
Identification of the Potential Role of the Rumen Microbiome in Milk Protein and Fat Synthesis in Dairy Cows Using Metagenomic Sequencing


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sample Collection
2.2. DNA Extraction and Metagenome Sequencing
2.3. Quality Control and Assembly of Metagenomic Data
2.4. Gene and Taxonomy Prediction
2.5. Comparative Analysis of Microorganism Abundance in the HPF and LPF Groups
2.6. Functional Annotation
2.7. Correlation Analysis
3. Results
3.1. Ruminal pH and VFA Concentrations
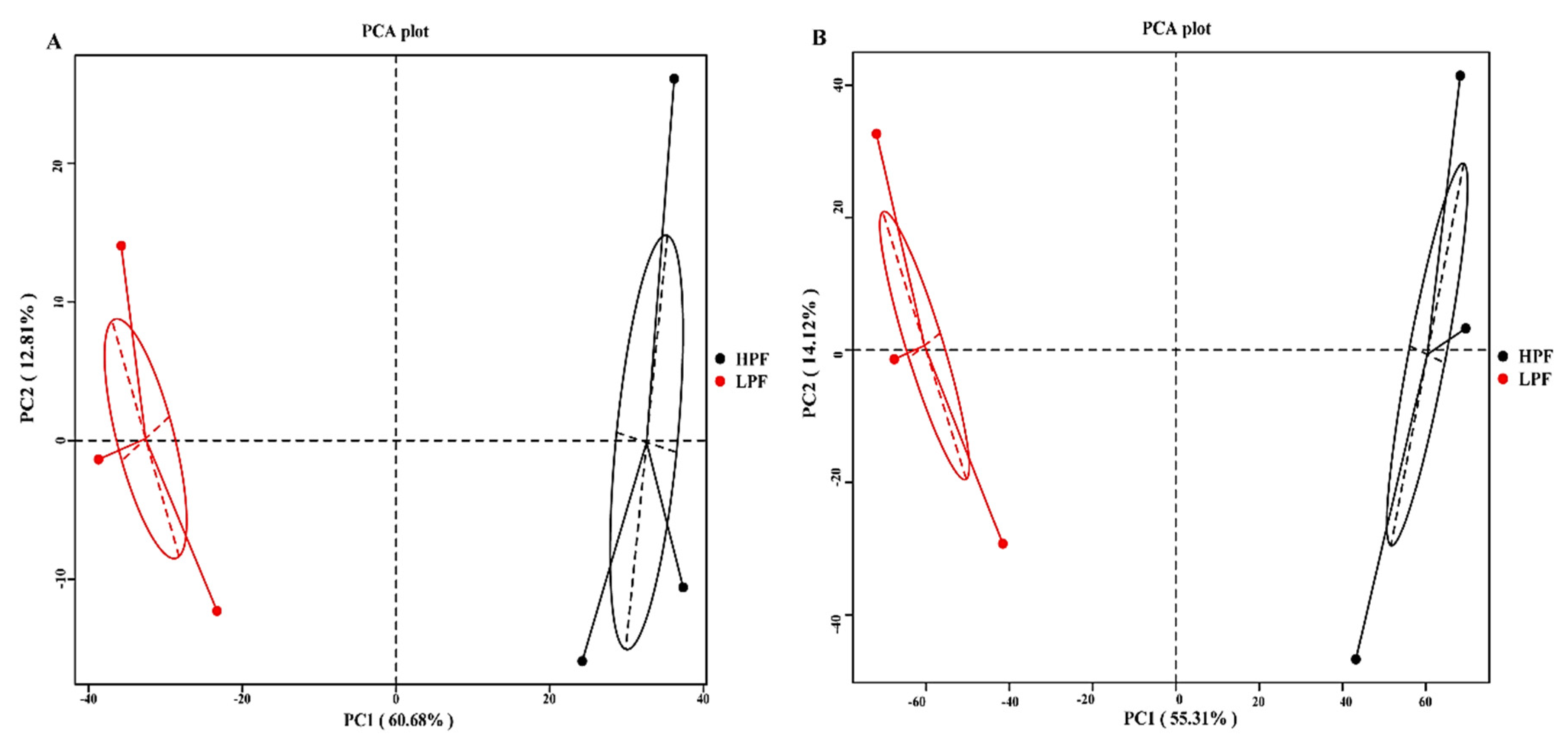
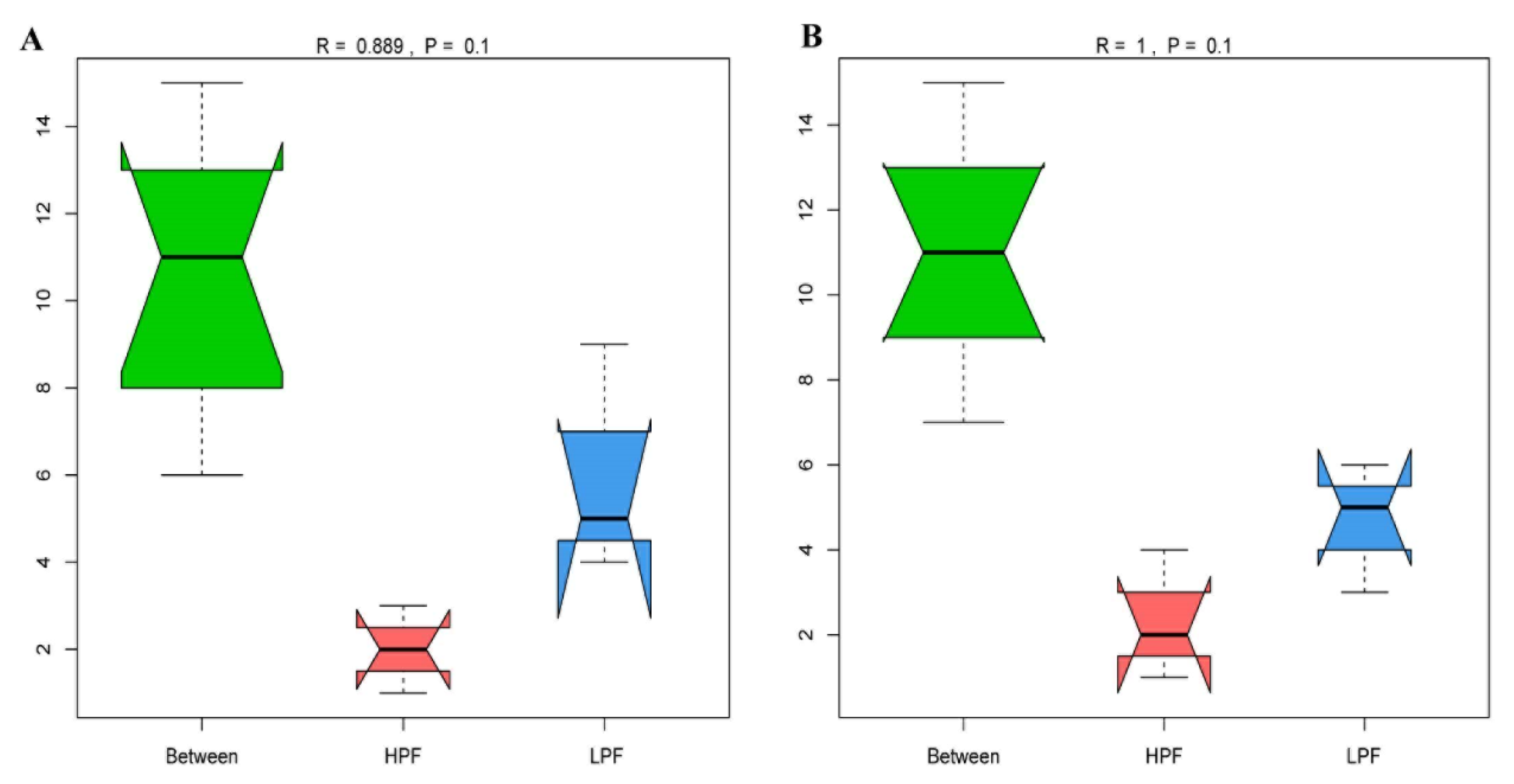
3.2. Sequencing of the Rumen Microbiota
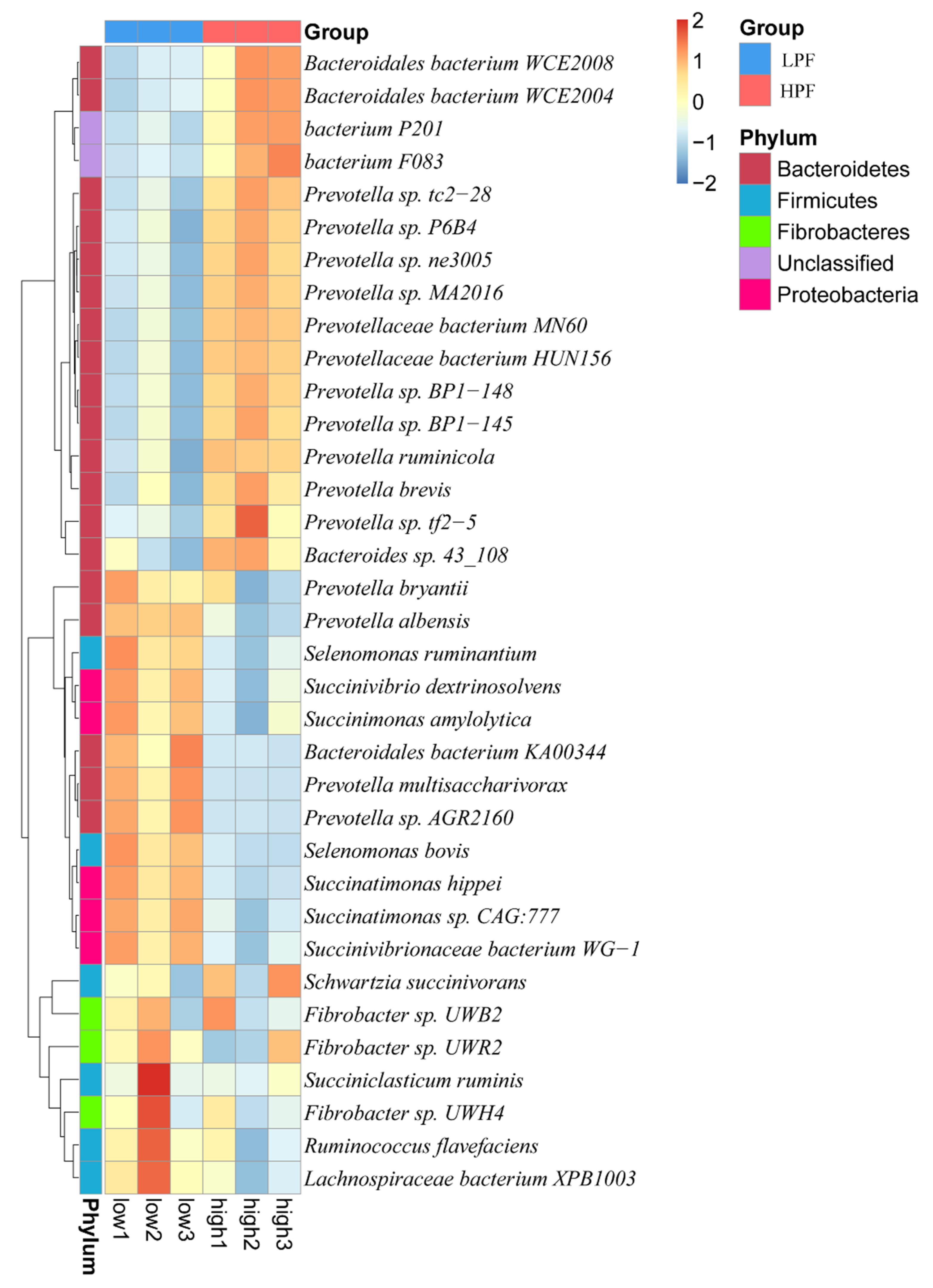
3.3. Taxonomic Composition of the Rumen Microbiota
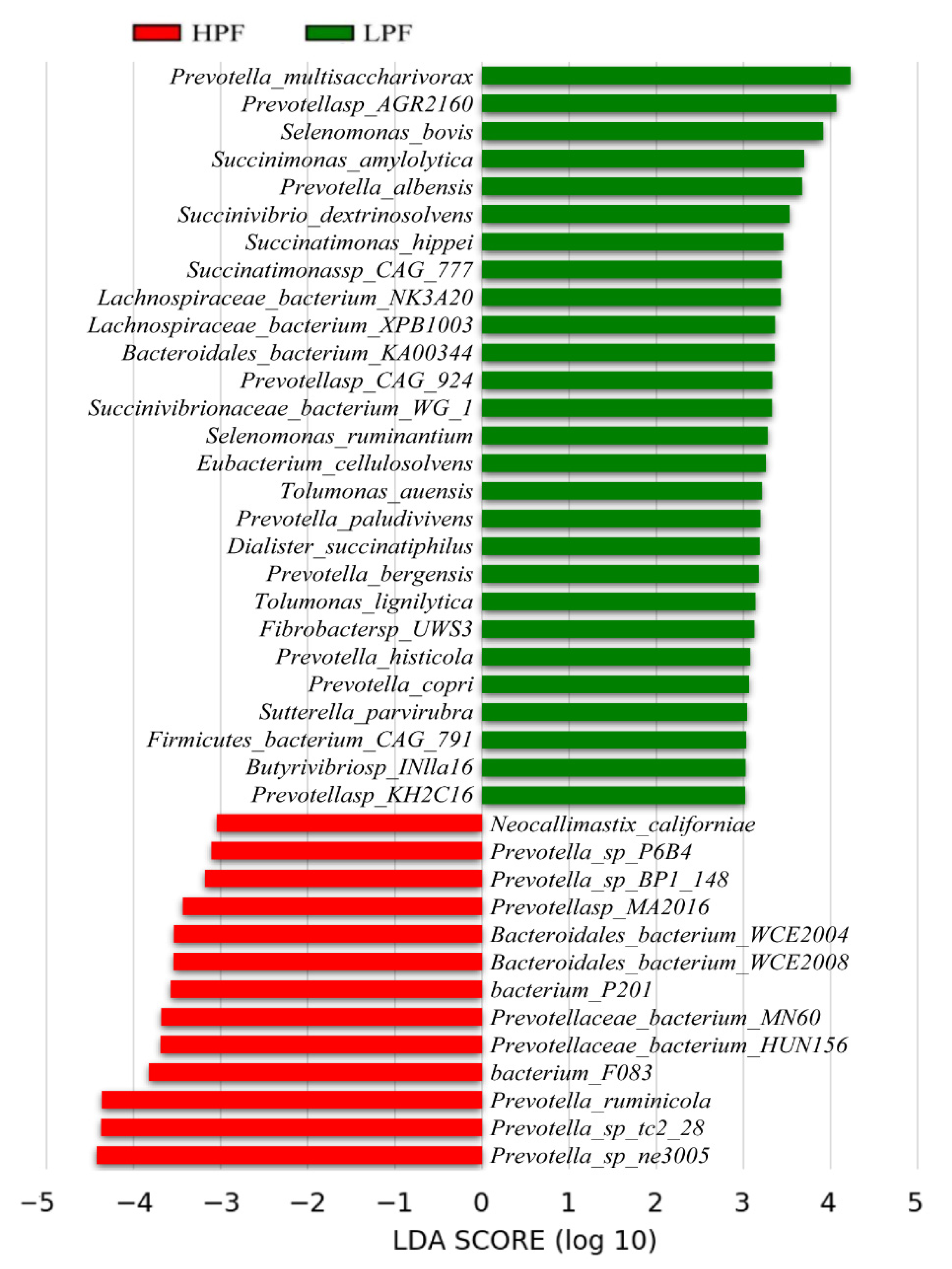
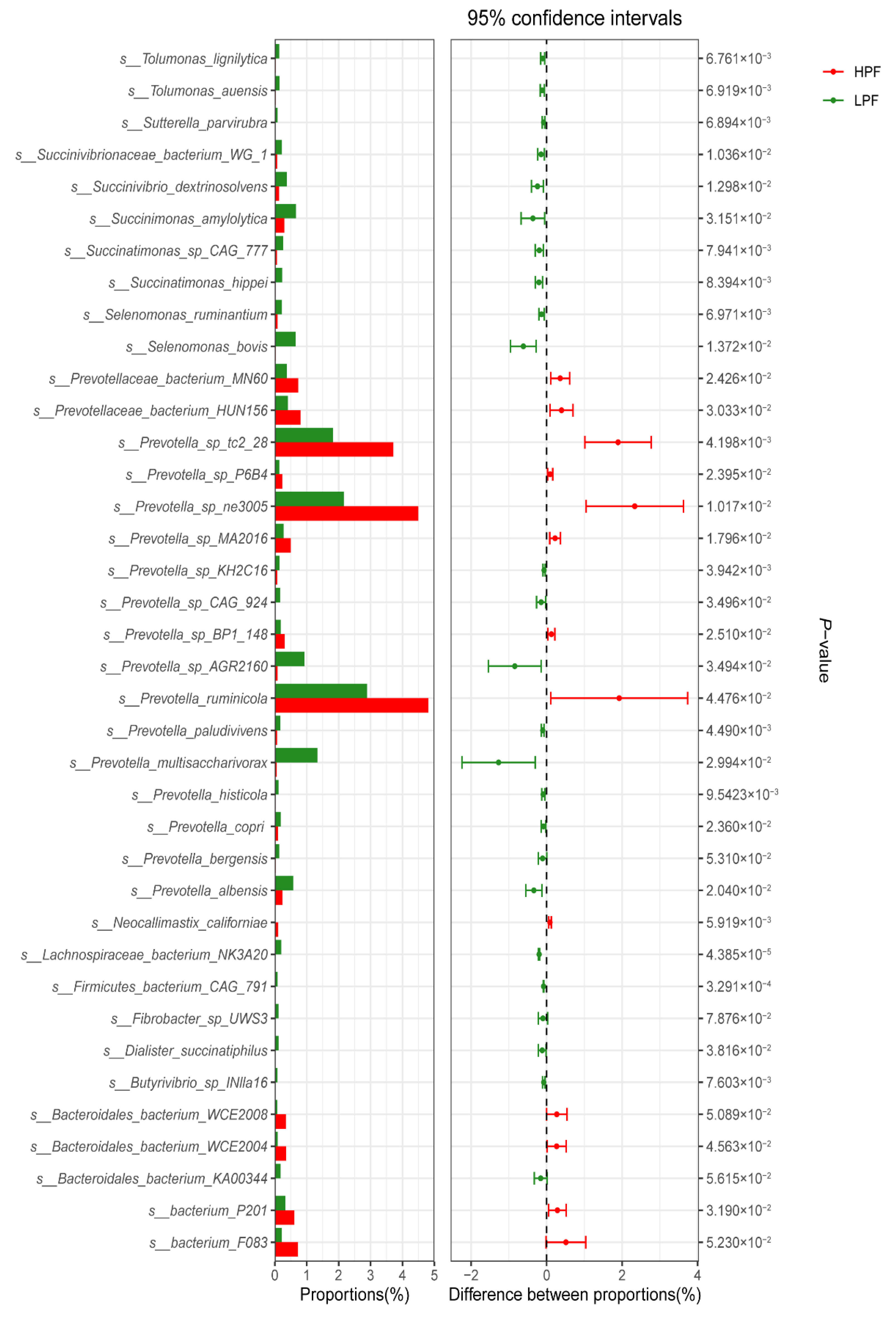
3.4. Differential Abundance of the Rumen Microbiome between the HPF and LPF Groups with PP and FP
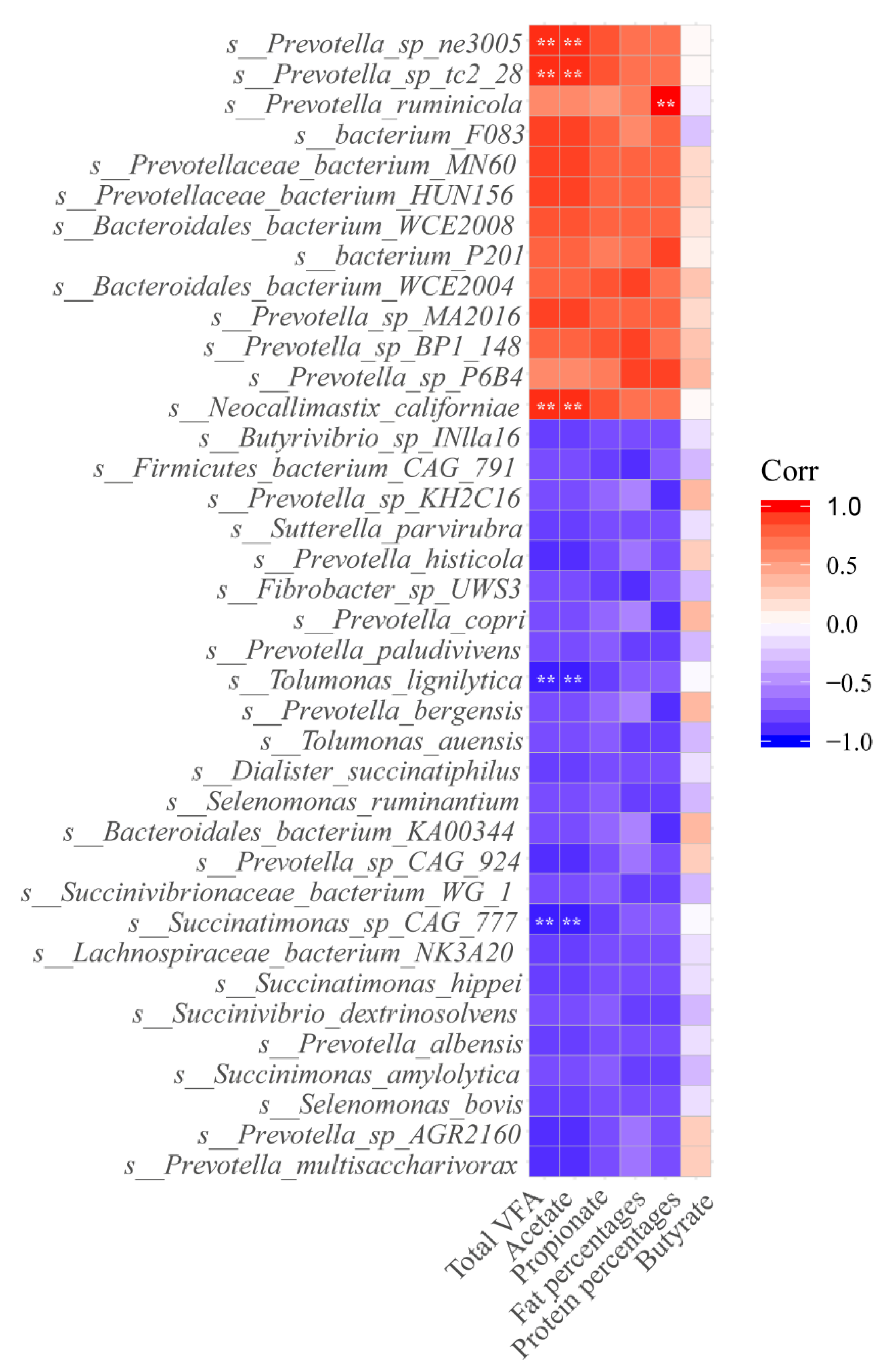
3.5. Correlation among Ruminal Microbiome, Ruminal Fermentation, and Milk Components
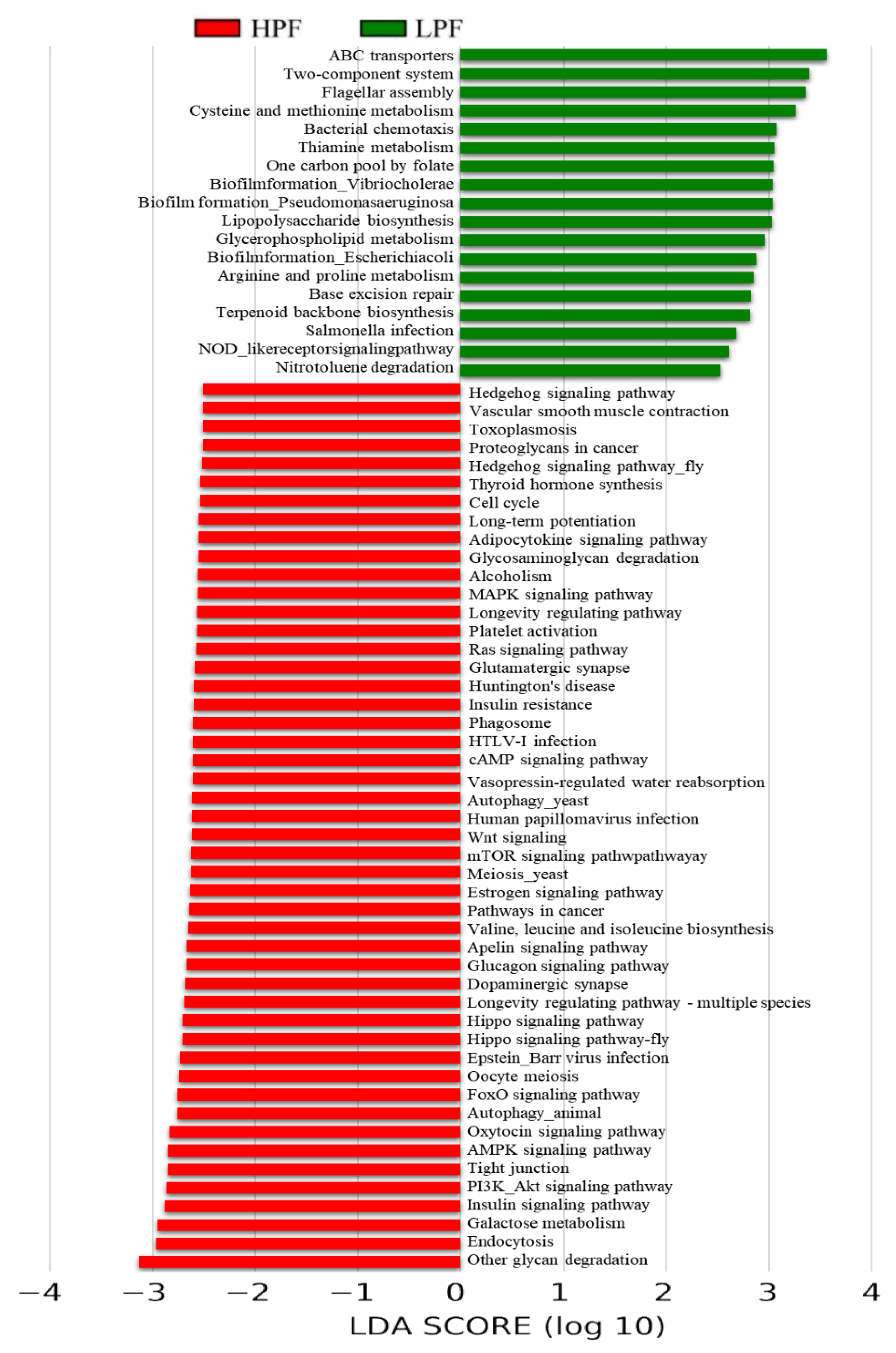
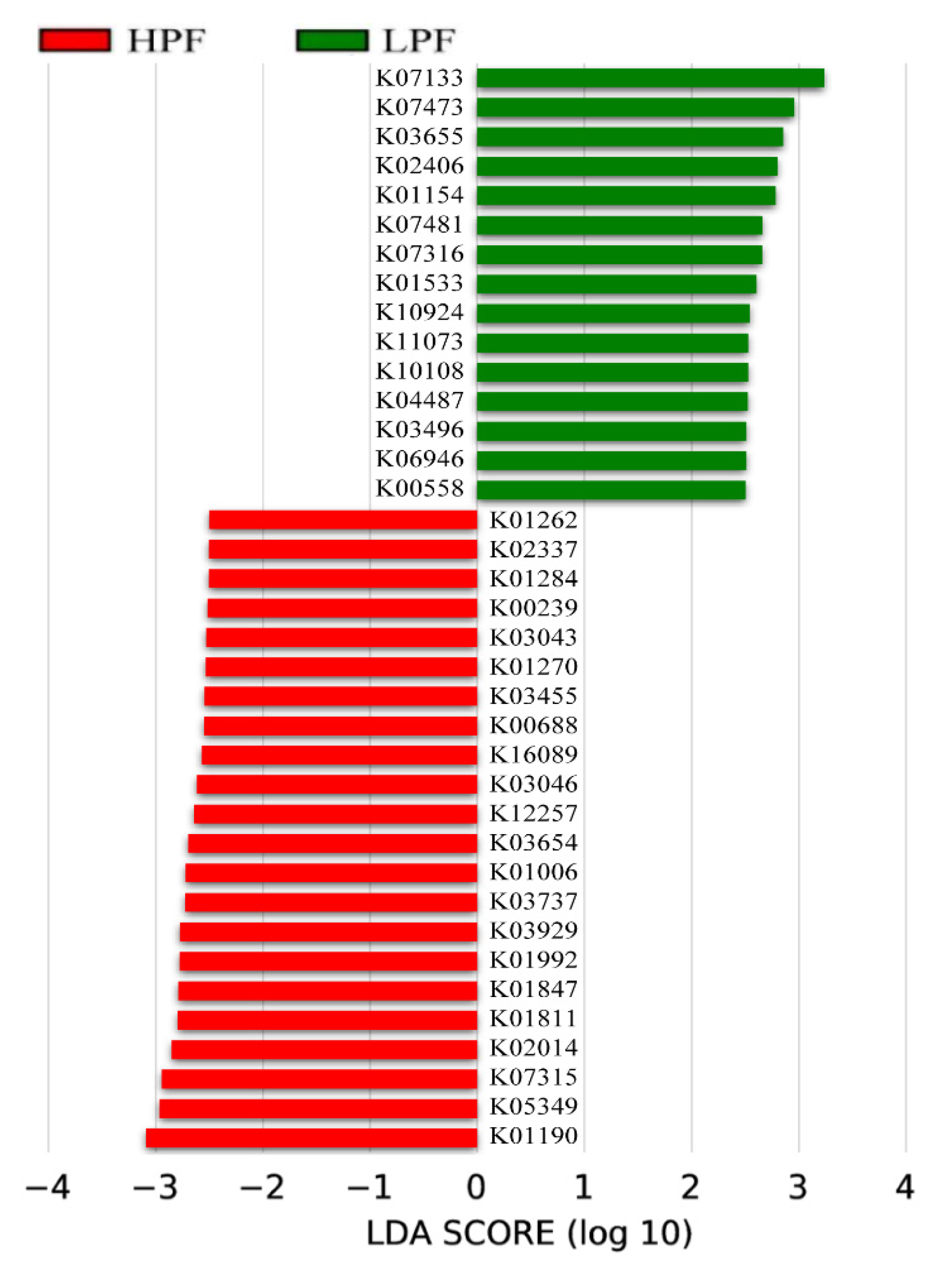
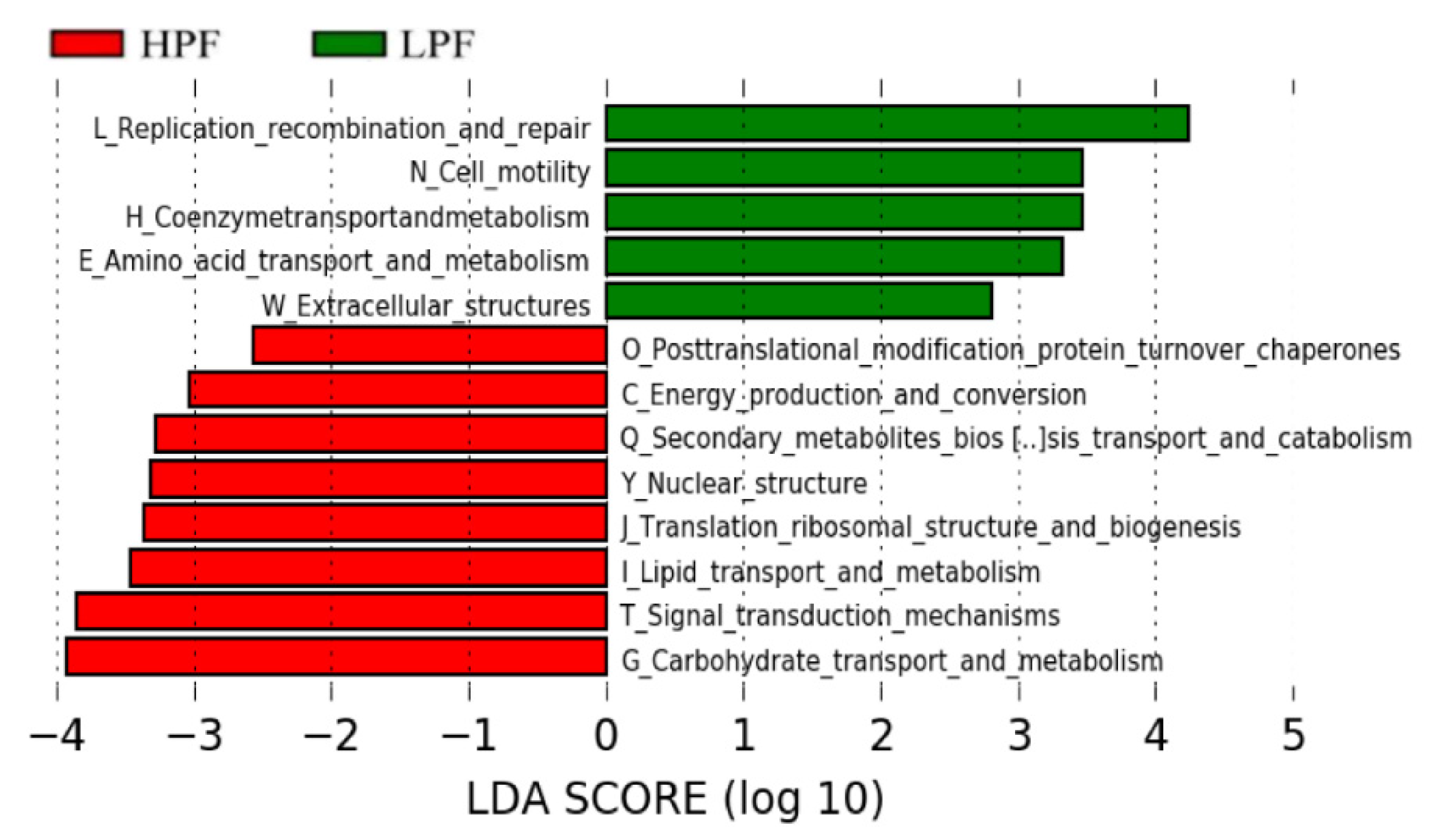
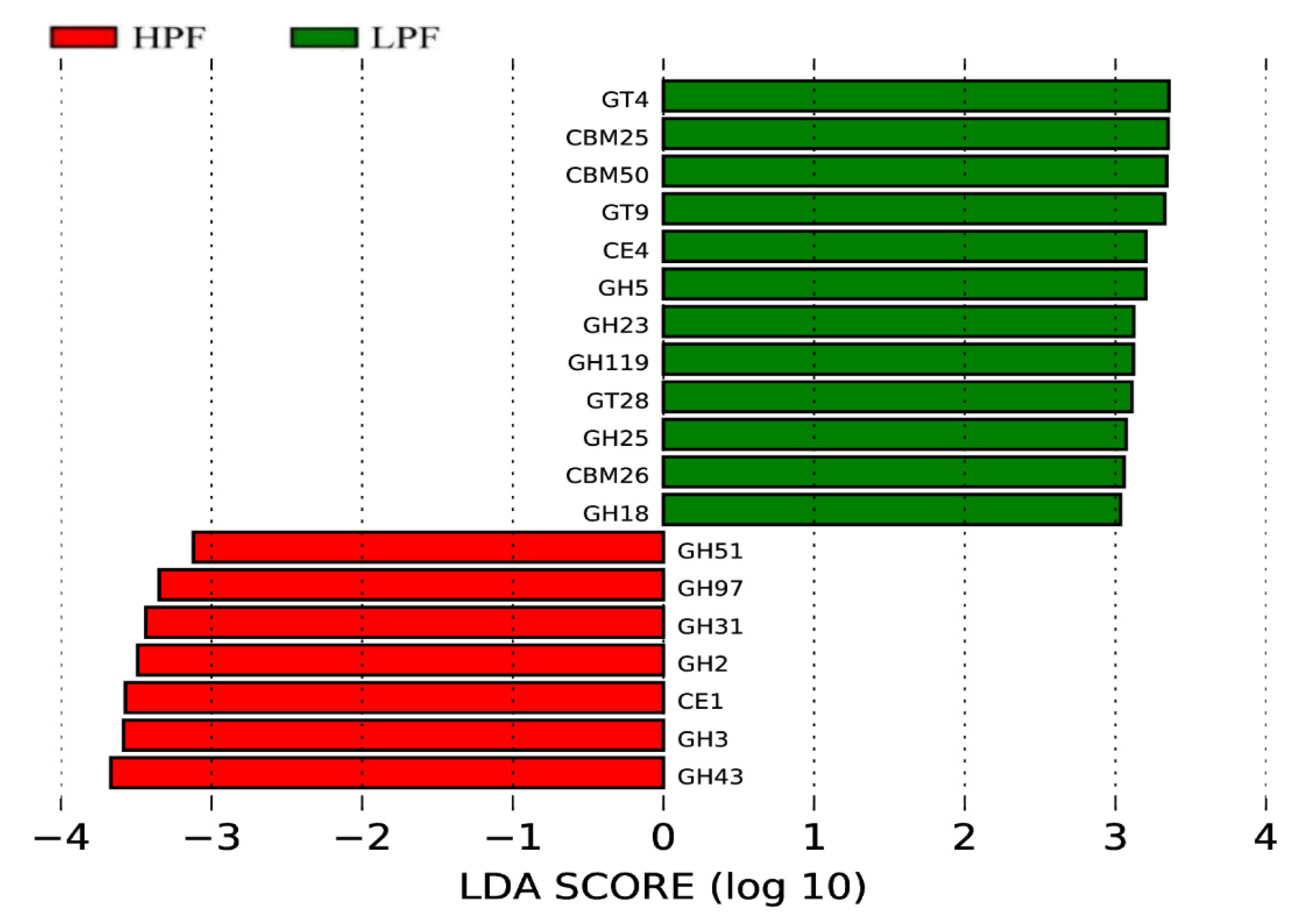
3.6. Functional Enrichment of the Rumen Microbiome Exhibiting Differential Abundances between the HPF and LPF Groups with PP and FP
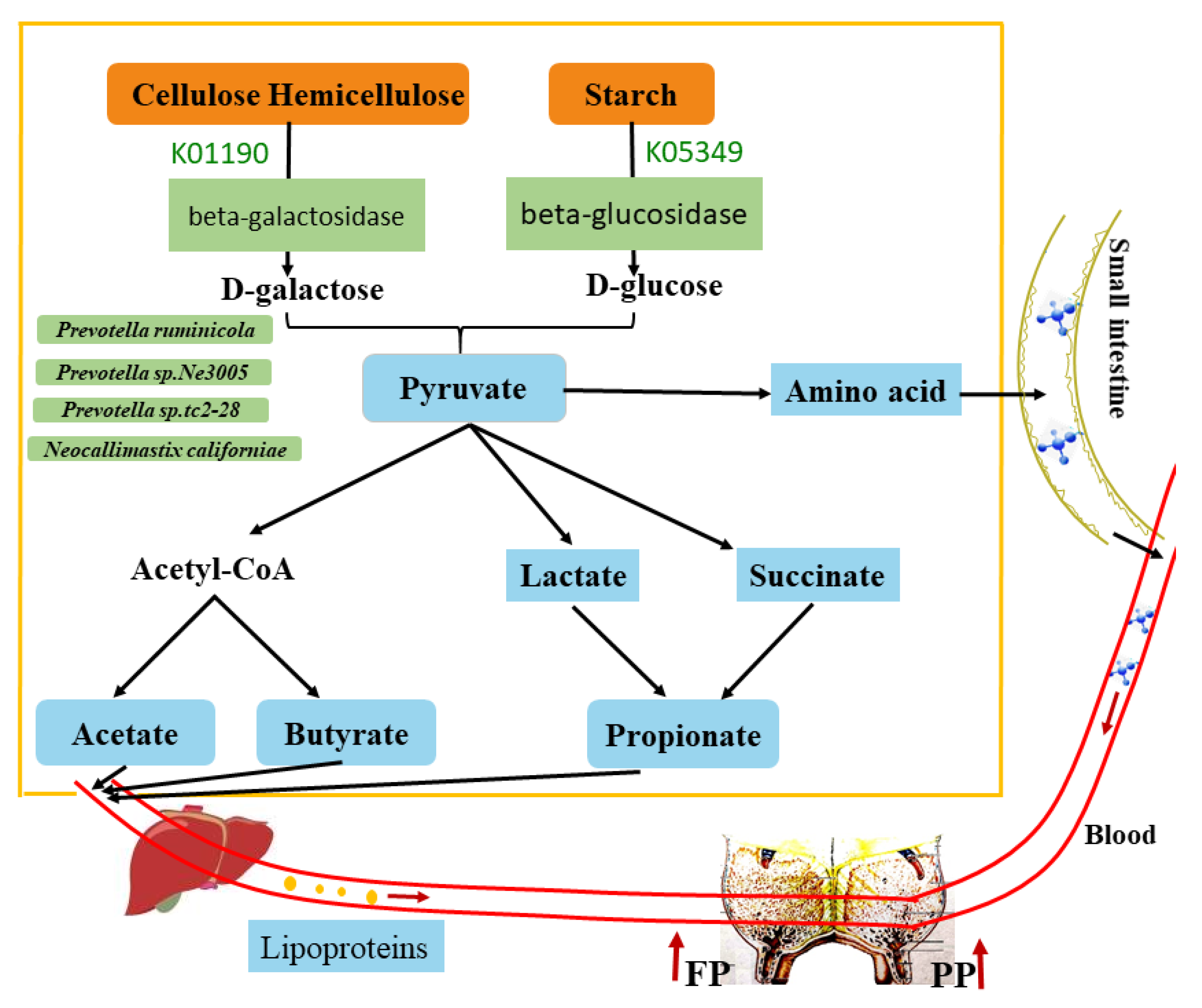
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pereira, P.C. Milk nutritional composition and its role in human health. Nutrition 2014, 30, 619–627. [Google Scholar] [CrossRef]
- Kearney, J. Food consumption trends and drivers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Ye, A.; Moughan, P.J.; Singh, H. Composition, structure, and digestive dynamics of milk from different species—A review. Front. Nutr. 2020, 7, 577759. [Google Scholar] [CrossRef] [PubMed]
- Givens, D.I. MILK symposium review: The importance of milk and dairy foods in the diets of infants, adolescents, pregnant women, adults, and the elderly. J. Dairy Sci. 2020, 103, 9681–9699. [Google Scholar] [CrossRef]
- Henderson, C. A study of the lipase produced by Anaerovibrio lipolytica, a rumen bacterium. J. Gen. Microbiol. 1971, 65, 81–89. [Google Scholar] [CrossRef][Green Version]
- Russell, J.B.; Rychlik, J.L. Factors that alter rumen microbial ecology. Science 2001, 292, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Herrero, M.; Havlik, P.; Valin, H.; Notenbaert, A.; Rufino, M.C.; Thornton, P.K.; Blummel, M.; Weiss, F.; Grace, D.; Obersteiner, M. Biomass use, production, feed efficiencies, and greenhouse gas emissions from global livestock systems. Proc. Natl. Acad. Sci. USA 2013, 110, 20888–20893. [Google Scholar] [CrossRef]
- Morgavi, D.P.; Kelly, W.J.; Janssen, P.H.; Attwood, G.T. Rumen microbial (meta)genomics and its application to ruminant production. Animal 2013, 7, 184–201. [Google Scholar] [CrossRef]
- Eisler, M.C.; Lee, M.R.; Tarlton, J.F.; Martin, G.B.; Beddington, J.; Dungait, J.A.; Greathead, H.; Liu, J.; Mathew, S.; Miller, H.; et al. Agriculture: Steps to sustainable livestock. Nature 2014, 507, 32–34. [Google Scholar] [CrossRef]
- Seshadri, R.; Leahy, S.C.; Attwood, G.T.; Teh, K.H.; Lambie, S.C.; Cookson, A.L.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Hadjithomas, M.; Varghese, N.J.; et al. Cultivation and sequencing of rumen microbiome members from the Hungate1000 collection. Nat. Biotechnol. 2018, 36, 359–367. [Google Scholar] [CrossRef]
- Baldwin, R.L.T.; Connor, E.E. Rumen function and development. Vet. Clin. N. Am. Food Anim. Pract. 2017, 33, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Bayer, E.A. Plant cell wall breakdown by anaerobic microorganisms from the Mammalian digestive tract. Ann. N. Y. Acad. Sci. 2008, 1125, 280–288. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Offermanns, S. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 2009, 30, 557–562. [Google Scholar] [CrossRef]
- Kondo, T.; Kishi, M.; Fushimi, T.; Kaga, T. Acetic acid upregulates the expression of genes for fatty acid oxidation enzymes in liver to suppress body fat accumulation. J. Agric. Food Chem. 2009, 57, 5982–5986. [Google Scholar] [CrossRef] [PubMed]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (D)-β-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Z.; Zhang, R.H.; Zhang, H.Y.; Fu, S.X.; Xia, C. Effect of NEFA and glucose levels on CPT-I mRNA expression and translation in cultured bovine hepatocytes. J. Vet. Med. Sci. 2011, 73, 97–101. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef]
- Smith, J.L.; Lear, S.R.; Forte, T.M.; Ko, W.; Massimi, M.; Erickson, S.K. Effect of pregnancy and lactation on lipoprotein and cholesterol metabolism in the rat. J. Lipid Res. 1998, 39, 2237–2249. [Google Scholar] [CrossRef]
- Brown, M.V.; Lauro, F.M.; DeMaere, M.Z.; Muir, L.; Wilkins, D.; Thomas, T.; Riddle, M.J.; Fuhrman, J.A.; Andrews-Pfannkoch, C.; Hoffman, J.M.; et al. Global biogeography of SAR11 marine bacteria. Mol. Syst. Biol. 2012, 8, 595. [Google Scholar] [CrossRef]
- Pasolli, E.; de Filippis, F.; Mauriello, I.E.; Cumbo, F.; Walsh, A.M.; Leech, J.; Cotter, P.D.; Segata, N.; Ercolini, D. Large-scale genome-wide analysis links lactic acid bacteria from food with the gut microbiome. Nat. Commun. 2020, 11, 2610. [Google Scholar] [CrossRef] [PubMed]
- Chanishvili, N. Phage therapy-history from Twort and d’Herelle through soviet experience to current approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Mao, Y.; Zhao, F.; Zhang, X.X.; Ju, F.; Ye, L.; Wang, Y.; Li, B.; Ren, H.; Zhang, T. Free-living bacteria and potential bacterial pathogens in sewage treatment plants. Appl. Microbiol. Biotechnol. 2018, 102, 2455–2464. [Google Scholar] [CrossRef]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Nunez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.W.; Chokhawala, H.; Schroth, G.; Luo, S.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef]
- Svartstrom, O.; Alneberg, J.; Terrapon, N.; Lombard, V.; de Bruijn, I.; Malmsten, J.; Dalin, A.M.; El Muller, E.; Shah, P.; Wilmes, P.; et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 2017, 11, 2538–2551. [Google Scholar] [CrossRef]
- Nudda, A.; Cannas, A.; Correddu, F.; Atzori, A.S.; Lunesu, M.F.; Battacone, G.; Pulina, G. Sheep and goats respond differently to feeding strategies directed to improve the fatty acid profile of milk fat. Animals 2020, 10, 1290. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Guan, L.L.; Liu, J.X. Assessment of rumen bacteria in dairy cows with varied milk protein yield. J. Dairy Sci. 2019, 102, 5031–5041. [Google Scholar] [CrossRef]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Matthews, C.; Crispie, F.; Lewis, E.; Reid, M.; O’Toole, P.W.; Cotter, P.D. The rumen microbiome: A crucial consideration when optimising milk and meat production and nitrogen utilisation efficiency. Gut Microbes 2019, 10, 115–132. [Google Scholar] [CrossRef]
- McGavin, M.D.; Morrill, J.L. Dissection technique for examination of the bovine ruminoreticulum. J. Anim. Sci. 1976, 42, 535–538. [Google Scholar] [CrossRef]
- Emery, R.S. Biosynthesis of milk fat. J. Dairy Sci. 1973, 56, 1187–1195. [Google Scholar] [CrossRef]
- Harvatine, K.J.; Bauman, D.E. Characterization of the acute lactational response to trans-10, cis-12 conjugated linoleic acid. J. Dairy Sci. 2011, 94, 6047–6056. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergstrom, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Backhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Mende, D.R.; Waller, A.S.; Sunagawa, S.; Jarvelin, A.I.; Chan, M.M.; Arumugam, M.; Raes, J.; Bork, P. Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE 2012, 7, e31386. [Google Scholar] [CrossRef]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.H.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Avershina, E.; Frisli, T.; Rudi, K. De novo semi-alignment of 16S rRNA gene sequences for deep phylogenetic characterization of next generation sequencing data. Microbes Environ. 2013, 28, 211–216. [Google Scholar] [CrossRef]
- Noval Rivas, M.; Burton, O.T.; Wise, P.; Zhang, Y.Q.; Hobson, S.A.; Garcia Lloret, M.; Chehoud, C.; Kuczynski, J.; DeSantis, T.; Warrington, J.; et al. A microbiota signature associated with experimental food allergy promotes allergic sensitization and anaphylaxis. J. Allergy Clin. Immunol. 2013, 131, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, r60. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Indugu, N.; Kumar, S.; Vecchiarelli, B.; Sinha, R.; Baker, L.D.; Bhukya, B.; Ferguson, J.D. Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe 2016, 38, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, H.; Wang, Y.; Li, S.; Cao, Z.; Ji, S.; He, Y.; Zhang, H. Effect of dietary forage to concentrate ratios on dynamic profile changes and interactions of ruminal microbiota and metabolites in Holstein heifers. Front. Microbiol. 2017, 8, 2206. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Liu, J.X.; Guan, L.L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 2020, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ji, S.; Yan, H.; Hao, Y.; Zhang, J.; Wang, Y.; Cao, Z.; Li, S. The day-to-day stability of the ruminal and fecal microbiota in lactating dairy cows. MicrobiologyOpen 2020, 9, e990. [Google Scholar] [CrossRef]
- Shah, H.N.; Collins, D.M. Prevotella, a new genus to include Bacteroides melaninogenicus and related species formerly classified in the genus Bacteroides. Int. J. Syst. Bacteriol. 1990, 40, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Metzler-Zebeli, B.U.; Schmitz-Esser, S.; Klevenhusen, F.; Podstatzky-Lichtenstein, L.; Wagner, M.; Zebeli, Q. Grain-rich diets differently alter ruminal and colonic abundance of microbial populations and lipopolysaccharide in goats. Anaerobe 2013, 20, 65–73. [Google Scholar] [CrossRef]
- Strobel, H.J. Vitamin B12-dependent propionate production by the ruminal bacterium Prevotella ruminicola 23. Appl. Environ. Microbiol. 1992, 58, 2331–2333. [Google Scholar] [CrossRef]
- Lamendella, R.; Domingo, J.W.; Ghosh, S.; Martinson, J.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef]
- Patel, D.D.; Patel, A.K.; Parmar, N.R.; Shah, T.M.; Patel, J.B.; Pandya, P.R.; Joshi, C.G. Microbial and carbohydrate active enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet. Gene 2014, 545, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Chiquette, J.; Allison, M.J.; Rasmussen, M.A. Prevotella bryantii 25A used as a probiotic in early-lactation dairy cows: Effect on ruminal fermentation characteristics, milk production, and milk composition. J. Dairy Sci. 2008, 91, 3536–3543. [Google Scholar] [CrossRef]
- Osborne, J.M.; Dehority, B.A. Synergism in degradation and utilization of intact forage cellulose, hemicellulose, and pectin by three pure cultures of ruminal bacteria. Appl. Environ. Microbiol. 1989, 55, 2247–2250. [Google Scholar] [CrossRef]
- Shabat, S.K.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef]
- Chang, J.; Park, H. Nucleotide and protein researches on anaerobic fungi during four decades. J. Anim. Sci. Technol. 2020, 62, 121–140. [Google Scholar] [CrossRef]
- Wang, X.; Ao, C.; Khas, E.; Liu, S.; Bai, C.; Zhang, F.; Zhang, Y.; Gao, P. Effects of infusing milk precursors into the artery on rumen fermentation in lactating cows. Anim. Nutr. 2016, 2, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Chibisa, G.E.; Gozho, G.N.; van Kessel, A.G.; Olkowski, A.A.; Mutsvangwa, T. Effects of peripartum propylene glycol supplementation on nitrogen metabolism, body composition, and gene expression for the major protein degradation pathways in skeletal muscle in dairy cows. J. Dairy Sci. 2008, 91, 3512–3527. [Google Scholar] [CrossRef] [PubMed]
- Raggio, G.; Lemosquet, S.; Lobley, G.E.; Rulquin, H.; Lapierre, H. Effect of casein and propionate supply on mammary protein metabolism in lactating dairy cows. J. Dairy Sci. 2006, 89, 4340–4351. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, C.; Guo, G.; Huo, W.J.; Zhang, S.L.; Pei, C.X.; Zhang, Y.L.; Wang, H. Effects of branched-chain volatile fatty acids on lactation performance and mRNA expression of genes related to fatty acid synthesis in mammary gland of dairy cows. Animal 2018, 12, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Allen, M.S. Dose-response effects of intrauminal infusion of propionate on feeding behavior of lactating cows in early or midlactation. J. Dairy Sci. 2003, 86, 2922–2931. [Google Scholar] [CrossRef]
- Lv, C.; Liu, S.; Xia, J.; Xu, L.; Cheng, Y.; Li, W.; Zhang, Y.; Wang, G.; Wei, W.; Shi, H.; et al. The mechanism of dietary protein modulation of bone metabolism via alterations in members of the GH/IGF axis. Curr. Protein Pept. Sci. 2019, 20, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Mazinani, M.; Naserian, A.A.; Rude, B.J.; Tahmasbi, A.M.; Valizadeh, R. Effects of feeding rumen-protected amino acids on the performance of feedlot calves. J. Adv. Vet. Anim. Res. 2020, 7, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Zhou, X.; Sun, Y.; Hu, L.; Zhu, J.; Shao, C.; Meng, Q.; Shan, A. Threonine, but not lysine and methionine, reduces fat accumulation by regulating lipid metabolism in obese mice. J. Agric. Food Chem. 2020, 68, 4876–4883. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Wykes, L.; Shum-Tim, D.; Nitschmann, E.; Bui, H.; Nakazawa, K.; Codere-Maruyama, T.; Schricker, T.; Hatzakorzian, R. Parenteral amino acid supplementation with high-dose insulin prevents hypoaminoacidemia during cardiac surgery. Nutrition 2020, 69, 110566. [Google Scholar] [CrossRef] [PubMed]
- Leal Yepes, F.A.; Mann, S.; Overton, T.R.; Ryan, C.M.; Bristol, L.S.; Granados, G.E.; Nydam, D.V.; Wakshlag, J.J. Effect of rumen-protected branched-chain amino acid supplementation on production- and energy-related metabolites during the first 35 days in milk in Holstein dairy cows. J. Dairy Sci. 2019, 102, 5657–5672. [Google Scholar] [CrossRef]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | HPF | LPF | SEM | p-Value |
|---|---|---|---|---|
| Milk, kg/d | 35.172 | 34.826 | 0.851 | 0.876 |
| DIM, days | 244.231 | 237.336 | 11.653 | 0.812 |
| Body weight, kg | 637.752 | 617.210 | 8.258 | 0.316 |
| Composition, % | ||||
| Fat | 4.393 | 1.940 | 0.252 | 0.004 ** |
| Protein | 3.943 | 2.923 | 0.102 | 0.002 ** |
| Groups | Protein Percentage | Fat Percentage |
|---|---|---|
| High1 | 4.02% | 4.66% |
| High2 | 3.86% | 4.68% |
| High3 | 3.95% | 3.84% |
| Average of 4000 cows | 3.60% | 3.10% |
| Low1 | 2.78% | 1.92% |
| Low2 | 3.04% | 1.86% |
| Low3 | 2.95% | 2.04% |
| Items | HPF | LPF | SEM | p-Value |
|---|---|---|---|---|
| pH | 6.466 | 6.606 | 0.083 | 0.458 |
| NH3-N, mg/dL | 17.956 | 17.701 | 0.102 | 0.221 |
| Proportion | - | - | - | - |
| Acetate | 67.104 | 47.508 | 4.599 | 0.003 ** |
| Propionate | 24.703 | 12.354 | 2.894 | 0.003 ** |
| Butyrate | 6.247 | 5.244 | 0.266 | 0.035 * |
| Isobutyrate | 0.703 | 0.519 | 0.072 | 0.235 |
| Valerate | 0.936 | 0.809 | 0.126 | 0.667 |
| Isovalerate | 0.378 | 0.345 | 0.019 | 0.447 |
| Total VFA, mmol/L | 100.071 | 66.780 | 7.754 | 0.002 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Huang, S.; Huang, J.; Peng, P.; Liu, Y.; Han, B.; Sun, D. Identification of the Potential Role of the Rumen Microbiome in Milk Protein and Fat Synthesis in Dairy Cows Using Metagenomic Sequencing. Animals 2021, 11, 1247. https://doi.org/10.3390/ani11051247
Wu X, Huang S, Huang J, Peng P, Liu Y, Han B, Sun D. Identification of the Potential Role of the Rumen Microbiome in Milk Protein and Fat Synthesis in Dairy Cows Using Metagenomic Sequencing. Animals. 2021; 11(5):1247. https://doi.org/10.3390/ani11051247
Chicago/Turabian StyleWu, Xin, Shuai Huang, Jinfeng Huang, Peng Peng, Yanan Liu, Bo Han, and Dongxiao Sun. 2021. "Identification of the Potential Role of the Rumen Microbiome in Milk Protein and Fat Synthesis in Dairy Cows Using Metagenomic Sequencing" Animals 11, no. 5: 1247. https://doi.org/10.3390/ani11051247
APA StyleWu, X., Huang, S., Huang, J., Peng, P., Liu, Y., Han, B., & Sun, D. (2021). Identification of the Potential Role of the Rumen Microbiome in Milk Protein and Fat Synthesis in Dairy Cows Using Metagenomic Sequencing. Animals, 11(5), 1247. https://doi.org/10.3390/ani11051247