260. [Google Scholar] [CrossRef]
- Gkatzis, K.; Taghizadeh, S.; Huh, D.; Stainier, D.Y.R.; Bellusci, S. Use of Three-Dimensional Organoids and Lung-on-a-Chip Methods to Study Lung Development, Regeneration and Disease. Eur. Respir. J. 2018, 52, 1800876. [Google Scholar] [CrossRef]
- Artzy-Schnirman, A.; Hobi, N.; Schneider-Daum, N.; Guenat, O.T.; Lehr, C.M.; Sznitman, J. Advanced in Vitro Lung-on-Chip Platforms for Inhalation Assays: From Prospect to Pipeline. Eur. J. Pharm. Biopharm. 2019, 144, 11–17. [Google Scholar] [CrossRef]
- Tse, J.R.; Engler, A.J. Preparation of Hydrogel Substrates with Tunable Mechanical Properties. Curr. Protoc. Cell Biol. 2010, 10, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Asano, S.; Ito, S.; Takahashi, K.; Furuya, K.; Kondo, M.; Sokabe, M.; Hasegawa, Y. Matrix Stiffness Regulates Migration of Human Lung Fibroblasts. Physiol. Rep. 2017, 5, e13281. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming Growth Factor (TGF)-Β1-Induced MiR-133a Inhibits Myofibroblast Differentiation and Pulmonary Fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Giménez, A.; Duch, P.; Puig, M.; Gabasa, M.; Xaubet, A.; Alcaraz, J. Dysregulated Collagen Homeostasis by Matrix Stiffening and TGF-Β1 in Fibroblasts from Idiopathic Pulmonary Fibrosis Patients: Role of FAK/Akt. Int. J. Mol. Sci. 2017, 18, 2431. [Google Scholar] [CrossRef] [Green Version]
- Wettlaufer, S.H.; Scott, J.P.; McEachin, R.C.; Peters-Golden, M.; Huang, S.K. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am. J. Respir. Cell Mol. Biol. 2016, 54, 114–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keerthisingam, C.B.; Jenkins, R.G.; Harrison, N.K.; Hernandez-Rodriguez, N.A.; Booth, H.; Laurent, G.J.; Hart, S.L.; Foster, M.L.; McAnulty, R.J. Cyclooxygenase-2 Deficiency Results in a Loss of the Anti-Proliferative Response to Transforming Growth Factor-β in Human Fibrotic Lung Fibroblasts and Promotes Bleomycin-Induced Pulmonary Fibrosis in Mice. Am. J. Pathol. 2001, 158, 1411–1422. [Google Scholar] [CrossRef]
- Coward, W.R.; Watts, K.; Feghali-Bostwick, C.A.; Knox, A.; Pang, L. Defective Histone Acetylation Is Responsible for the Diminished Expression of Cyclooxygenase 2 in Idiopathic Pulmonary Fibrosis. Mol. Cell. Biol. 2009, 29, 4325–4339. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback Amplification of Fibrosis through Matrix Stiffening and COX-2 Suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Berhan, A.; Harris, T.; Jaffar, J.; Jativa, F.; Langenbach, S.; Lönnstedt, I.; Alhamdoosh, M.; Ng, M.; Lee, P.; Westall, G.; et al. Cellular Microenvironment Stiffness Regulates Eicosanoid Production and Signaling Pathways. Am. J. Respir. Cell Mol. Biol. 2020, 63, 819–830. [Google Scholar] [CrossRef]
- Chen, H.; Qu, J.; Huang, X.; Kurundkar, A.; Zhu, L.; Yang, N.; Venado, A.; Ding, Q.; Liu, G.; Antony, V.B.; et al. Mechanosensing by the A6-Integrin Confers an Invasive Fibroblast Phenotype and Mediates Lung Fibrosis. Nat. Commun. 2016, 7, 12564. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. The Leading Role of Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Cell. Signal. 2020, 66, 109482. [Google Scholar] [CrossRef] [PubMed]
- Cedilak, M.; Banjanac, M.; Belamarić, D.; Paravić Radičević, A.; Faraho, I.; Ilić, K.; Čužić, S.; Glojnarić, I.; Eraković Haber, V.; Bosnar, M. Precision-Cut Lung Slices from Bleomycin Treated Animals as a Model for Testing Potential Therapies for Idiopathic Pulmonary Fibrosis. Pulm. Pharmacol. Ther. 2019, 55, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Marudamuthu, A.S.; Bhandary, Y.P.; Fan, L.; Radhakrishnan, V.; MacKenzie, B.A.; Maier, E.; Shetty, S.K.; Nagaraja, M.R.; Gopu, V.; Tiwari, N.; et al. Caveolin-1-Derived Peptide Limits Development of Pulmonary Fibrosis. Sci. Transl. Med. 2019, 11, eaat2848. [Google Scholar] [CrossRef] [PubMed]
- Alsafadi, H.N.; Staab-Weijnitz, C.A.; Lehmann, M.; Lindner, M.; Peschel, B.; Königshoff, M.; Wagner, D.E. An Ex Vivo Model to Induce Early Fibrosis-like Changes in Human Precision-Cut Lung Slices. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L896–L902. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, M.; Buhl, L.; Alsafadi, H.N.; Klee, S.; Hermann, S.; Mutze, K.; Ota, C.; Lindner, M.; Behr, J.; Hilgendorff, A.; et al. Differential Effects of Nintedanib and Pirfenidone on Lung Alveolar Epithelial Cell Function in Ex Vivo Murine and Human Lung Tissue Cultures of Pulmonary Fibrosis 11 Medical and Health Sciences 1102 Cardiorespiratory Medicine and Haematology 06 Biologica. Respir. Res. 2018, 19. [Google Scholar] [CrossRef]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic Drugs Target Alveolar Epithelial Cell Function and Attenuate Experimental Lung Fibrosis Ex Vivo. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.S.; Kwapiszewska, G.; et al. Metformin Induces Lipogenic Differentiation in Myofibroblasts to Reverse Lung Fibrosis. Nat. Commun. 2019, 10, 2987. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin Reverses Established Lung Fibrosis in a Bleomycin Model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Tsoyi, K.; Liang, X.; De Rossi, G.; Ryter, S.W.; Xiong, K.; Chu, S.G.; Liu, X.; Ith, B.; Celada, L.J.; Romero, F.; et al. Cd148 Deficiency in Fibroblasts Promotes the Development of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 204, 312–325. [Google Scholar] [CrossRef]
- Decaris, M.L.; Schaub, J.R.; Chen, C.; Cha, J.; Lee, G.G.; Rexhepaj, M.; Ho, S.S.; Rao, V.; Marlow, M.M.; Kotak, P.; et al. Dual Inhibition of Avβ6 and Avβ1 Reduces Fibrogenesis in Lung Tissue Explants from Patients with IPF. Respir. Res. 2021, 22, 265. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human Organoids: Model Systems for Human Biology and Medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Papaspyropoulos, A.; Ommen, D.D.Z.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-Term Expanding Human Airway Organoids for Disease Modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Sette, G.; Cicero, S.L.; Blaconà, G.; Pierandrei, S.; Bruno, S.M.; Salvati, V.; Castelli, G.; Falchi, M.; Fabrizzi, B.; Cimino, G.; et al. Theratyping Cystic Fibrosis in Vitro in ALI Culture and Organoid Models Generated from Patient-Derived Nasal Epithelial Conditionally Reprogrammed Stem Cells. Eur. Respir. J. 2021, 58. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 Promote Surfactant-Protein-C-Positive Alveolar Progenitor Cell Renewal and Prevent Severe Pulmonary Fibrosis in Mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef]
- Moiseenko, A.; Vazquez-Armendariz, A.I.; Kheirollahi, V.; Chu, X.; Tata, A.; Rivetti, S.; Günther, S.; Lebrigand, K.; Herold, S.; Braun, T.; et al. Identification of a Repair-Supportive Mesenchymal Cell Population during Airway Epithelial Regeneration. Cell Rep. 2020, 33. [Google Scholar] [CrossRef]
- Vazquez-Armendariz, A.I.; Heiner, M.; El Agha, E.; Salwig, I.; Hoek, A.; Hessler, M.C.; Shalashova, I.; Shrestha, A.; Carraro, G.; Mengel, J.P.; et al. Multilineage Murine Stem Cells Generate Complex Organoids to Model Distal Lung Development and Disease. EMBO J. 2020, 39. [Google Scholar] [CrossRef]
- Moore, B.B.; Moore, T.A. Viruses in Idiopathic Pulmonary Fibrosis Etiology and Exacerbation. Ann. Am. Thorac. Soc. 2015, 12, S186–S192. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 Alveolar Cells Are Stem Cells in Adult Lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Vazquez-Armendariz, A.I.; Seeger, W.; Herold, S.; El Agha, E. Protocol for the Generation of Murine Bronchiolospheres. STAR Protoc. 2021, 2, 100594. [Google Scholar] [CrossRef]
- Wilkinson, D.C.; Alva-Ornelas, J.A.; Sucre, J.M.S.; Vijayaraj, P.; Durra, A.; Richardson, W.; Jonas, S.J.; Paul, M.K.; Karumbayaram, S.; Dunn, B.; et al. Development of a Three-Dimensional Bioengineering Technology to Generate Lung Tissue for Personalized Disease Modeling. Stem Cells Transl. Med. 2017, 6, 622–633. [Google Scholar] [CrossRef]
- Surolia, R.; Li, F.J.; Wang, Z.; Li, H.; Liu, G.; Zhou, Y.; Luckhardt, T.; Bae, S.; Liu, R.M.; Rangarajan, S.; et al. 3D Pulmospheres Serve as a Personalized and Predictive Multicellular Model for Assessment of Antifibrotic Drugs. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strikoudis, A.; Cieślak, A.; Loffredo, L.; Chen, Y.W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 3709–3723.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-Chip: Recent Breakthroughs and Future Prospects. Biomed. Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Azadi, S.; Aboulkheyr Es, H.; Razavi Bazaz, S.; Thiery, J.P.; Asadnia, M.; Ebrahimi Warkiani, M. Upregulation of PD-L1 Expression in Breast Cancer Cells through the Formation of 3D Multicellular Cancer Aggregates under Different Chemical and Mechanical Conditions. Biochim. Biophys. Acta-Mol. Cell Res. 2019, 1866, 118526. [Google Scholar] [CrossRef] [PubMed]
- Neužil, P.; Giselbrecht, S.; Länge, K.; Huang, T.J.; Manz, A. Revisiting Lab-on-a-Chip Technology for Drug Discovery. Nat. Rev. Drug Discov. 2012, 11, 620–632. [Google Scholar] [CrossRef]
- Si, L.; Bai, H.; Oh, C.Y.; Jin, L.; Prantil-Baun, R.; Ingber, D.E. Clinically Relevant Influenza Virus Evolution Reconstituted in a Human Lung Airway-on-a-Chip. Microbiol. Spectr. 2021, 9, e00257-21. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Liu, T.; Liao, J.; Maharjan, S.; Xie, X.; Pérez, M.; Anaya, I.; Wang, S.; Mayer, A.T.; Kang, Z.; et al. Reversed-Engineered Human Alveolar Lung-on-a-Chip Model. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Plebani, R.; Potla, R.; Soong, M.; Bai, H.; Izadifar, Z.; Jiang, A.; Travis, R.N.; Belgur, C.; Dinis, A.; Cartwright, M.J.; et al. Modeling Pulmonary Cystic Fibrosis in a Human Lung Airway-on-a-Chip: Cystic Fibrosis Airway Chip. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Barkal, L.J.; Procknow, C.L.; Álvarez-Garciá, Y.R.; Niu, M.; Jiménez-Torres, J.A.; Brockman-Schneider, R.A.; Gern, J.E.; Denlinger, L.C.; Theberge, A.B.; Keller, N.P.; et al. Microbial Volatile Communication in Human Organotypic Lung Models. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Mejías, J.C.; Nelson, M.R.; Liseth, O.; Roy, K. A 96-Well Format Microvascularized Human Lung-on-a-Chip Platform for Microphysiological Modeling of Fibrotic Diseases. Lab Chip 2020, 20, 3601–3611. [Google Scholar] [CrossRef]
- Varone, A.; Nguyen, J.K.; Leng, L.; Barrile, R.; Sliz, J.; Lucchesi, C.; Wen, N.; Gravanis, A.; Hamilton, G.A.; Karalis, K.; et al. A Novel Organ-Chip System Emulates Three-Dimensional Architecture of the Human Epithelia and the Mechanical Forces Acting on It. Biomaterials 2021, 275, 120957. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Ask, K.; Warburton, D.; Gauldie, J.; Kolb, M. The Bleomycin Animal Model: A Useful Tool to Investigate Treatment Options for Idiopathic Pulmonary Fibrosis? Int. J. Biochem. Cell Biol. 2008, 40, 362–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, G.M.; D’Agnano, V.; Piloni, D.; Saracino, L.; Lettieri, S.; Mariani, F.; Lancia, A.; Bortolotto, C.; Rinaldi, P.; Falanga, F.; et al. The Oncogenic Landscape of the Idiopathic Pulmonary Fibrosis: A Narrative Review. Transl. Lung Cancer Res. 2022, 11, 472–496. [Google Scholar] [CrossRef] [PubMed]
| 3D Culture System | Strengths | Weaknesses |
|---|---|---|
| Hydrogels |
|
|
| PCLS |
|
|
| Lung organoids |
|
|
| Lung-on-chip |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vazquez-Armendariz, A.I.; Barroso, M.M.; El Agha, E.; Herold, S. 3D In Vitro Models: Novel Insights into Idiopathic Pulmonary Fibrosis Pathophysiology and Drug Screening. Cells 2022, 11, 1526. https://doi.org/10.3390/cells11091526
Vazquez-Armendariz AI, Barroso MM, El Agha E, Herold S. 3D In Vitro Models: Novel Insights into Idiopathic Pulmonary Fibrosis Pathophysiology and Drug Screening. Cells. 2022; 11(9):1526. https://doi.org/10.3390/cells11091526
Chicago/Turabian StyleVazquez-Armendariz, Ana Ivonne, Margarida Maria Barroso, Elie El Agha, and Susanne Herold. 2022. "3D In Vitro Models: Novel Insights into Idiopathic Pulmonary Fibrosis Pathophysiology and Drug Screening" Cells 11, no. 9: 1526. https://doi.org/10.3390/cells11091526
APA StyleVazquez-Armendariz, A. I., Barroso, M. M., El Agha, E., & Herold, S. (2022). 3D In Vitro Models: Novel Insights into Idiopathic Pulmonary Fibrosis Pathophysiology and Drug Screening. Cells, 11(9), 1526. https://doi.org/10.3390/cells11091526