
CAPE and Neuroprotection: A Review





Abstract
1. Introduction
2. Methodology
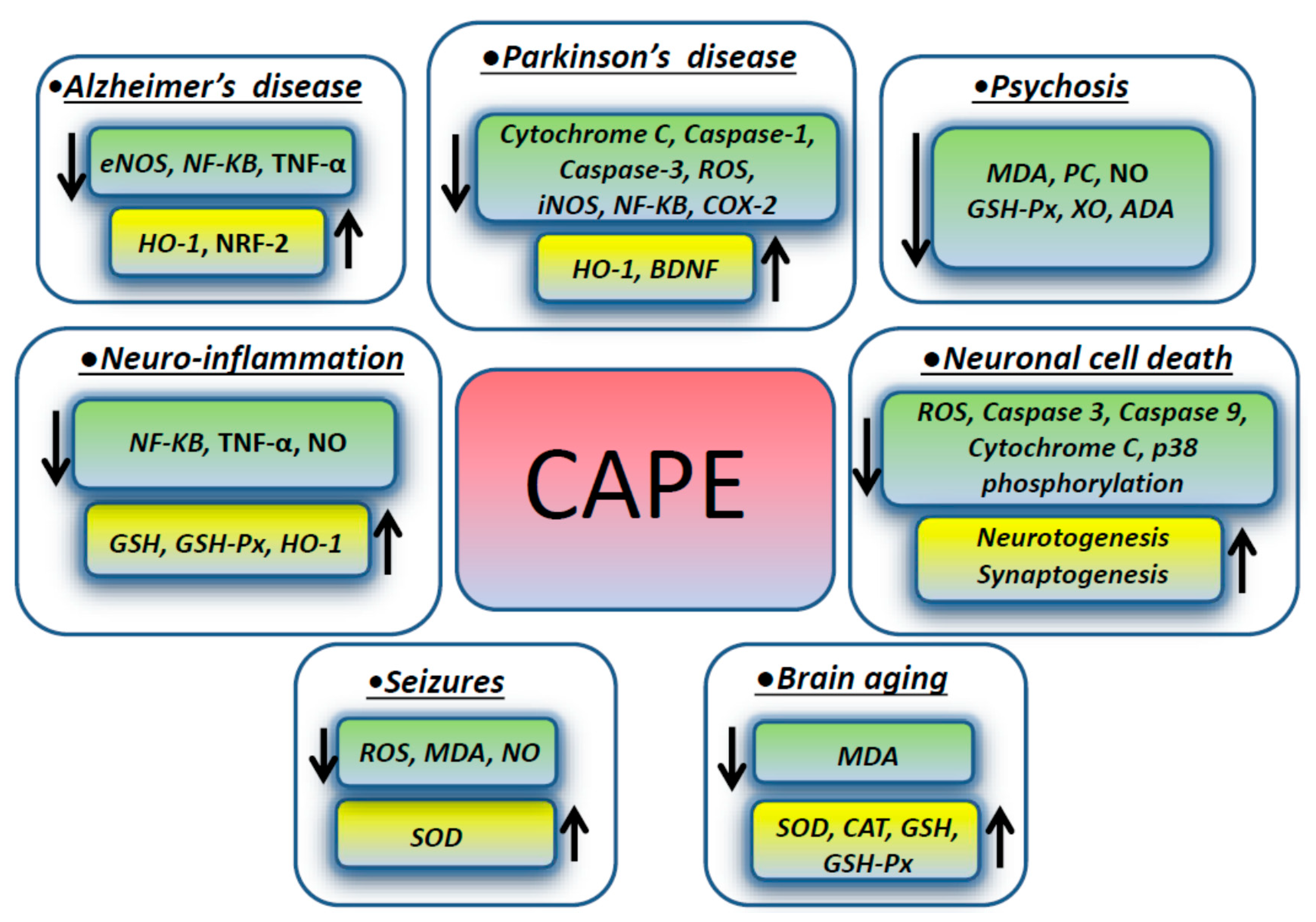
3. CAPE Effects on Different Neurologic Disorders
3.1. Apoptosis

{kind=link}
{kind=link}
| Stimulus | Parameters Measured | CAPE Dose(s) | Experimental Model | Ref. |
|---|---|---|---|---|
| Aging: 18 month old rats | -Histopathological assessment -MDA (−) -SOD (+) -CAT (+) -GSH-Px (+) -GSH (+) | 15 mg/kg/day, i.p., for 95 days | Male Sprague Dawley rats | [13] |
| Apoptosis: serum-free medium with low K+ (5 mM KCl) | -Apoptosis (−) -ROS (−) -Ca2+ (=) -NF-κB (−) -Caspase-3 and -9 (−) | 10 µg/mL | Primary CGNs from 8-day-old Wistar rats | [14] |
| SE: PTZ 40 mg/kg followed by 10 mg/kg every 10 min until SE occurrence, i.p. | -Histopathological assessment -Caspase-3 (−) | 30 mg/kg /day, i.p., for 5 days starting 40 min after the SE tonic phase | Dams reared Wistar male rats | [17] |
| Excitotoxicity: -Cells: glutamate 30 µM/24 h -Isolated mitochondria: glutamate and maleate (both 5 mM) | -Cell viability (+) -Caspase-3 (−) -Cytochrome c (−) -Glutamate-evoked currents | 0 µM–200 µM, pre- and co-treatment | -CGNs from 8-day-old Sprague Dawley rats -Mitochondria from CGNs and livers | [18] |
| Cytotoxicity: MPP+ 100, 500 or 1000 µM | -Cell differentiation (+) -Cell viability (+) -Protein content (+) -Synaptophysin (+) -GAP-43 (+) -Synapsin I (+) | 1, 5 or 10 µM | PC-12 cells | [19] |
| Neuroinflammation: IFN-γ and LPS | -NF-κB (−) -TNF-α (−) -NOS-2 (iNOS) (−) -CREB (+) | 4 to 100 µM, 30 min before and during LPS exposure | Organotypic hippocampal cultures from the hippocampi of 5–7- day-old Wistar rats | [20] |
| Neuroinflammation: TNF-α 10 ng/mL/6 h | -CCL-2 (−) -CXCL-8 (−) -ICAM-1 (−) -Monocyte Adhesion (-) -DNA-binding activity of NF-κB and AP-1 (−) -IκBα -IKK -TRAF2 -TAK1 -MKK4 -JNKs -c-Jun | 30 µM, pretreatment | -CRT-MG human astroglial cells -U937 human monocytic cells | [21] |
| XALD: human skin fibroblasts derived from XALD (GM04932, GM04934), and AMN (GM07531) patients | -TNF-α (−) -ROS (−) -NO (−) -Fatty acids (−) | 1–5 µM | -Fibroblasts -Mouse primary mixed glia and astrocytes | [22] |
| EAE: 50 µg of guinea pig MBP and 7 mg/mL heat-killed Mycobacterium tuberculosis, intradermally. | -Neurological assessment -MDA (−) -NO (−) -XO -GSH-Px -ADA -SOD | 25 µmol/kg/day, i.p., for 14 days after immunization | Female Wistar rats | [23] |
| ALS: transfection with pIRESneo and/or SOD1 mutants | -In silico analysis -DCF -Cell viability (+) -Nrf2 (+) -5- LO -NF-κB (−) | 10 µM, co-treatment | NSC34 mouse motor neurons | [24] |
| ALS: SOD1G93A mutated mice | -CAPE level -Behavioural assessment - pp38 (−) | 10 mg/kg/day, orally, for 7 days after disease onset | SOD1G93A mice | [25] |
| -Neuroinflammation, in vitro: 200 ng/mL LPS -in vivo, a single intraperitoneal injection of 20 mg/kg LPS, i.p., 2 h after the last CAPE injection | -Cell viability -ERK2 -Akt -p38, -pERK1/2 -pp38 -pAKT -pJNK -EPO (+) -HO-1 (+) -iNOS (−) -COX-2 (−) -pAMPKα (+) | -0.1 to 1.75 µM 30 min before LPS treatment, or co-treatment -1 or 5 mg/kg once daily for 3 days | -BV-2 murine microglial cell line -Eight-week-old male ICR mice | [26] |
| PD: 6-OHDA 70 µM for 6 h, on day 8–10 | -Cell viability (+) -Cytochrome c (−) -Caspase-3 (−) -Ca2+ | 10 to 100 µM, pre-treatment for 4 h | -Primary CGNs from 8-day-old Wistar rats -Rat liver mitochondria from 7-day-old Sprague–Dawley rats | [27] |
| Dopaminergic neurodegeneration: 6-OHDA, 40 μM for RMN and 70 μM for CGN | -Free radicals (−) -Peroxynitrite (−) | 10 μM, pre-treatment for 2 h | -Rat RMN -Primary CGNs | [28] |
| PD: 6-OHDA 8 mg/mL, s.i. | -Fe, Cu, Zn and Mn (−) -ROS (−) -Protein content -TH -Mitochondrial functions: Ca2+-induced swelling, Ca2+ uptake and respiration | -In Vivo: 10 μmol/kg/day, i.p., 5 days -In Vitro: 0.5 or 10 µM | Wistar rats | [29] |
| -Dopaminergic neurodegeneration, in vitro: LPS/72 h -PD, in vivo: LPS 3 µg/µL, intranigral, or 6-hydroxydopamine 2 µg/µL, intrastriatal, 30 min after first CAPE injection. | -NO (−) -ERK -p38 MAPK -HO-1 (+) -BDNF (+) -Nrf2 | -In Vitro: 3–30 µM -In Vivo: 10 or 30 mg/kg/day, i.p., for 4 days | -In Vitro: rat organotypic midbrain slice cultures -In Vivo: mouse model of dopaminergic neurodegeneration | [30] |
| PD: rotenone1 mg/kg, s.c., every 48 h, 9 injections | -Behavioural assessment -Histopathological assessment -CD11b -COX-2 (−) -iNOS (−) -NF-κB (−) -Dopamine level (+) -TNF-α (−) -IL-1β | 2.5, 5 or 10 mg/kg/day. orally, every 48 h, 9 doses | Male Swiss albino mice | [31] |
| PD: CPF 80 mg/kg, s.c. | -PON1 activity (+) -Lipid profile -TSA (+) -TAC (+) -TOC (−) -Histopathological assessment | 10 μmol/kg/day, i.p., 21 days | Male Swiss albino mice | [32] |
| PD: MPTP–HCl 20 mg/kg, i.p., four in at 2 h intervals | -TH-positive neurons (+) -Cell viability (+) -CAPE and MPP+ levels -DA (+) -MAO (−) -i- and nNOS (−) -Caspase-1 (−) -Cytochrome c (−) -AIF (−) -Free radicals (−) -Peroxynitrite (−) | 2, 5, or 10 mg/kg/day, 7days | Eight-week-old male C57BL/6 mice | [33] |
| Loss of memory (AD): STZ 3 mg/kg, bilaterally on day 1 and 3 | -TBARS (−) -GSH (+) -SOD (+) -CAT (+) -Nitrite (−) -AChE (−) -TNF-α (−) -eNOS (+) -NF-κB (−) -Behavioural assessment -Histopathological assessment | 6 mg/kg/day, i.p., 28 days | Wistar rats | [34] |
| Dementia (AD type): STZ; 3 mg/kg, on day 1 and 3, ICV | -MDA (−) -GSH (+) -TNF-α (−) -Behavioural tests | 3, 6 mg/kg/day, i.p., 28 days | Wistar rats | [35] |
| Dementia (AD type): Aβ1-42O, unilateral stereotaxic, ICV | -Behavioural assessment -ROS (−) -Nrf2 (+) -GSH -pGSK3α/β -Caspase-9 | 10 mg/kg/day, i.p., 1 h after brain lesion, 10 days | Male C57Bl/6 mice | [36] |
| Seizures: 60 mg/kg PTZ, i.p., single dose | -Neurological assessment -MDA (−) -NO (−) -XO -SOD (+) | 100 µmol/kg, i.p., 2 days prior to PTZ injection | Female Swiss albino mice | [15] |
| Psychosis: dizocilpine maleate (MK-801), 0.5 mg/kg/day for 5 days, i.p. | -Behavioural assessment -Histopathological assessment -MDA (−) -PC (−) -NO (−) -SOD -GSH-Px (−) -XO (−) -ADA (−) -CAT (=) | 10 μmol/kg, 6 days, started one day before MK-801, i.p. | Wistar rats | [37] |
| Diabetes: STZ 45 mg/kg, i.p., single dose | -NO (−) -SOD -GSH-Px (−) -GSH -XO (−) -CAT (−) -MDA (−) -iNOS (−) -TNF-α (−) -IFN-γ (−) -IL-10 | 25 µM/kg/day, two days after STZ treatment for 60 days | Male Wistar rats | [38] |
| Endotoxic shock: LPS, 20 mg/kg, i.p, | -TNF-α (−) -IL-1α, -1β, -6 (−) -IL-4, -10 (+) -sICAM-1 (−) -Histopathological assessment | 10 μmol/kg/day, 14 days before shock induction and a single dose 30 min after induction | Male Wister rats | [39] |
| Hepatic encephalopathy: thioacetamide:600 mg/kg, i.p., two doses (0 and 24 h) | -Behavioural and motor assessment -Blood ammonia (=) -ALT (−) -AST (−) | 10 µmol/kg/day, i.p., starting 1 day before the first dose of thioacetamide | Male Wistar rats | [40] |
| Optic nerve crushing, 10 s | -Apoptosis (−) -Astrocyte migration -Cell viability (+) -NF-κB (−) -IL-6 and -8 (−) -iNOS (−) -COX-2 (−) -TNF-α (−) -CCL-2 (−) | 10 μmol/kg, i.p., 10 min after the surgery | Male Sprague Dawley rats | [41] |
3.2. Neuro-Inflammation
3.3. Parkinson’s Disease
3.4. Alzheimer’s Disease
3.5. Seizures and Psychosis
3.6. Other Diseases
4. CAPE Protective Effects against Different Neurotoxic Substances
5. CAPE Protective Effects against Ischemia
6. CAPE Protective Effects against Injury
7. CAPE Anti-Tumoral Effects in CNS
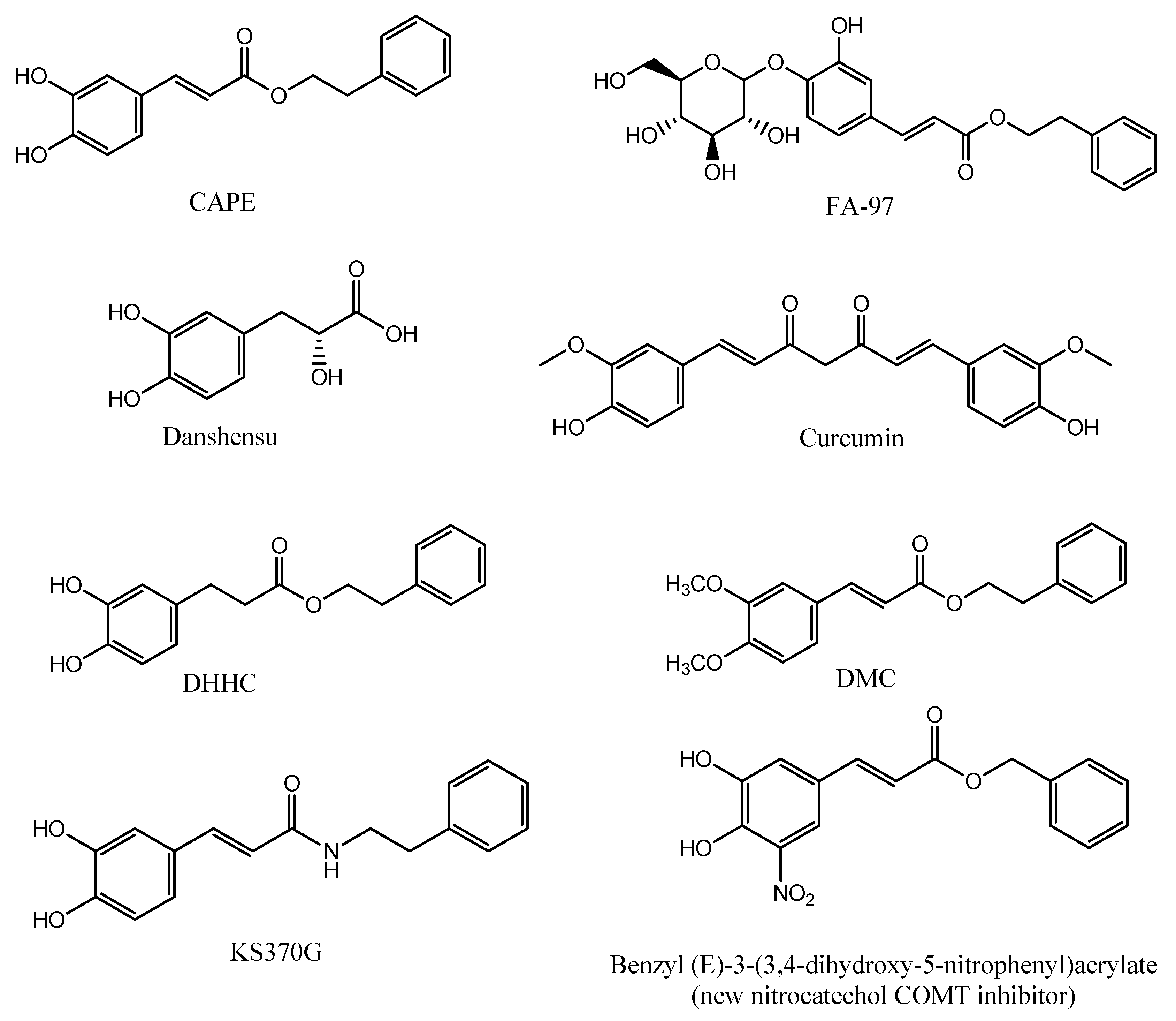
8. CAPE Derivatives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-LO | 5-lipoxygenase |
| 6-OHDA | 6-Hydroxydopamine |
| AChE | acetylcholinesterase |
| AD | Alzheimer’s disease |
| ADA | adenosine deaminase |
| AIF | Apoptosis-inducing factor |
| ALS | Amyotrophic lateral sclerosis |
| ALT | alanine transaminase |
| AST | aspartate transaminase |
| AMN | adrenomyeloneuropathy |
| AMPK | 5′-adenosine monophosphate-activated protein kinase |
| Bax | Bcl-2-associated X protein |
| Bad | Bcl-2-associated agonist of cell death |
| BBB | blood brain barrier |
| Bcl-2 | B cell CCL/lymphoma 2 |
| Bcl-xL | Bcl-2-like 1 |
| BDNF | brain-derived neurotrophic factor |
| CAPE | caffeic acid phenethyl ester |
| CAT | Catalase |
| CCA | common carotid arteries |
| CCI | controlled cortical impact |
| CCL- 2 | C-C.motif ligand-2 |
| CGNs | cerebellar granule neurons |
| CNS | central nervous system |
| COMT | catechol O-methyltransferase |
| COX-2 | cyclooxygenase-2 |
| CPF | Clorpyrifos-ethyl |
| CREB | cAMP-responsive element binding protein |
| DA | dopamine |
| DCF | dichlorofluorescein assayDHHC: dihydroxydihydrocinnamic acid phenethylester |
| DMC | dimethoxycinnamic acid phenethyl ester |
| EAE | experimental autoimmune encephalomyelitis |
| ED1 | marker of activated macrophage/microglia |
| eNOS | endothelial nitric oxide synthase |
| ERK | extracellular signal-regulated kinase |
| EPO | erythropoietin |
| ETM | ethambutol |
| FAPE | ferulic acid esterGSH: glutathione |
| GSH-Px | glutathione peroxidase |
| GSNO | S-nitrosoglutathione |
| HE | encephalopathy |
| HIF-1α | hypoxia inducing factor-1α |
| HO-1 | heme oxygenase |
| ICAM-1 | Intercellular adhesion molecule-1 |
| ICV | intracerebroventricular |
| IDO | indoleamine 2,3-dioxegenase |
| IFN- γ | interferon-γ |
| IFOS | ifosfamide |
| IκB | inhibitor κB |
| IKK | inhibitor of nuclear factor-κB (IκB) kinase |
| IL-1β | interleukine-1β |
| INH | isoniazid |
| iNOS | inducible nitric oxide synthase |
| JNK | c-Jun N-terminal kinase |
| LPS | lipopolysaccharide |
| MAO | monoamine oxidase |
| MAPK | mitogen-activated protein kinases |
| MBP | Myelin Basic Protein |
| MCA | middle cerebral artery |
| MCP-1 | monocyte chemoattractant protein-1 |
| MDA | Malondialdehyde |
| MK-801 | dizocilipine maleate |
| MKK4 | Mitogen-Activated Protein Kinase Kinase 4 |
| MMP | matrix metalloproteinase |
| MP | Methylprednisolone |
| MPO | myeloperoxidase |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MS | multiple sclerosis |
| MTX | methotrexate |
| NF | neurofibromatosis |
| NF-κB | nuclear factor-κB |
| NGF | nerve growth factor |
| NMDA | N-methyl-D-aspartate |
| NO | nitric oxide |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PAMPA | parallel artificial membrane permeability assay (PAMPA)PC: protein carbonyl |
| PD | Parkinson’s disease |
| PFC | prefrontal cortex |
| PI3K | phosphoinositide 3-kinase |
| PLD1 | phospholipase D1 |
| PMNL | polymorphonuclear leukocytes |
| PON1 | paraoxonase |
| PTZ | pentylenetetrazole |
| RMN | rostral mesencephalic neurons |
| ROS | reactive oxygen species |
| SAH | Subarachnoid hemorrhage |
| SCI | spinal cord injury |
| SE | status epilepticus |
| SOD | superoxide dismutase |
| STZ | streptozotocin |
| TAC | total antioxidant capacity |
| TAK1 | transforming growth factor-β-activated kinase 1 |
| TAR | total antioxidant response |
| TAS | total antioxidant status |
| TBARS | thiobarbituric acid reactive substances |
| TBI | traumatic brain injury |
| TH | tyrosine hydroxylase |
| TNF-α | tumor necrosis factor-α |
| TOA | total oxidant activity |
| TOC | total oxidant capacity |
| TOS | Total oxidant status |
| TRAF2 | TNF Receptor Associated Factor 2 |
| TSA | total sialic acid |
| VLCFA | very long chain fatty acids |
| X-ALD | X-linked adrenoleukodystrophy |
| XO | xanthine oxidase |
References
- Menezes da Silveira, C.C.; Luz, D.A.; da Silva, C.C.; Prediger, R.D.; Martins, M.D.; Martins, M.A.; Fontes-Júnior, E.A.; Maia, C.S. Propolis: A useful agent on psychiatric and neurological disorders? A focus on CAPE and pinocembrin components. Med. Res. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ağrı, İ.; Erdal Ağrı, A.; Özdemir, D.; Özgür, A. Chapter 31—CAPE and Tympanosclerosis. In Polyphenols: Mechanisms of Action in Human Health and Disease, 2nd ed.; Watson, R.R., Preedy, V.R., Zibadi, S., Eds.; Academic Press: Amsterdam, The Netherlands; Elsevier: Amsterdam, The Netherlands, 2018; pp. 421–430. [Google Scholar] [CrossRef]
- Sforcin, J.M.; Bankova, V. Propolis: Is there a potential for the development of new drugs? J. Ethnopharmacol. 2011, 133, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Toreti, V.C.; Sato, H.H.; Pastore, G.M.; Park, Y.K. Recent progress of propolis for its biological and chemical compositions and its botanical origin. Evid.-Based Complement. Altern. Med. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grunberger, D.; Banerjee, R.; Eisinger, K.; Oltz, E.M.; Efros, L.; Caldwell, M.; Estevez, V.; Nakanishi, K. Preferential cytotoxicity on tumor cells by caffeic acid phenethyl ester isolated from propolis. Cell. Mol. Life Sci. 1988, 44, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Akyol, S.; Armutcu, F.; Yigitoglu, M. The medical usage of caffeic acid phenethyl ester (CAPE), an active compound of propolis, in neurological disorders and emergencies. Spatula DD 2011, 1, 37–42. [Google Scholar] [CrossRef]
- Göçer, H.; Gülçin, İ. Caffeic acid phenethyl ester (CAPE): Correlation of structure and antioxidant properties. Int. J. Food Sci. Nutr. 2011, 62, 821–825. [Google Scholar] [CrossRef]
- Celli, N.; Dragani, L.K.; Murzilli, S.; Pagliani, T.; Poggi, A. In Vitro and in vivo stability of caffeic acid phenethyl ester, a bioactive compound of propolis. J. Agric. Food Chem. 2007, 55, 3398–3407. [Google Scholar] [CrossRef]
- Tolba, M.F.; Azab, S.S.; Khalifa, A.E.; Abdel-Rahman, S.Z.; Abdel-Naim, A.B. Caffeic acid phenethyl ester, a promising component of propolis with a plethora of biological activities: A review on its anti-inflammatory, neuroprotective, hepatoprotective, and cardioprotective effects. IUBMB Life 2013, 65, 699–709. [Google Scholar] [CrossRef]
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Bin Asad, M.H.H. Caffeic acid phenethyl ester and therapeutic potentials. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Ali, M.A.; Menze, E.T.; Tadros, M.G.; Tolba, M.F. Caffeic acid phenethyl ester counteracts doxorubicin-induced chemobrain in Sprague-Dawley rats: Emphasis on the modulation of oxidative stress and neuroinflammation. Neuropharmacology 2020, 181, 108334. [Google Scholar] [CrossRef]
- Çetin, A.; Deveci, E. Evaluation of PECAM-1 and p38 MAPK expressions in cerebellum tissue of rats treated with caffeic acid phenethyl ester: A biochemical and immunohistochemical study. Folia Morphol. 2019, 78, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Eşrefoğlu, M.; Gül, M.; Ateş, B.; Yılmaz, İ. The ultrastructural and biochemical evidences of the beneficial effects of chronic caffeic acid phenethyl ester and melatonin administration on brain and cerebellum of aged rats. Fundam. Clin. Pharmacol. 2010, 24, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Amodio, R.; De Ruvo, C.; Di Matteo, V.; Poggi, A.; Di Santo, A.; Martelli, N.; Lorenzet, R.; Rotilio, D.; Cacchio, M.; Esposito, E. Caffeic acid phenethyl ester blocks apoptosis induced by low potassium in cerebellar granule cells. Int. J. Dev. Neurosci. 2003, 21, 379–389. [Google Scholar] [CrossRef]
- Ilhan, A.; Iraz, M.; Gurel, A.; Armutcu, F.; Akyol, O. Caffeic acid phenethyl ester exerts a neuroprotective effect on CNS against pentylenetetrazol-induced seizures in mice. Neurochem. Res. 2004, 29, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-W.; Yang, S.-R.; Huang, M.-H.; Wu, S.-N. Stimulatory actions of caffeic acid phenethyl ester, a known inhibitor of NF-κB activation, on Ca2+-activated K+ current in pituitary GH3 cells. J. Biol. Chem. 2004, 279, 26885–26892. [Google Scholar] [CrossRef] [PubMed]
- Yiş, U.; Topçu, Y.; Özbal, S.; Tuğyan, K.; Bayram, E.; Karakaya, P.; Yilmaz, O.; Kurul, S.H. Caffeic acid phenethyl ester prevents apoptotic cell death in the developing rat brain after pentylenetetrazole-induced status epilepticus. Epilepsy Behav. 2013, 29, 275–280. [Google Scholar] [CrossRef]
- Wei, X.; Ma, Z.; Fontanilla, C.; Zhao, L.; Xu, Z.; Taggliabraci, V.; Johnstone, B.; Dodel, R.; Farlow, M.; Du, Y. Caffeic acid phenethyl ester prevents cerebellar granule neurons (CGNs) against glutamate-induced neurotoxicity. Neuroscience 2008, 155, 1098–1105. [Google Scholar] [CrossRef]
- dos Santos, N.A.G.; Martins, N.M.; de Barros Silva, R.; Ferreira, R.S.; Sisti, F.M.; dos Santos, A.C. Caffeic acid phenethyl ester (CAPE) protects PC12 cells from MPP+ toxicity by inducing the expression of neuron-typical proteins. Neurotoxicology 2014, 45, 131–138. [Google Scholar] [CrossRef]
- Montpied, P.; de Bock, F.; Rondouin, G.; Niel, G.; Briant, L.; Courseau, A.-S.; Lerner-Natoli, M.; Bockaert, J. Caffeic acid phenethyl ester (CAPE) prevents inflammatory stress in organotypic hippocampal slice cultures. Mol. Brain Res. 2003, 115, 111–120. [Google Scholar] [CrossRef]
- Choi, K.; Choi, C. Differential regulation of c-Jun N-terminal kinase and NF-κB pathway by caffeic acid phenethyl ester in astroglial and monocytic cells. J. Neurochem. 2008, 105, 557–564. [Google Scholar] [CrossRef]
- Singh, J.; Khan, M.; Singh, I. Caffeic acid phenethyl ester induces adrenoleukodystrophy (Abcd2) gene in human X-ALD fibroblasts and inhibits the proinflammatory response in Abcd1/2 silenced mouse primary astrocytes. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2013, 1831, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, A.; Akyol, O.; Gurel, A.; Armutcu, F.; Iraz, M.; Oztas, E. Protective effects of caffeic acid phenethyl ester against experimental allergic encephalomyelitis-induced oxidative stress in rats. Free Radic. Biol. Med. 2004, 37, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Higginbottom, A.; Mead, R.J.; Barber, S.; Shaw, P.J. An in vitro screening cascade to identify neuroprotective antioxidants in ALS. Free Radic. Biol. Med. 2009, 46, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Fontanilla, C.; Wei, X.; Zhao, L.; Johnstone, B.; Pascuzzi, R.; Farlow, M.; Du, Y. Caffeic acid phenethyl ester extends survival of a mouse model of amyotrophic lateral sclerosis. Neuroscience 2012, 205, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-F.; Kuo, Y.-H.; Yeh, W.-L.; Wu, C.Y.-J.; Lin, H.-Y.; Lai, S.-W.; Liu, Y.-S.; Wu, L.-H.; Lu, J.-K.; Lu, D.-Y. Regulatory effects of caffeic acid phenethyl ester on neuroinflammation in microglial cells. Int. J. Mol. Sci. 2015, 16, 5572–5589. [Google Scholar] [CrossRef]
- Noelker, C.; Bacher, M.; Gocke, P.; Wei, X.; Klockgether, T.; Du, Y.; Dodel, R. The flavanoide caffeic acid phenethyl ester blocks 6-hydroxydopamine-induced neurotoxicity. Neurosci. Lett. 2005, 383, 39–43. [Google Scholar] [CrossRef]
- Ma, Z.; Wei, X.; Fontanilla, C.; Noelker, C.; Dodel, R.; Hampel, H.; Du, Y. Caffeic acid phenethyl ester blocks free radical generation and 6-hydroxydopamine-induced neurotoxicity. Life Sci. 2006, 79, 1307–1311. [Google Scholar] [CrossRef]
- Silva, R.B.; Santos, N.; Martins, N.; Ferreira, D.; Barbosa, F., Jr.; Souza, V.O.; Kinoshita, A.; Baffa, O.; Del-Bel, E.; Santos, A. Caffeic acid phenethyl ester protects against the dopaminergic neuronal loss induced by 6-hydroxydopamine in rats. Neuroscience 2013, 233, 86–94. [Google Scholar] [CrossRef]
- Kurauchi, Y.; Hisatsune, A.; Isohama, Y.; Mishima, S.; Katsuki, H. Caffeic acid phenethyl ester protects nigral dopaminergic neurons via dual mechanisms involving haem oxygenase-1 and brain-derived neurotrophic factor. Br. J. Pharmacol. 2012, 166, 1151–1168. [Google Scholar] [CrossRef]
- Zaitone, S.A.; Ahmed, E.; Elsherbiny, N.M.; Mehanna, E.T.; El-Kherbetawy, M.K.; ElSayed, M.H.; Alshareef, D.M.; Moustafa, Y.M. Caffeic acid improves locomotor activity and lessens inflammatory burden in a mouse model of rotenone-induced nigral neurodegeneration: Relevance to Parkinson’s disease therapy. Pharmacol. Rep. 2019, 71, 32–41. [Google Scholar] [CrossRef]
- Deveci, H.A.; Karapehlivan, M. Chlorpyrifos-induced parkinsonian model in mice: Behavior, histopathology and biochemistry. Pestic. Biochem. Physiol. 2018, 144, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Fontanilla, C.; Ma, Z.; Wei, X.; Klotsche, J.; Zhao, L.; Wisniowski, P.; Dodel, R.; Farlow, M.; Oertel, W.; Du, Y. Caffeic acid phenethyl ester prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration. Neuroscience 2011, 188, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Bansal, N. Caffeic acid phenethyl ester rescued streptozotocin-induced memory loss through PI3-kinase dependent pathway. Biomed. Pharmacother. 2018, 101, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kaur, D.; Bansal, N. Caffeic acid phenethyl ester (CAPE) prevents development of STZ-ICV induced dementia in rats. Pharmacogn. Mag. 2017, 13, S10. [Google Scholar]
- Morroni, F.; Sita, G.; Graziosi, A.; Turrini, E.; Fimognari, C.; Tarozzi, A.; Hrelia, P. Neuroprotective effect of caffeic acid phenethyl ester in a mouse model of Alzheimer’s disease involves Nrf2/HO-1 pathway. Aging Dis. 2018, 9, 605. [Google Scholar] [CrossRef]
- Ozyurt, B.; Ozyurt, H.; Akpolat, N.; Erdogan, H.; Sarsilmaz, M. Oxidative stress in prefrontal cortex of rat exposed to MK-801 and protective effects of CAPE. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 832–838. [Google Scholar] [CrossRef]
- Celik, S.; Erdogan, S. Caffeic acid phenethyl ester (CAPE) protects brain against oxidative stress and inflammation induced by diabetes in rats. Mol. Cell. Biochem. 2008, 312, 39–46. [Google Scholar] [CrossRef]
- Korish, A.A.; Arafa, M.M. Propolis derivatives inhibit the systemic inflammatory response and protect hepatic and neuronal cells in acute septic shock. Braz. J. Infect. Dis. 2011, 15, 332–338. [Google Scholar] [CrossRef]
- Fadillioglu, E.; Gursul, C.; Iraz, M. Effects of caffeic acid phenethyl ester on thioacetamide-induced hepatic encephalopathy in rats. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 1440–1445. [Google Scholar] [CrossRef]
- Jia, Y.; Jiang, S.; Chen, C.; Lu, G.; Xie, Y.; Sun, X.; Huang, L. Caffeic acid phenethyl ester attenuates nuclear factor-κB-mediated inflammatory responses in Müller cells and protects against retinal ganglion cell death. Mol. Med. Rep. 2019, 19, 4863–4871. [Google Scholar]
- Uzar, E.; Sahin, O.; Koyuncuoglu, H.R.; Uz, E.; Bas, O.; Kilbas, S.; Yilmaz, H.R.; Yurekli, V.A.; Kucuker, H.; Songur, A. The activity of adenosine deaminase and the level of nitric oxide in spinal cord of methotrexate administered rats: Protective effect of caffeic acid phenethyl ester. Toxicology 2006, 218, 125–133. [Google Scholar] [CrossRef]
- Uzar, E.; Koyuncuoglu, H.R.; Uz, E.; Yilmaz, H.R.; Kutluhan, S.; Kilbas, S.; Gultekin, F. The activities of antioxidant enzymes and the level of malondialdehyde in cerebellum of rats subjected to methotrexate: Protective effect of caffeic acid phenethyl ester. Mol. Cell. Biochem. 2006, 291, 63–68. [Google Scholar] [CrossRef]
- Uzar, E.; Koyuncuoğlu, H.R.; Yılmaz, H.R.; Uz, E.; Songur, A.; Şahin, Ö.; Yürekli, V.A.; Yılmaz, M.; Kılbaş, S.; Kutluhan, S. Ameliorating Role of Caffeic Acid Phenethyl Ester (CAPE) Against Methotrexate-Induced Oxidative Stress in the Sciatic Nerve, Spinal Cord and Brain Stem Tissues of Rats. Turk. J. Neurol./Turk Noroloji Derg. 2010, 16, 12–20. [Google Scholar]
- Ginis, Z.; Ozturk, G.; Albayrak, A.; Kurt, S.N.; Albayrak, M.; Fadillioglu, E. Protective effects of caffeic acid phenethyl ester on ifosfamide-induced central neurotoxicity in rats. Toxicol. Ind. Health 2016, 32, 337–343. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Dos Santos, N.A.G.; Martins, N.M.; Fernandes, L.S.; Dos Santos, A.C. Caffeic Acid Phenethyl Ester (CAPE) Protects PC12 Cells from Cisplatin-Induced Neurotoxicity by Activating the NGF-Signaling Pathway. Neurotox. Res. 2018, 34, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; dos Santos, N.A.G.; Bernardes, C.P.; Sisti, F.M.; Amaral, L.; Fontana, A.C.; dos Santos, A.C. Caffeic acid phenethyl ester (CAPE) protects PC12 cells against cisplatin-induced neurotoxicity by activating the AMPK/SIRT1, MAPK/Erk, and PI3k/Akt signaling pathways. Neurotox. Res. 2019, 36, 175–192. [Google Scholar] [CrossRef]
- Fadillioglu, E.; Erdogan, H.; Iraz, M.; Yagmurca, M. Effects of caffeic acid phenethyl ester against doxorubicin-induced neuronal oxidant injury. Neurosci. Res. Commun. 2003, 33, 132–138. [Google Scholar] [CrossRef]
- Eser, O.; Cosar, M.; Sahin, O.; Mollaoglu, H.; Sezer, M.; Yaman, M.; Songur, A. The neuroprotective effects of caffeic acid phenethyl ester (CAPE) in the hippocampal formation of cigarette smoke exposed rabbits. Pathology 2007, 39, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Başarslan, S.K.; Ösün, A.; Şenol, S.; Korkmaz, M.; Özkan, Ü.; Kaplan, I. Protective effects of intralipid and caffeic acid phenyl esther (CAPE) on neurotoxicity induced by ethanol in rats. Turk. Neurosurg. 2015, 27. [Google Scholar] [CrossRef]
- Huang, Y.; Jin, M.; Pi, R.; Zhang, J.; Chen, M.; Ouyang, Y.; Liu, A.; Chao, X.; Liu, P.; Liu, J. Protective effects of caffeic acid and caffeic acid phenethyl ester against acrolein-induced neurotoxicity in HT22 mouse hippocampal cells. Neurosci. Lett. 2013, 535, 146–151. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Abd El-Twab, S.M. Caffeic acid phenethyl ester protects the brain against hexavalent chromium toxicity by enhancing endogenous antioxidants and modulating the JAK/STAT signaling pathway. Biomed. Pharmacother. 2017, 91, 303–311. [Google Scholar] [CrossRef]
- Hao, R.; Song, X.; Li, F.; Tan, X.; Sun-Waterhouse, D.; Li, D. Caffeic acid phenethyl ester reversed cadmium-induced cell death in hippocampus and cortex and subsequent cognitive disorders in mice: Involvements of AMPK/SIRT1 pathway and amyloid-tau-neuroinflammation axis. Food Chem. Toxicol. 2020, 144, 111636. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, U.; Osun, A.; Basarslan, K.; Senol, S.; Kaplan, I.; Alp, H. Effects of intralipid and caffeic acid phenethyl ester on neurotoxicity, oxidative stress, and acetylcholinesterase activity in acute chlorpyriphos intoxication. Int. J. Clin. Exp. Med. 2014, 7, 837. [Google Scholar] [PubMed]
- Uzar, E.; Varol, S.; Acar, A.; Firat, U.; Basarslan, S.; Evliyaoglu, O.; Yucel, Y.; Alp, H.; Gökalp, O. Assesment the role of oxidative stress and efficacy of caffeic acid phenethyl ester (CAPE) on neurotoxicity induced by isoniazid and ethambutol in a rat model. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2953–2959. [Google Scholar] [PubMed]
- Wang, L.Y.; Tang, Z.J.; Han, Y.Z. Neuroprotective effects of caffeic acid phenethyl ester against sevoflurane-induced neuronal degeneration in the hippocampus of neonatal rats involve MAPK and PI3K/Akt signaling pathways. Mol. Med. Rep. 2016, 14, 3403–3412. [Google Scholar] [CrossRef]
- Irmak, M.K.; Fadillioglu, E.; Sogut, S.; Erdogan, H.; Gulec, M.; Ozer, M.; Yagmurca, M.; Gozukara, M.E. Effects of caffeic acid phenethyl ester and alpha-tocopherol on reperfusion injury in rat brain. Cell Biochem. Funct. Cell. Biochem. Modul. Act. Agents Dis. 2003, 21, 283–289. [Google Scholar] [CrossRef]
- Tsai, S.-K.; Lin, M.-J.; Liao, P.-H.; Yang, C.-Y.; Lin, S.-M.; Liu, S.-M.; Lin, R.-H.; Chih, C.-L.; Huang, S.-S. Caffeic acid phenethyl ester ameliorates cerebral infarction in rats subjected to focal cerebral ischemia. Life Sci. 2006, 78, 2758–2762. [Google Scholar] [CrossRef]
- Khan, M.; Elango, C.; Ansari, M.A.; Singh, I.; Singh, A.K. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. J. Neurochem. 2007, 102, 365–377. [Google Scholar] [CrossRef]
- Altuğ, M.E.; Serarslan, Y.; Bal, R.; Kontaş, T.; Ekici, F.; Melek, I.M.; Aslan, H.; Duman, T. Caffeic acid phenethyl ester protects rabbit brains against permanent focal ischemia by antioxidant action: A biochemical and planimetric study. Brain Res. 2008, 1201, 135–142. [Google Scholar] [CrossRef]
- Serarslan, Y.; Bal, R.; Altuğ, M.E.; Kontaş, T.; Melek, I.M. Caffeic acid phenethyl ester decreases the level of S-100B protein after middle cerebral AFTER occusion in rabbits. Pak. J. Pharm. Sci. 2009, 22. [Google Scholar]
- Cengiz, N.; Colakoglu, N.; Kavakli, A.; Sahna, E.; Parlakpinar, H.; Acet, A. Effects of caffeic acid phenethyl ester on cerebral cortex: Structural changes resulting from middle cerebral artery ischemia reperfusion. Clin. Neuropathol. 2007, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Kim, C.D.; Lee, W.S. Caffeic acid phenethyl ester protects against photothrombotic cortical ischemic injury in mice. Korean J. Physiol. Pharmacol. 2018, 22, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Lu, Y.-W.; Xu, P.-H.; Long, Y.; Wu, W.-M.; Li, W.; Wang, R. Caffeic acid phenethyl ester and its related compounds limit the functional alterations of the isolated mouse brain and liver mitochondria submitted to in vitro anoxia–reoxygenation: Relationship to their antioxidant activities. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2008, 1780, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhao, L.; Ma, Z.; Holtzman, D.M.; Yan, C.; Dodel, R.C.; Hampel, H.; Oertel, W.; Farlow, M.R.; Du, Y. Caffeic acid phenethyl ester prevents neonatal hypoxic–ischaemic brain injury. Brain 2004, 127, 2629–2635. [Google Scholar] [CrossRef] [PubMed]
- Aladag, M.A.; Turkoz, Y.; Ozcan, C.; Sahna, E.; Parlakpınar, H.; Akpolat, N.; Cigremis, Y. Caffeic acid phenethyl ester (CAPE) attenuates cerebral vasospasm after experimental subarachnoidal haemorrhage by increasing brain nitric oxide levels. Int. J. Dev. Neurosci. 2006, 24, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Palaz, M.N.; Akcay, E. The Impact of Propolis Factor Caffeic Acid Phenethyl-Ester on the Cerebral Vasospasm and Early Brain Damage in the Exparimentally Induced Subarachnoid Hemorrhage on Rats. World Neurosurg. 2020, 138, e736–e742. [Google Scholar] [CrossRef]
- Ilhan, A.; Koltuksuz, U.; Ozen, S.; Uz, E.; Ciralik, H.; Akyol, O. The effects of caffeic acid phenethyl ester (CAPE) on spinal cord ischemia/reperfusion injury in rabbits. Eur. J. Cardio-Thoracic Surg. 1999, 16, 458–463. [Google Scholar] [CrossRef]
- Ak, H.; Gulsen, I.; Karaaslan, T.; Alaca, İ.; Candan, A.; Koçak, H.; Atalay, T.; Çelikbilek, A.; Demir, İ.; Yılmaz, T. The effects of caffeic acid phenethyl ester on inflammatory cytokines after acute spinal cord injury. Ulus Travma Acil Cerrahi Derg 2015, 21, 96–101. [Google Scholar] [CrossRef][Green Version]
- Aydin, H.E.; Ozkara, E.; Ozbek, Z.; Vural, M.; Burukoglu, D.; Arslantas, A.; Atasoy, M.A. Histopathological evaluation of the effects of CAPE in experimental spinal cord injury. Turk. Neurosurg. 2016, 26, 437–444. [Google Scholar]
- Akgun, B.; Ozturk, S.; Artas, G.; Erol, F.S. Effects of intrathecal caffeic acid phenethyl ester (CAPE) on IL-6 and TNF-α levels and local inflammatory responses in spinal cord injuries. Turk. Neurosurg. 2018, 28, 625–629. [Google Scholar] [CrossRef]
- Gocmez, C.; Celik, F.; Kamasak, K.; Kaplan, M.; Uzar, E.; Arıkanoglu, A.; Evliyaoglu, O. Effects of intrathecal caffeic acid phenethyl ester and methylprednisolone on oxidant/antioxidant status in traumatic spinal cord injuries. J. Neurol. Surg. Part A Central Eur. Neurosurg. 2015, 76, 20–24. [Google Scholar]
- Kasai, M.; Fukumitsu, H.; Soumiya, H.; Furukawa, S. Caffeic acid phenethyl ester reduces spinal cord injury-evoked locomotor dysfunction. Biomed. Res. 2011, 32, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nasution, R.A.; Islam, A.A.; Hatta, M. Decreased neutrophil levels in mice with traumatic brain injury after cape administration. Ann. Med. Surg. 2020, 54, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Pati, S.; Redell, J.B.; Zhang, M.; Moore, A.N.; Dash, P.K. Caffeic acid phenethyl ester protects blood–brain barrier integrity and reduces contusion volume in rodent models of traumatic brain injury. J. Neurotrauma 2012, 29, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Kerman, M.; Kanter, M.; Coşkun, K.K.; Erboga, M.; Gurel, A. Neuroprotective effects of caffeic acid phenethyl ester on experimental traumatic brain injury in rats. J. Mol. Histol. 2012, 43, 49–57. [Google Scholar] [CrossRef]
- Khan, M.; Shunmugavel, A.; Dhammu, T.S.; Khan, H.; Singh, I.; Singh, A.K. Combined treatment with GSNO and CAPE accelerates functional recovery via additive antioxidant activities in a mouse model of TBI. J. Neurosci. Res. 2018, 96, 1900–1913. [Google Scholar] [CrossRef]
- National Cancer Institute (Cancer Stat Facts: Brain and Other Nervous System Cancer). Available online: https://seer.cancer.gov/statfacts/html/brain.html (accessed on 3 November 2020).
- Choi, K.; Han, Y.-H.; Choi, C. N-Acetyl cysteine and caffeic acid phenethyl ester sensitize astrocytoma cells to Fas-mediated cell death in a redox-dependent manner. Cancer Lett. 2007, 257, 79–86. [Google Scholar] [CrossRef]
- Park, M.H.; Kang, D.W.; Jung, Y.; Choi, K.-Y. Caffeic acid phenethyl ester downregulates phospholipase D1 via direct binding and inhibition of NFκB transactivation. Biochem. Biophys. Res. Commun. 2013, 442, 1–7. [Google Scholar] [CrossRef]
- Morin, P.; St-Coeur, P.-D.; Doiron, J.A.; Cormier, M.; Poitras, J.J.; Surette, M.E.; Touaibia, M. Substituted caffeic and ferulic acid phenethyl esters: Synthesis, leukotrienes biosynthesis inhibition, and cytotoxic activity. Molecules 2017, 22, 1124. [Google Scholar] [CrossRef]
- Demestre, M.; Messerli, S.; Celli, N.; Shahhossini, M.; Kluwe, L.; Mautner, V.; Maruta, H. CAPE (caffeic acid phenethyl ester)-based propolis extract (Bio 30) suppresses the growth of human neurofibromatosis (NF) tumor xenografts in mice. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Product Deriv. 2009, 23, 226–230. [Google Scholar] [CrossRef]
- Wu, B.; Hao, Y.; Chen, Y.; Liu, Q.; Tian, C.; Zhang, Z.; Liu, J.; Wang, X. Studies on the structure-activity relationship of caffeate derivatives as neuroprotective agents. J. Chin. Pharm. Sci. 2019, 28, 615–626. [Google Scholar] [CrossRef]
- Shi, H.; Xie, D.; Yang, R.; Cheng, Y. Synthesis of caffeic acid phenethyl ester derivatives, and their cytoprotective and neuritogenic activities in PC12 cells. J. Agric. Food Chem. 2014, 62, 5046–5053. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wang, H.; Ren, X.; Yu, H.; Lei, Y.; Chen, Q. Neuroprotective effect of three caffeic acid derivatives via ameliorate oxidative stress and enhance PKA/CREB signaling pathway. Behav. Brain Res. 2017, 328, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Wan, T.; Wang, Z.; Luo, Y.; Zhang, Y.; He, W.; Mei, Y.; Xue, J.; Li, M.; Pan, H.; Li, W. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Protects against Oxidative Stress-Mediated Neuronal Cell Apoptosis and Scopolamine-Induced Cognitive Impairment by Activating Nrf2/HO-1 Signaling. Oxidative Med. Cell. Longev. 2019, 2019, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Guo, Y.; Ma, X.; Zhu, R.; Tian, C.; Zhang, Z.; Wang, X.; Ma, Z.; Liu, J. Design, synthesis and pharmacological evaluation of (E)-3, 4-dihydroxy styryl sulfonamides derivatives as multifunctional neuroprotective agents against oxidative and inflammatory injury. Bioorganic Med. Chem. 2013, 21, 5589–5597. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.-Y.; Huang, B.-R.; Yeh, W.-L.; Lin, H.-Y.; Huang, S.-S.; Liu, Y.-S.; Kuo, Y.-H. Anti-neuroinflammatory effect of a novel caffeamide derivative, KS370G, in microglial cells. Mol. Neurobiol. 2013, 48, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Mohamed, T.; Shakeri, A.; Rao, P.P.; Martínez-González, L.; Pérez, D.I.; Martínez, A.; Valente, M.J.o.; Garrido, J.; Uriarte, E. Development of blood–brain barrier permeable nitrocatechol-based catechol O-methyltransferase inhibitors with reduced potential for hepatotoxicity. J. Med. Chem. 2016, 59, 7584–7597. [Google Scholar] [CrossRef]
- Silva, T.; Mohamed, T.; Shakeri, A.; Rao, P.P.; da Silva, P.S.; Remião, F.; Borges, F. Repurposing nitrocatechols: 5-Nitro-α-cyanocarboxamide derivatives of caffeic acid and caffeic acid phenethyl ester effectively inhibit aggregation of tau-derived hexapeptide AcPHF6. Eur. J. Med. Chem. 2019, 167, 146–152. [Google Scholar] [CrossRef]


| Neurotoxic Substance | Parameters Measured | CAPE Dose | Animal/Cell Used | Ref. |
|---|---|---|---|---|
| MTX: 20 mg/kg, i.p., single dose | -Histopathological assessment -ADA (−) -NO (−) | 10 µmol/kg/day, i.p., for 7 days | Male rats | [42] |
| MTX: 20 mg/kg, i.p., single dose on day 2 | -MDA (−) -SOD (−) -CAT (−) | 10 µmol/kg/day, i.p., for 7 days | Male rats (cerebellum) | [43] |
| MTX: 20 mg/kg, i.p., single dose on day 2. | -MDA (−) -SOD (−) -GSH-Px (−) -CAT (−) | 10 μmol/kg/day, i.p., for 7 days | Wistar male rats (spinal cord, sciatic nerve, brain stem) | [44] |
| IFOS: 300 and 500 mg/kg, i.p., two doses | -Carbonyl content (−) -CAT -MDA (−) -Caspase-3 (−) | 10 µmol/kg/days, i.p., for 2 days, starting 1 day before injection of IFOS | Wistar male rats | [45] |
| Cisplatin: 5 and 32 μM | -Cell viability (+) -Neurite outgrowth (+) -GAP-43 (+) -Synapsin I (+) -Synaptophysin (+) | 1, 5, 10, 25, 50, and 100 μM for 24 h | -PC12 cells -SH-SY5Y cells | [46] |
| Cisplatin: 5 μM | -Protein content (+) -Glucose uptake (+) -Glutamate uptake -ROS (−) -F-Actin (+) -β-III-Tubulin (+) -SIRT 1 (+) -AMPK α and pAMPK α (+) | 10 μM | -PC12 cells -transfected COS-7 cells -transfected HEK cells -glial cells | [47] |
| Doxorubicin: 20 mg/kg i.p., single dose | -MDA (−) -NO (−) -GSH-Px -CAT (+) -SOD | 10 μmol/kg/day, i.p., for 12 days starting 2 days before doxorubicin | Male Sprague Dawley rats | [48] |
| Cigarette smoke: 1 h daily for 4 weeks | -MDA (−) -SOD (+) -Apoptosis (−) | 10 mmol/kg/day, i.p., for 4 weeks before the exposure to cigarette smoke | Rabbits | [49] |
| Ethanol: 3 mg/kg, oral | -TOS (−) -TAS (=) -Histopathological assessment | 10 μmol/kg, i.p., immediately after ehanol administration | Rats | [50] |
| Acrolein: 1 M | -Cell viability (+) -ROS (−) -GSH (+) -MAPKs -Akt/GSK3 -α/β-secretase | 0-90 μM, pretreatment for 30 min | HT22 mouse hippocampal cells | [51] |
| K2CrO4: 2 mg/kg/day, i.p., for 30 days | -SOCS3, JAK2 and STAT3 -NO (−) -GSH (+) -SOD (+) -GSH-Px -AChE -TNF-α (−) -IL-6 (−) | 20 mg/kg/day, orally, for 30 days | Wistar male rats | [52] |
| CdCl2: 1.5 mg/kg | -Neurobehavioural assessment -Histopathological assessment -AMPK and pAMPK -SIRT1 -Bcl-2 (+) -Bax (−) -Caspase-3 (−) -p-Tau -TLR4 -IL-6 (−) -IL1-β (−) -TNF-α (−) | 10 μmol/kg/ day, for 4 weeks | 7 weeks old Kunming mice | [53] |
| Chlorpyriphos: 10 mg/kg, oral | -AChE (−) -TOS (−) -TAR -Histopathological assessment -Caspase-3 -Bcl-2 -Bax | 10 μmol/kg, i.p., immediately after chlorpyriphos admnistration | Wistar rats | [54] |
| INH and ETM: 50 mg/kg/day, orally, for 30 days | -Histopathological assessment -MDA (−) -TOS (−) -TAC (+) -SOD (+) -PON-1 (+) -NO (−) | 10 mol/kg/day, i.p., for 30 days | Male Sprague-Dawley rats | [55] |
| Sevoflurane: (2.9%) for 6 h at day 7 | -Caspase-3, -8 and -9 (−) -Bax (−) -Bcl-2 (+) -Bcl-xL (+) -Bad (−) -MAPK (−) -JNK -ERK -PI3K (−) | 10, 20 or 40 mg/kg, from postnatal day 1 to day15 | Rat pups | [56] |
| Stimulus | Parameters Measured | CAPE Dose | Animal Used | Ref. |
|---|---|---|---|---|
| Bilateral CCA occlusion (20 min) then reperfusion (20 min) | -ADA -XO -SOD -GSH-Px -CAT -NO -MDA (-) | 10 µmol/kg, i.p., 10 min. after placing the occlusive vascular clamps | Sprague–Dawley rats | [57] |
| Cerebral infarction: right MCA occlusion and bilateral CCA clipping, 60 min | -NO (+) -Histopathological assessment | 0.01, 0.1, 1 and 10 µg/kg, i.v. 15 min before MCA occlusion | Male Long–Evans rats | [58] |
| MCA occlusion, 20 or 90 min | -Histopathological assessment -Neurological assessment -TNF-α (−) -IL-1β (−) -iNOS (−) -ED1 (−) -Bcl-xL (+) -Caspase-3 (−) -NO (+) -TBARS -GSH (+) -NF-κB (−) -MDA (−) -ICAM-1 (−) -E-selectin (−) -Nitrotyrosine (−) | 1–10 mg/kg, i.v., either at or after reperfusion | Male Sprague–Dawley rats | [59] |
| Right permanent MCA occlusion | -Histopathological assessment -Neurological assessment -MDA (−) -GSH (+) -CAT (−) -NO (+) -XO (−) | 10 µmol/kg/day, i.p., after occlusion for 7 days | Male New Zealand rabbits | [60] |
| Permanent MCA occlusion | -Serum S-100B (−) | 10 µg/kg/day, i.p., for 7 days after occlusion | Male New Zealand rabbits | [61] |
| MCA occlusion (60 min), followed by 24 h reperfusion | -Structural changes | 50 µM/kg, i.p., once before occlusion | Wistar rats | [62] |
| Cortical ischaemia: skull irradiation with cold light laser in combination with systemic administration of rose bengal | -Histopathological assessment -TNF-α (−) -HIF-1α (−) -MCP-1 (−) -IDO (−) -HO-1(+) -IL-1α (−) -IL-10 (+) | 0.5–5 mg/kg, i.p., 1 and 6 h after ischaemic insult | Male C57BL/6 mice | [63] |
| Anoxia-reoxygenation | -Mitochondrial oxygen consumption -Mitochondrial anisotropy -Mitochondrial TBARS -Mitochondrial protein concentrations -Protein carbonylation (−) -CL and Cytochrome c release (−) | 10–10−5 μM before the anoxia or just at reoxygenation | Male Kunming mice | [64] |
| CCA ligation followed by exposure to hypoxia | -i- and nNOS (−) -Cytochrome c (−) -Caspase-1 and -3 (−) | 40 mg/kg/day, 4 hrs before and/or after the stimulus | 7-day-old Sprague–Dawley rats | [65] |
| SAH | -MDA (−) -GSH (+) -NO (+) -Histopathological assessment | 10 µmol/kg, i.p., twice daily for 5 days after SAH | 15-week-old male Wistar rats | [66] |
| SAH | -Histopathological assessment | 10 mg/kg/day, twice a day, for 3 days starting 6 h after SAH. | Wistar rats | [67] |
| Aortic occlusion, 21 min | -MDA (−) -SOD -CAT -Histopathological assessment -Neurologic assessment | 10 µmol/kg, i.p. 30 min before the stimulus | New Zealand rabbits | [68] |
| Stimulus/Injury | Parameters Measured | CAPE Dose | Animal/Cell Used | Ref. |
|---|---|---|---|---|
| SCI: aneurysm clip | -IL-1β (−) -TNF-α (−) -Histopathological parameters | 10 μg/kg, i.p., 30 min after trauma | Male Wistar rats | [69] |
| Paraplegia: epidural clip application for 60 s. | Histopathological parameters | 10 μmol/kg, i.p. | Female Sprague-Dawley rats | [70] |
| SCI: Yasargil aneurysm clips | -IL-6 (−) -TNF-α (=) -Histopathological parameters | 1 μg/kg, following SCI induction | Female Wistar rats | [71] |
| SCI: Yasargil aneurysm | -MDA (−) -TOA (−) -TAC (+) -SOD (−) -GSH-Px | 1 μg/kg, single dose | Female Wistar rats | [72] |
| SCI: hemitransection | -Locomotor function -Histopathological parameters -IL-1β (−) -iNOS (−) -COX-2 (−) | 2 or 10 μmol/kg/day, i.p., for 28 days | Female Wistar rats | [73] |
| Head trauma with marmarou model | MPO activity (−) | 10 mg/kg, i.p., 24 h before trauma and 30 min after trauma and every day for 7 days | Male Sprague mice | [74] |
| Brain trauma: CCI injury model | -Blood–brain barrier (BBB) integrity (+) -Claudin-5 expression (+) -Neurobehavioural assessment | 10 mg/kg, i.p., -30 min following injury and/or daily for the next 4 days | -Male Sprague-Dawley rats -C57BL/6 mice | [75] |
| TBI: using cranial impact to the skull from a height of 7 cm at a point just in front of the coronal suture and over the right hemisphere. | -MDA (−) -SOD (+) -GSH-Px (+) -CAT (=) -Histological examinations -Caspase- 3 | 10 μmol /kg/i.p., single dose15 min after trauma | Male Sprague–Dawley rats | [76] |
| TBI: focal CCI technique | -Neurobehavioural parameters -Histopathological parameters -AMPK and pAMPK (+) -Fission (Drp1 and Fis 1) and fusion (Opa1)-associated proteins (+) -Mitochondrial factor PGC1α -HO-1 -MnSOD (+) | 5 mg/kg, plus GSNO 0.05 mg/kg, 2 h after CCI, i.v. and then daily orally | Young adult male wild type C57BL/6 mice | [77] |
| Stimulus/Tumor | Parameters Measured | CAPE Dose | Ref. |
|---|---|---|---|
| Human astrocytoma (CRT-MG cells) | -Cell viability (−) -ROS (−) -Caspase-3 and -8 (+) -NOX4 -DEVDase activity | 0–25 µg/mL pre-treatment for 1 h | [79] |
| Malignant brain tumor: human U87MG glioma cells | -PLD (−) -PLD1 (−) -PLD2 -NF-κB-binding motif (−) -α-tubulin -MMP-2 (−) -Invasion assay (−) -Gelatin zymography | 10, 20 µM for 24 h | [80] |
| -HEK293 cells stably co-transfected with a pcDNA3.1 vector expressing 5-LO and a pBUDCE4.1 vector expressing 5-LO activating protein -Human glioma cells: Hs683 and LN319. | -Cell viability (−) -Molecular Docking -5-LO activity (−) | -1 μM, pre-incubation -10 µM, for 3 days | [81] |
| Tumor xenografts in nu/nu mice: SC injection of NF1-deficient MPNST (S-462) cells or NF2-deficient Schwannoma (HEI-193) cells | -Cell viability (−) -Tumor size (−) | Bio 30 (a CAPE-rich extract), 100–300 mg/kg, i.p., twice a week | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balaha, M.; De Filippis, B.; Cataldi, A.; di Giacomo, V. CAPE and Neuroprotection: A Review. Biomolecules 2021, 11, 176. https://doi.org/10.3390/biom11020176
Balaha M, De Filippis B, Cataldi A, di Giacomo V. CAPE and Neuroprotection: A Review. Biomolecules. 2021; 11(2):176. https://doi.org/10.3390/biom11020176
Chicago/Turabian StyleBalaha, Marwa, Barbara De Filippis, Amelia Cataldi, and Viviana di Giacomo. 2021. "CAPE and Neuroprotection: A Review" Biomolecules 11, no. 2: 176. https://doi.org/10.3390/biom11020176
APA StyleBalaha, M., De Filippis, B., Cataldi, A., & di Giacomo, V. (2021). CAPE and Neuroprotection: A Review. Biomolecules, 11(2), 176. https://doi.org/10.3390/biom11020176