
Genome-Wide DNA Methylation Analysis of a Cohort of 41 Patients Affected by Oculo-Auriculo-Vertebral Spectrum (OAVS)

 , , , , , ,
, , , , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. OAVS Patients and Controls
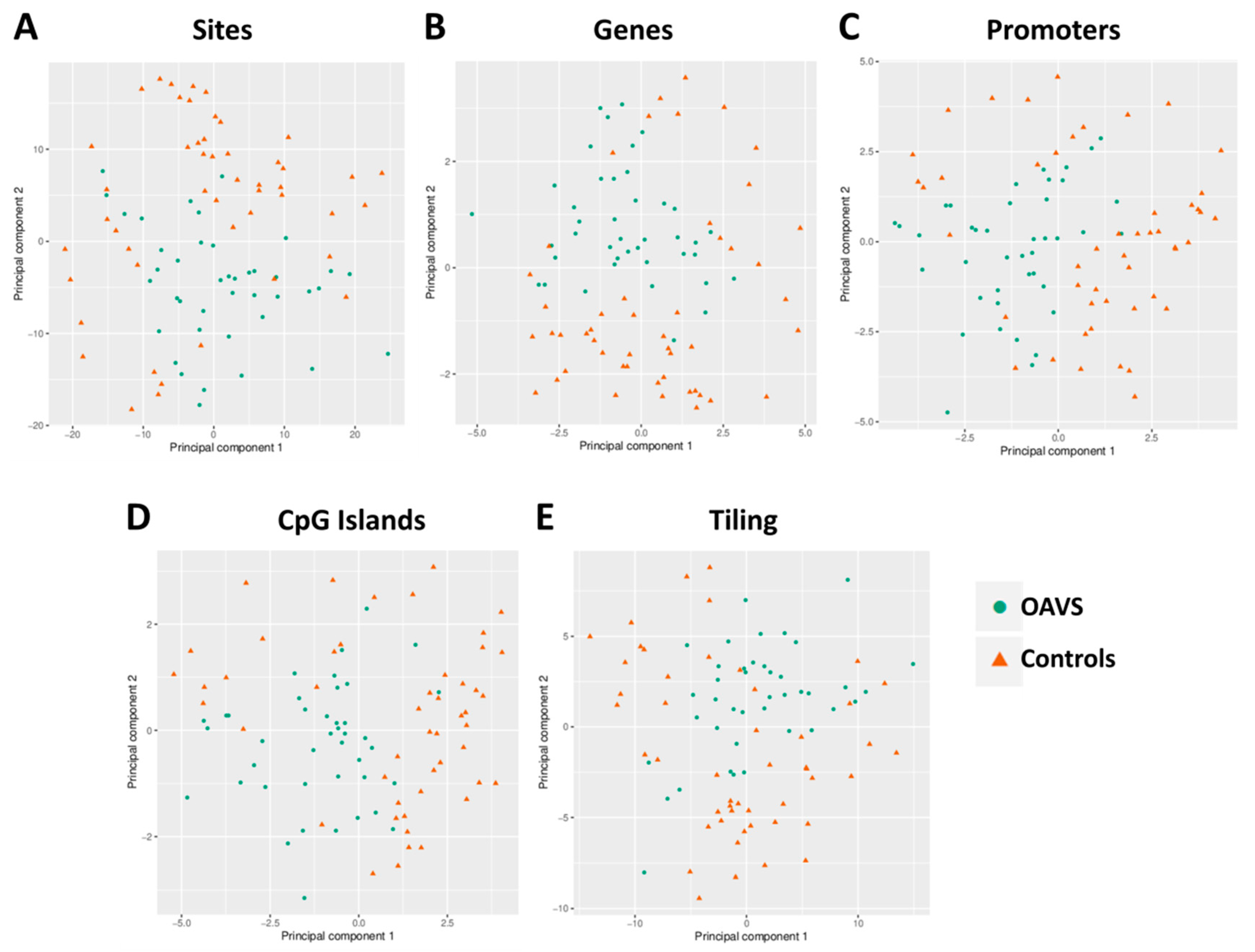
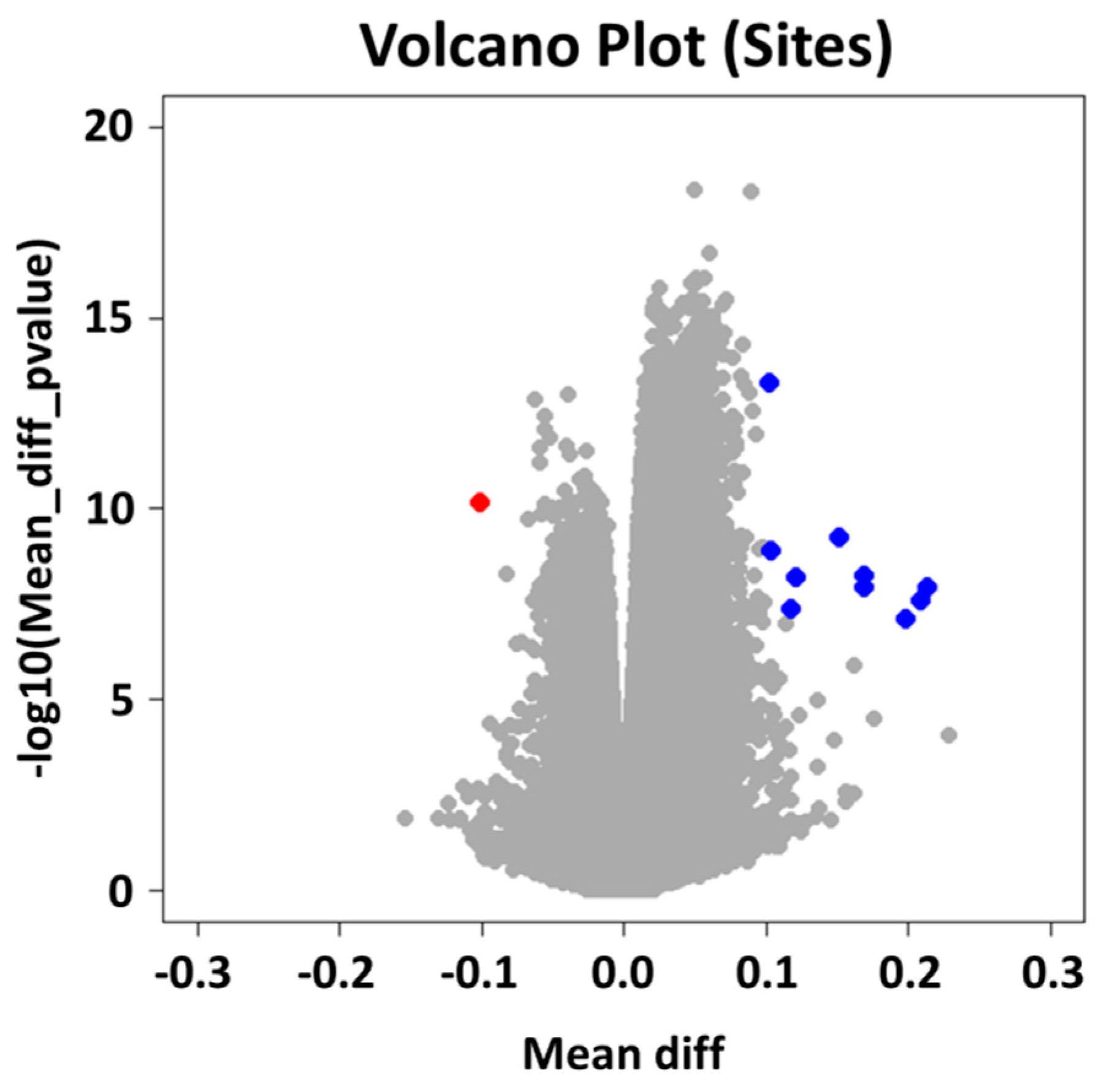
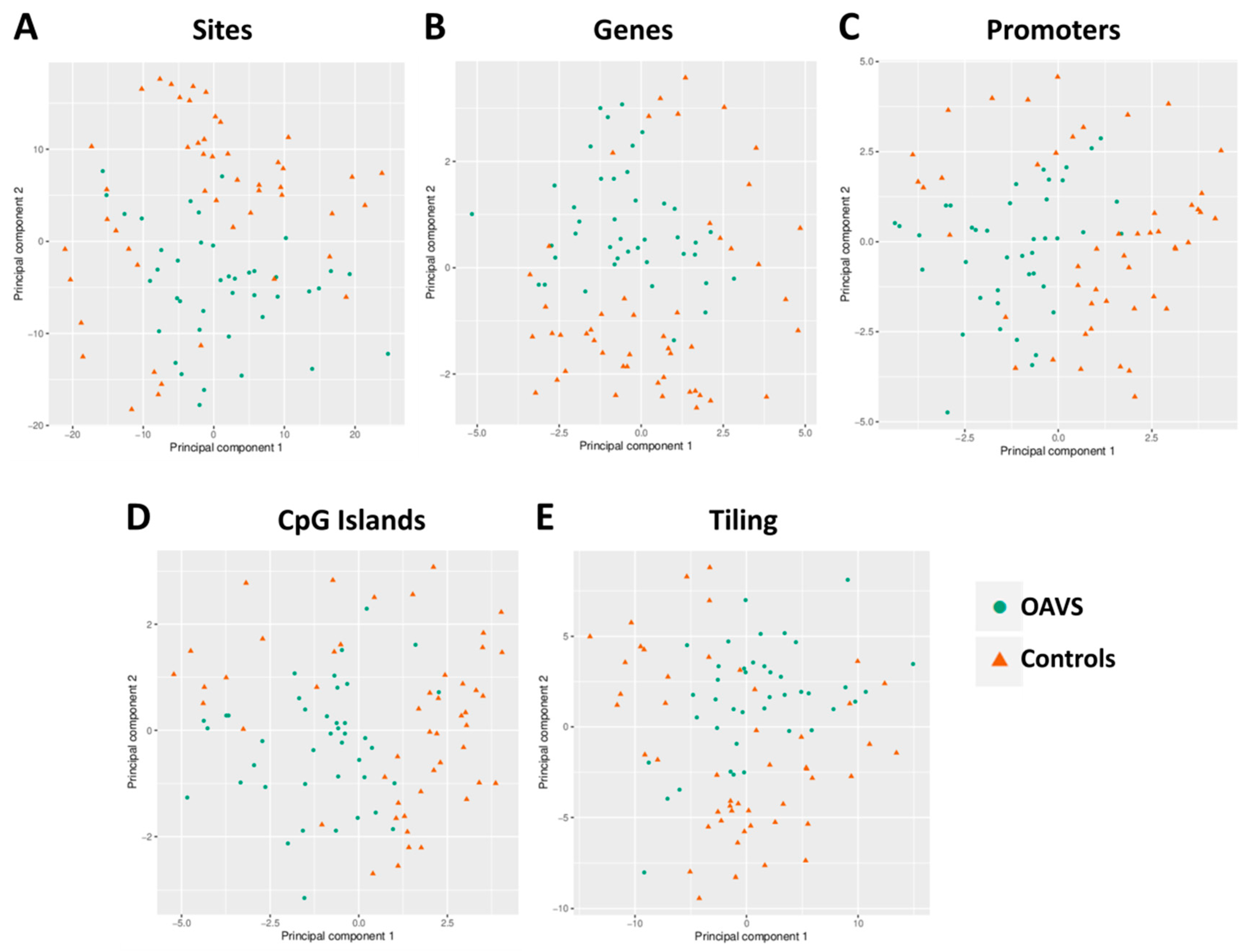
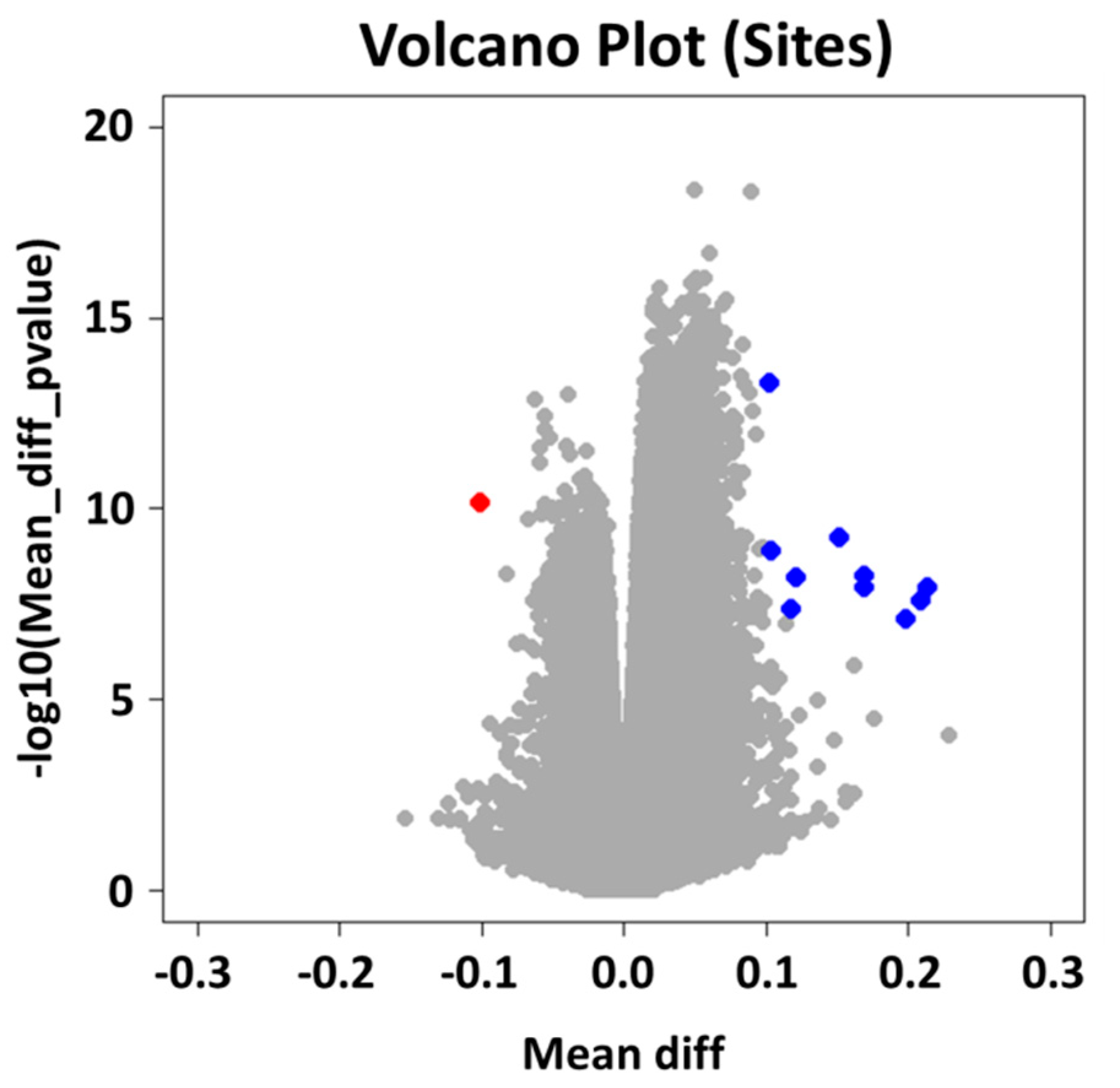
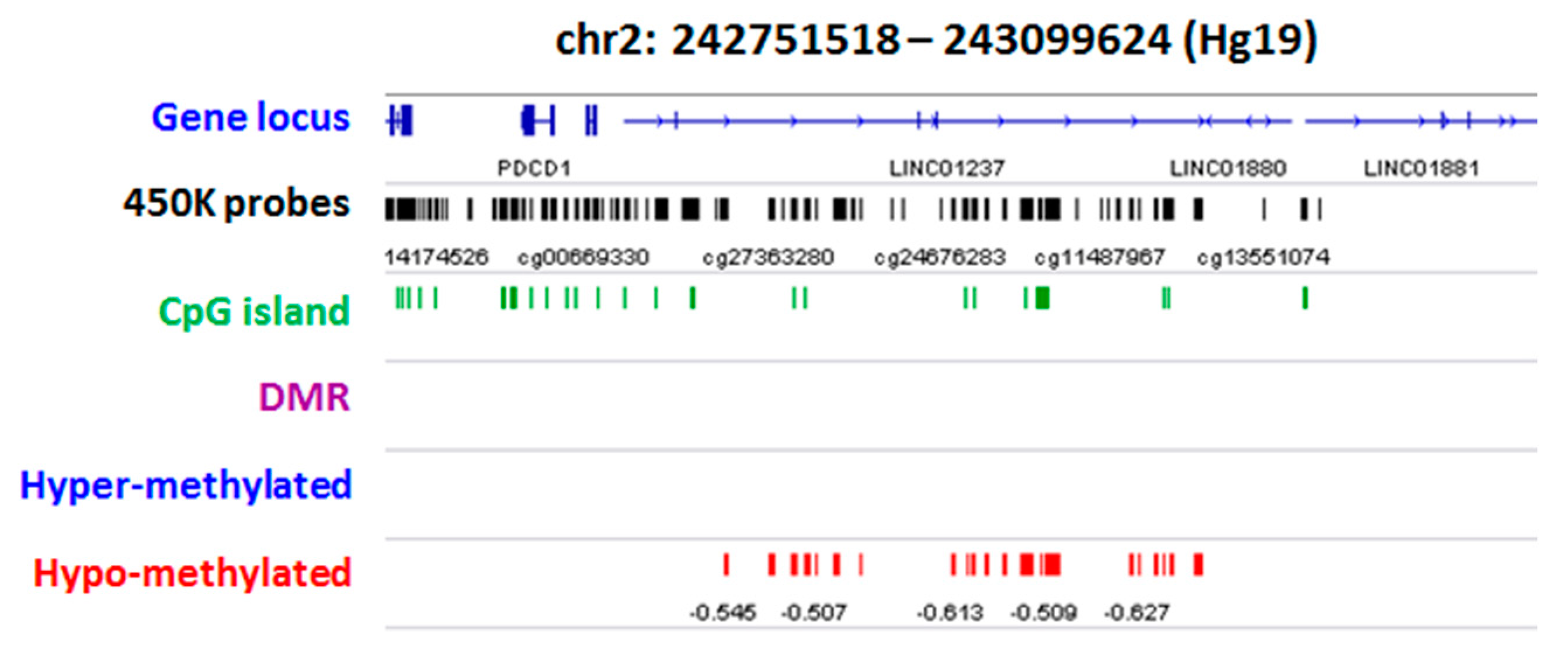
2.2. Group Level: Differential Methylation Analysis
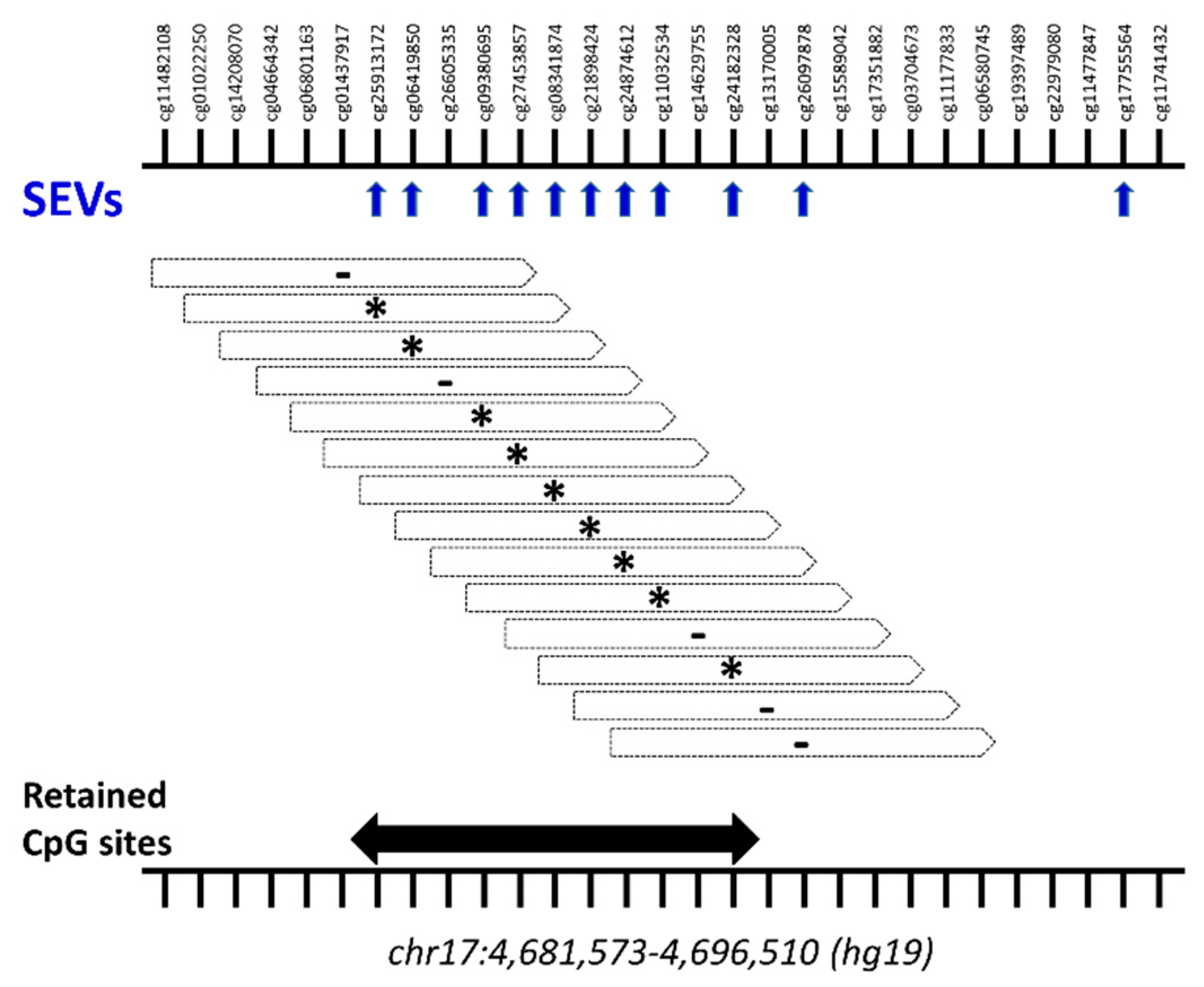
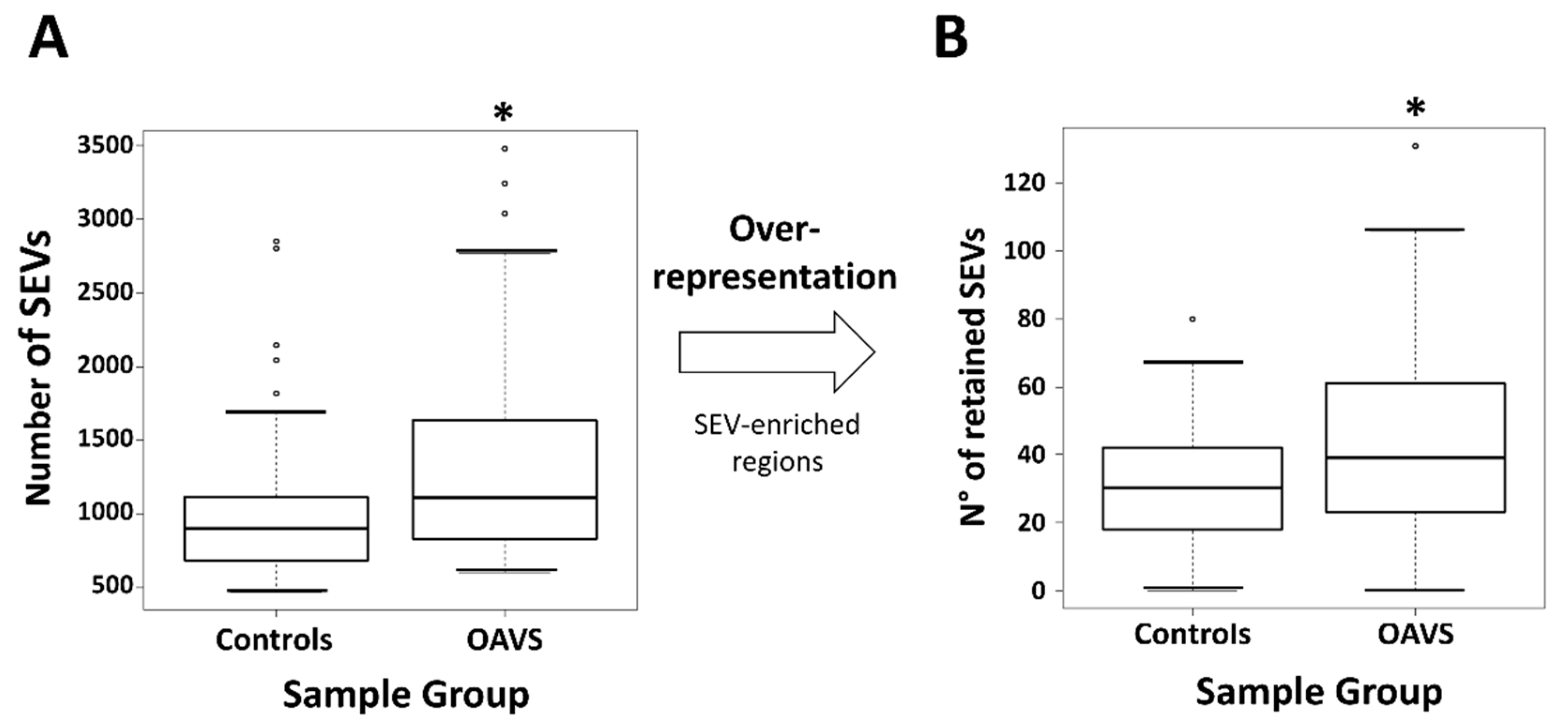
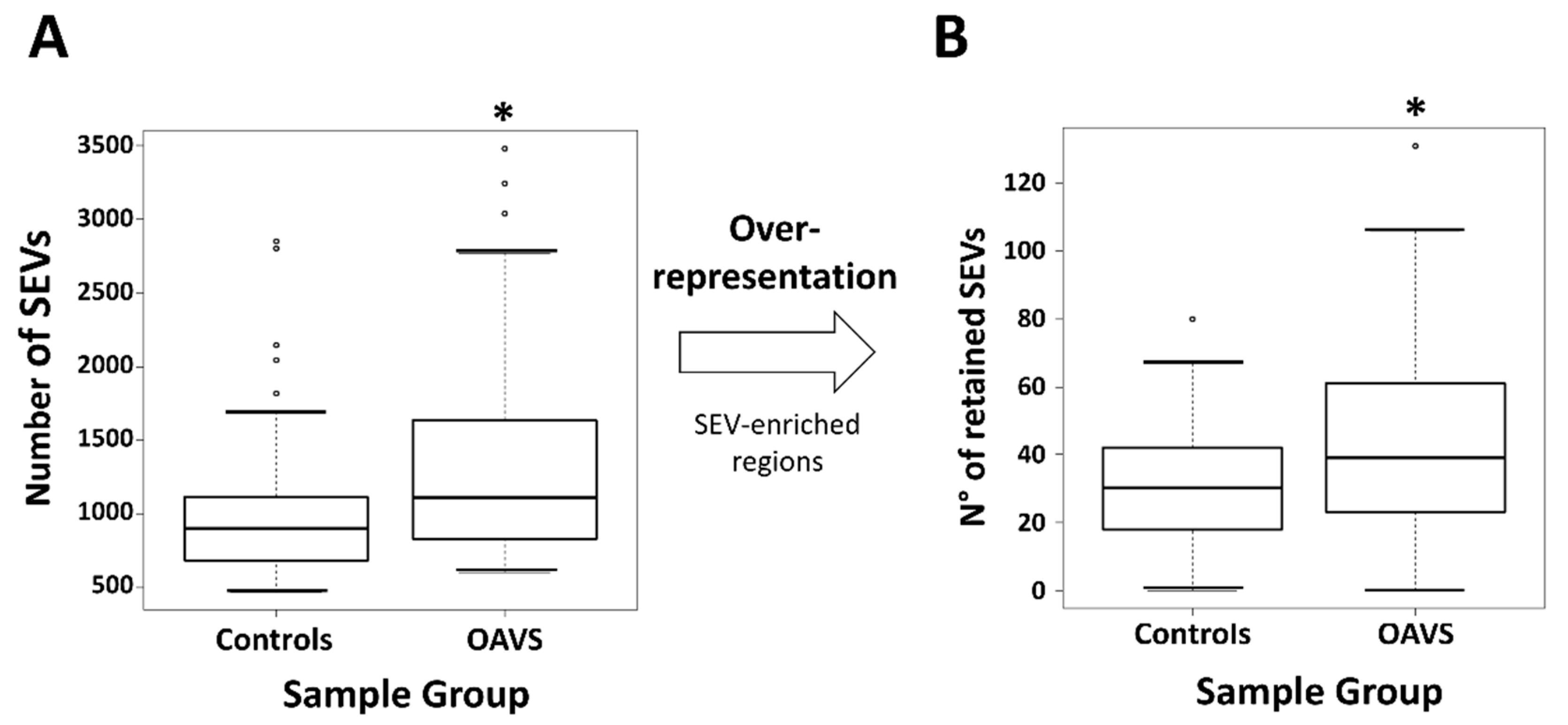
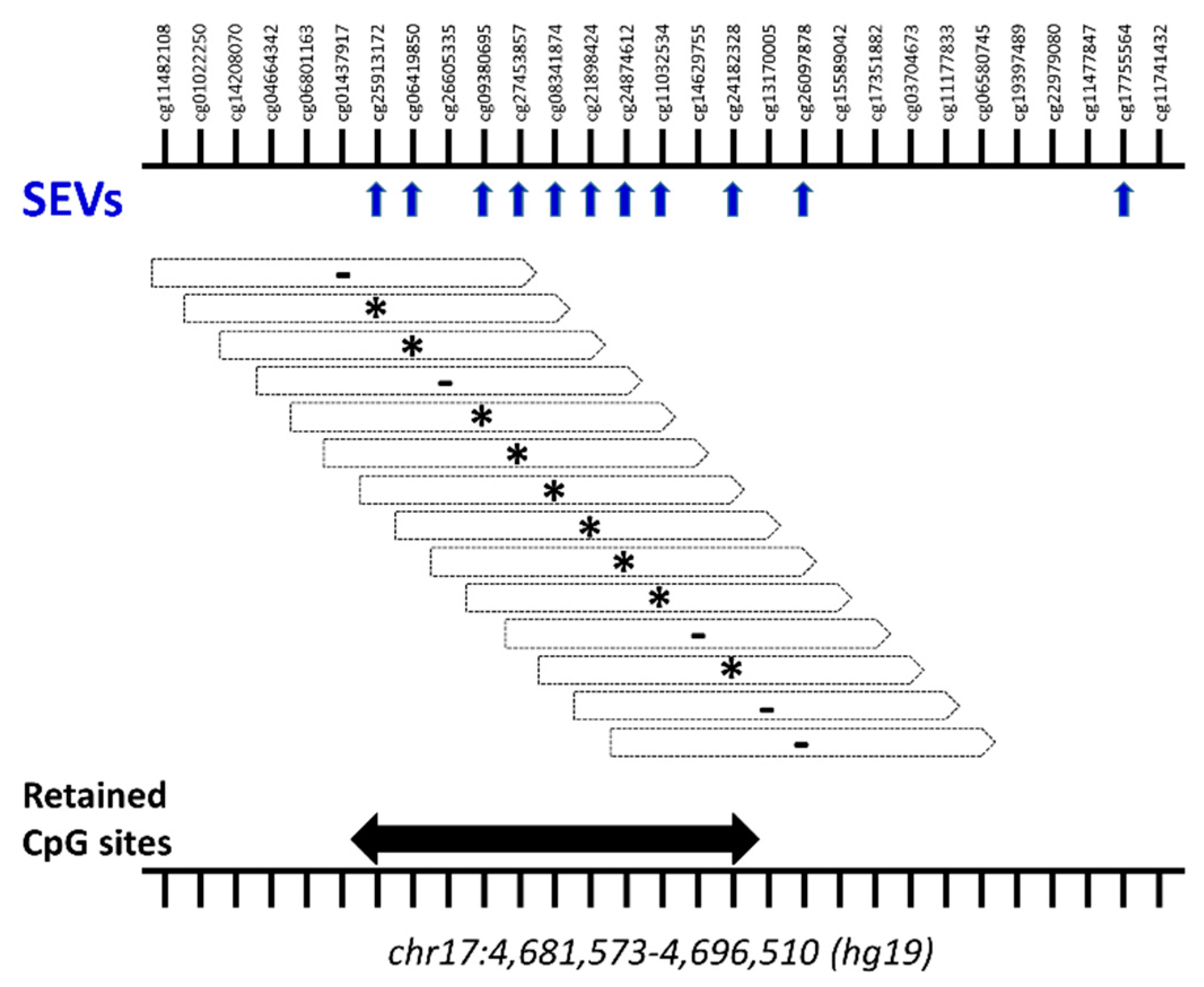
2.3. Differential Methylation at Single Case Level: Stochastic Epigenetic Variants (SEV)
Annotation and Prioritization Analysis
3. Discussion
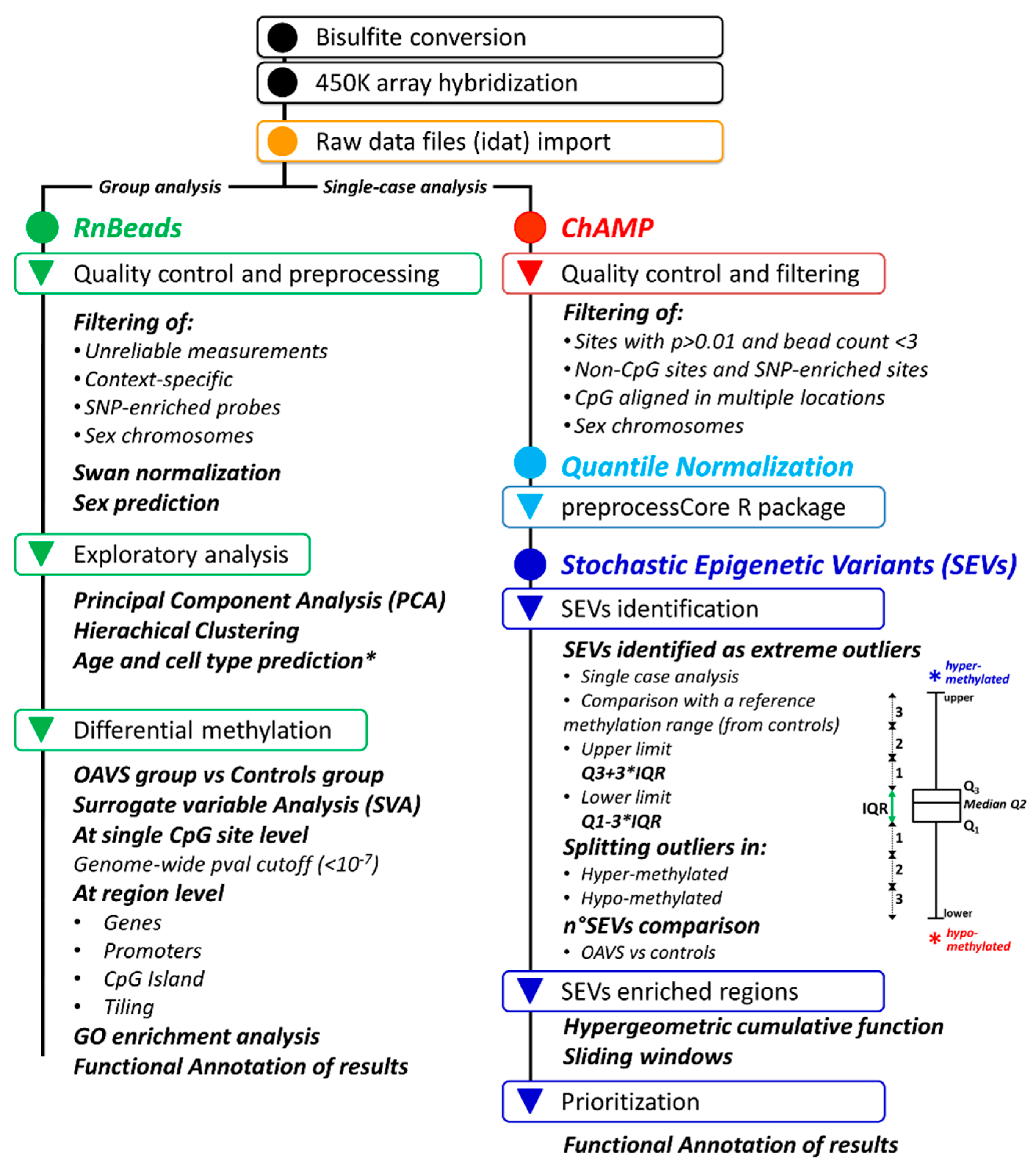
4. Materials and Methods
4.1. Study Design and Population
4.2. DNA Extraction
4.3. Bisulfite Conversion
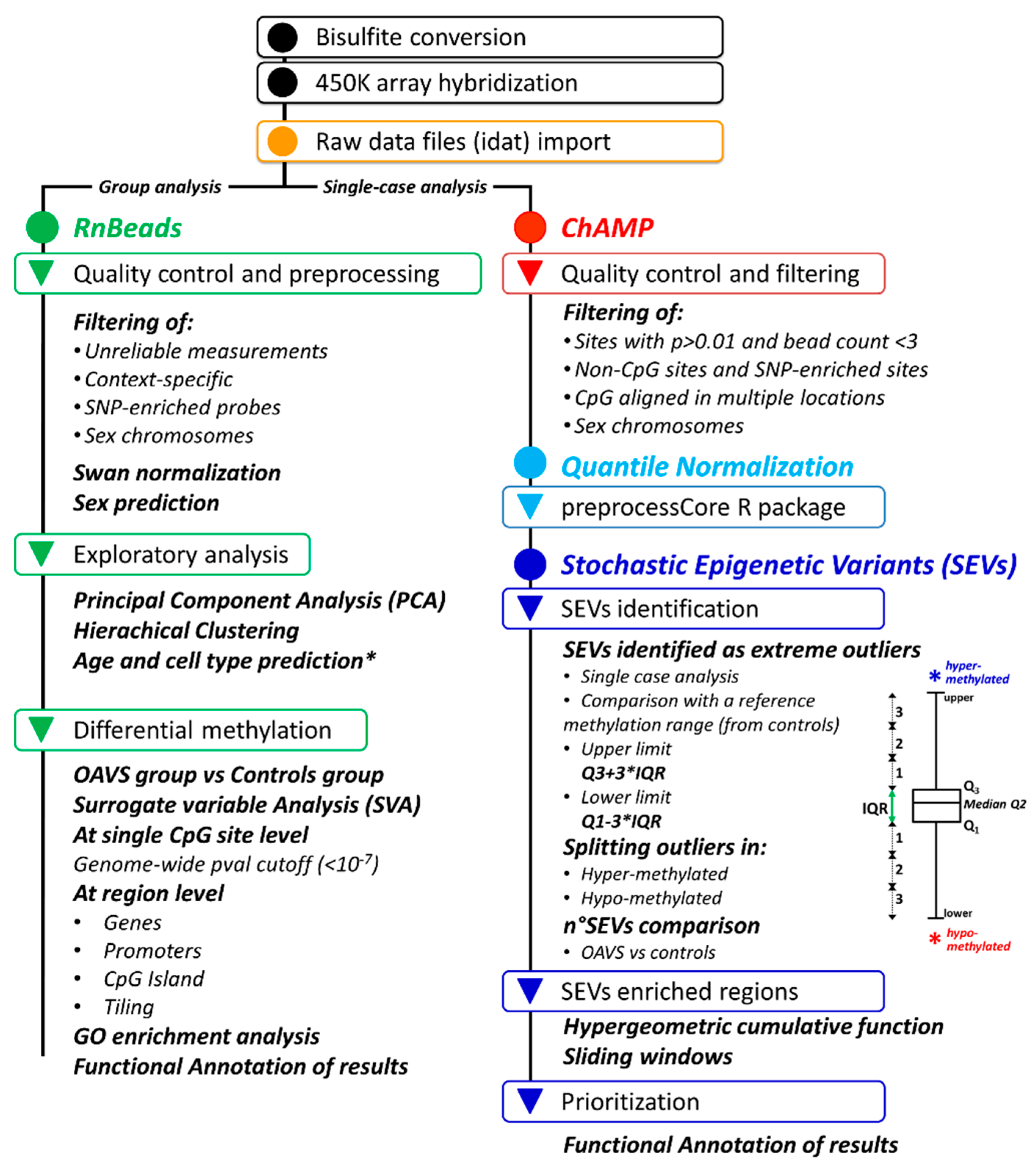
4.4. Genome-Wide Methylation Analysis
4.4.1. Differential Methylation Analysis at the Group Level
4.4.2. Stochastic Epigenetic Variants (SEVs)
4.5. DNA Methylation Age and Cell Types
4.6. Statistics
4.7. Data Visualization
5. Data Repository
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| OAVS | Oculo-Auriculo-Vertebral-Spectrum |
| SEV | Stochastic Epigenetic Variant |
| PCA | Principal Component Analysis |
| IQR | Interquartile Range |
| SVA | Surrogate Variable Analysis |
Appendix A
Appendix B
References
- Barisic, I.; Odak, L.; Loane, M.; Garne, E.; Wellesley, D.; Calzolari, E.; Dolk, H.; Addor, M.-C.; Arriola, L.; Bergman, J.E.H.; et al. Prevalence, prenatal diagnosis and clinical features of oculo-auriculo-vertebral spectrum: A registry-based study in Europe. Eur. J. Hum. Genet. 2014, 22, 1026–1033. [Google Scholar] [CrossRef]
- Alasti, F.; Van Camp, G. Genetics of microtia and associated syndromes. J. Med. Genet. 2009, 46, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Beleza-Meireles, A.; Hart, R.; Clayton-Smith, J.; De Oliveira, R.; Reis, C.F.; Venâncio, M.; Ramos, F.; Sá, J.; Ramos, L.; Cunha, E.; et al. Oculo-auriculo-vertebral spectrum: Clinical and molecular analysis of 51 patients. Eur. J. Med. Genet. 2015, 58, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J.; Cohen, M.M.; Hennekam, R.C.M. Syndromes of the Head and Neck, 4th ed.; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Gorlin, R.J.; Jue, K.L.; Jacobsen, U.; Goldschmidt, E. Oculoauriculovertebral dysplasia. J. Pediatr. 1963, 63, 991–999. [Google Scholar] [CrossRef]
- Rooryck, C.; Souakri, N.; Cailley, D.; Bouron, J.; Goizet, C.; Delrue, M.-A.; Marlin, S.; Lacombe, F.D.; Arveiler, B. Array-CGH analysis of a cohort of 86 patients with oculoauriculovertebral spectrum. Am. J. Med. Genet. Part A 2010, 152A, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Tasse, C.; Böhringer, S.; Fischer, S.; Lüdecke, H.-J.; Albrecht, B.; Horn, D.; Janecke, A.; Kling, R.; König, R.; Lorenz, B.; et al. Oculo-auriculo-vertebral spectrum (OAVS): Clinical evaluation and severity scoring of 53 patients and proposal for a new classification. Eur. J. Med. Genet. 2005, 48, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ballesta-Martínez, M.J.; López-González, V.; Dulcet, L.A.; Rodríguez-Santiago, B.; Garcia-Miñaúr, S.; Guillén-Navarro, E. Autosomal dominant oculoauriculovertebral spectrum and 14q23.1 microduplication. Am. J. Med. Genet. Part A 2013, 161, 2030–2035. [Google Scholar] [CrossRef]
- Lammer, E.J.; Chen, D.T.; Hoar, R.M.; Agnish, N.D.; Benke, P.J.; Braun, J.T.; Curry, C.J.; Fernhoff, P.M.; Grix, A.W.; Lott, I.T.; et al. Retinoic Acid Embryopathy. N. Engl. J. Med. 1985, 313, 837–841. [Google Scholar] [CrossRef]
- Wang, R.Y.; Martínez-Frías, M.L.; Graham, J.M. Infants of diabetic mothers are at increased risk for the oculo-auriculo-vertebral sequence: A case-based and case-control approach. J. Pediatr. 2002, 141, 611–617. [Google Scholar] [CrossRef]
- Kaye, C.I.; Martin, A.O.; Rollnick, B.R.; Rollnick, R.; Nagatoshi, K.; Israel, J.; Hermanoff, M.; Tropea, B.; Richtsmeier, J.T.; Morton, N.E. Oculoauriculovertebral anomaly: Segregation analysis. Am. J. Med. Genet. 1992, 43, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Tasse, C.; Majewski, F.; Böhringer, S.; Fischer, S.; Lüdecke, H.-J.; Gillessen-Kaesbach, G.; Wieczorek, D. A family with autosomal dominant oculo-auriculo-vertebral spectrum. Clin. Dysmorphol. 2007, 16, 1–7. [Google Scholar] [CrossRef]
- Vendramini-Pittoli, S.; Kokitsu-Nakata, N.M. Oculoauriculovertebral spectrum: Report of nine familial cases with evidence of autosomal dominant inheritance and review of the literature. Clin. Dysmorphol. 2009, 18, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kelberman, D.; Tyson, J.; Chandler, D.C.; McInerney, A.M.; Slee, J.; Albert, D.; Aymat, A.; Botma, M.; Calvert, M.; Goldblatt, J.; et al. Hemifacial microsomia: Progress in understanding the genetic basis of a complex malformation syndrome. Qual. Life Res. 2001, 109, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Ala-Mello, S.; Siggberg, L.; Knuutila, S.; Von Koskull, H.; Taskinen, M.; Peippo, M. Further evidence for a relationship between the 5p15 chromosome region and the oculoauriculovertebral anomaly. Am. J. Med. Genet. Part A 2008, 146A, 2490–2494. [Google Scholar] [CrossRef] [PubMed]
- Bragagnolo, S.; Colovati, M.E.S.; Souza, M.Z.; Dantas, A.G.; De Soares, M.F.F.; Melaragno, M.I.; Perez, A.B. Clinical and cytogenomic findings in OAV spectrum. Am. J. Med. Genet. Part A 2018, 176, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Descartes, M. Oculoauriculovertebral spectrum with 5p15.33-pter deletion. Clin. Dysmorphol. 2006, 15, 153–154. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; McDonald-McGinn, D.M.; Heike, C.; Catania, C.; Dallapiccola, B.; Marino, B.; Zackai, E.H. Three patients with oculo-auriculo-vertebral spectrum and microdeletion 22q11.2. Am. J. Med. Genet. Part A 2009, 149A, 2860–2864. [Google Scholar] [CrossRef] [Green Version]
- Garavelli, L.; Virdis, R.; Donadio, A.; Sigorini, M.; Banchini, G.; Balestrazzi, P.; Fryns, J.P. Oculo-auriculo-vertebral spectrum in Klinefelter syndrome. Genet. Couns. (Geneva, Switzerland) 1999, 10, 321–324. [Google Scholar]
- Huang, X.-S.; Li, X.; Tan, C.; Xiao, L.; Jiang, H.-O.; Zhang, S.-F.; Wang, D.-M.; Zhang, J. Genome-Wide Scanning Reveals Complex Etiology of Oculo-Auriculo-Vertebral Spectrum. Tohoku J. Exp. Med. 2010, 222, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Ou, Z.; Martin, D.M.; Bedoyan, J.K.; Cooper, M.L.; Chinault, A.C.; Stankiewicz, P.; Cheung, S.W. Branchiootorenal syndrome and oculoauriculovertebral spectrum features associated with duplication ofSIX1,SIX6, andOTX2resulting from a complex chromosomal rearrangement. Am. J. Med. Genet. Part A 2008, 146A, 2480–2489. [Google Scholar] [CrossRef] [Green Version]
- Berenguer, M.; Tingaud-Sequeira, A.; Colovati, M.; Melaragno, M.I.; Bragagnolo, S.; Perez, A.B.A.; Arveiler, B.; Lacombe, D.; Rooryck, C. A novel de novo mutation in MYT1, the unique OAVS gene identified so far. Eur. J. Hum. Genet. 2017, 25, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, E.; Berenguer, M.; Tingaud-Sequeira, A.; Marlin, S.; Toutain, A.; Denoyelle, F.; Picard, A.; Charron, S.; Mathieu, G.; De Belvalet, H.; et al. Mutations inMYT1, encoding the myelin transcription factor 1, are a rare cause of OAVS. J. Med. Genet. 2016, 53, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Venugopalan, S.R.; Farrow, E.; Sanchez–Lara, P.A.; Yen, S.; Lypka, M.; Jiang, S.; Allareddy, V. A novel nonsense substitution identified in the AMIGO 2 gene in an Occulo-Auriculo-Vertebral spectrum patient. Orthod. Craniofacial Res. 2019, 22, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Tingaud-Sequeira, A.; Trimouille, A.; Marlin, S.; Lopez, E.; Berenguer, M.; Gherbi, S.; Arveiler, B.; Lacombe, D.; Rooryck, C. Functional and genetic analyses of ZYG11B provide evidences for its involvement in OAVS. Mol. Genet. Genom. Med. 2020, 8, e1375. [Google Scholar] [CrossRef] [PubMed]
- Boles, D.J.; Bodurtha, J.; Nance, W.E.; Reynolds, J.F. Goldenhar complex in discordant monozygotic twins: A case report and review of the literature. Am. J. Med. Genet. 1987, 28, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, S.; Silengo, M.; Ponzone, A.; Perugini, L. Goldenhar anomaly in one of triplets derived from in vitro fertilization. Am. J. Med. Genet. 1999, 84, 167–168. [Google Scholar] [CrossRef]
- Gittins, V.; Kasraie, J. Case report: Goldenhar syndrome following donor oocyte IVF. J. Assist. Reprod. Genet. 2010, 27, 561–563. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Lüdecke, H.-J.; Wieczorek, D.; Böhringer, S.; Gillessen-Kaesbach, G.; Horsthemke, B. Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Hum. Mol. Genet. 2006, 15, 581–587. [Google Scholar] [CrossRef] [Green Version]
- Tribioli, C.; Lufkin, T. The murine Bapx1 homeobox gene plays a critical role in embryonic development of the axial skeleton and spleen. Development 1999, 126, 5699–5711. [Google Scholar]
- Tucker, A.S.; Lumsden, A. Neural crest cells provide species-specific patterning information in the developing branchial skeleton. Evol. Dev. 2004, 6, 32–40. [Google Scholar] [CrossRef]
- Haberland, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009, 23, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef] [PubMed]
- Zamariolli, M.; Colovati, M.; Moyses-Oliveira, M.; Nunes, N.; Caires Dos Santos, L.; Alvarez Perez, A.B.; Bragagnolo, S.; Melaragno, M.I. Rare single-nucleotide variants in oculo-auriculo-vertebral spectrum (oavs). Mol. Genet. Genom. Med. 2019, 7, e00959. [Google Scholar] [CrossRef] [Green Version]
- Guida, V.; Sinibaldi, L.; Pagnoni, M.; Bernardini, L.; Loddo, S.; Margiotti, K.; Digilio, M.C.; Fadda, M.T.; Dallapiccola, B.; Iannetti, G.; et al. A de novo proximal 3q29 chromosome microduplication in a patient with oculo auriculo vertebral spectrum. Am. J. Med. Genet. Part A 2015, 167, 797–801. [Google Scholar] [CrossRef]
- Katsanis, S.H.; Jabs, E.W. Treacher collins syndrome. In Genereviews((r)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Splendore, A.; Passos-Bueno, M.; Jabs, E.W.; Van Maldergem, L.; Wulfsberg, E.A. TCOF1 mutations excluded from a role in other first and second branchial arch-related disorders. Am. J. Med. Genet. 2002, 111, 324–327. [Google Scholar] [CrossRef]
- Lalani, S.R.; Safiullah, A.M.; Fernbach, S.D.; Harutyunyan, K.G.; Thaller, C.; Peterson, L.E.; McPherson, J.D.; Gibbs, R.A.; White, L.D.; Hefner, M.; et al. Spectrum of CHD7 Mutations in 110 Individuals with CHARGE Syndrome and Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2006, 78, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Lalani, S.R.; Safiullah, A.M.; Molinari, L.M.; Fernbach, S.D.; Martin, D.M.; Belmont, J.W. SEMA3E mutation in a patient with CHARGE syndrome. J. Med. Genet. 2004, 41, e94. [Google Scholar] [CrossRef] [Green Version]
- Kohlhase, J. SALL1 mutations in Townes-Brocks syndrome and related disorders. Hum. Mutat. 2000, 16, 460–466. [Google Scholar] [CrossRef]
- Kochhar, A.; Fischer, S.M.; Kimberling, W.J.; Smith, R.J.H. Branchio-oto-renal syndrome. Am. J. Med. Genet. Part A 2007, 143A, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.H. Branchiootorenal spectrum disorder. In Genereviews((r)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Müller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Gentilini, D.; Garagnani, P.; Pisoni, S.; Bacalini, M.G.; Calzari, L.; Mari, D.; Vitale, G.; Franceschi, C.; Di Blasio, A.M. Stochastic epigenetic mutations (DNA methylation) increase exponentially in human aging and correlate with X chromosome inactivation skewing in females. Aging 2015, 7, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Gentilini, D.; Somigliana, E.; Pagliardini, L.; Rabellotti, E.; Garagnani, P.; Bernardinelli, L.; Papaleo, E.; Candiani, M.; Di Blasio, A.M.; Viganò, P. Multifactorial analysis of the stochastic epigenetic variability in cord blood confirmed an impact of common behavioral and environmental factors but not of in vitro conception. Clin. Epigenetics 2018, 10, 1–13. [Google Scholar] [CrossRef]
- Fiorito, G.; Polidoro, S.; Dugué, P.-A.; Kivimäki, M.; Ponzi, E.; Matullo, G.; Guarrera, S.; Assumma, M.B.; Georgiadis, P.; Kyrtopoulos, S.A.; et al. Social adversity and epigenetic aging: A multi-cohort study on socioeconomic differences in peripheral blood DNA methylation. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Spada, E.; Calzari, L.; Corsaro, L.; Fazia, T.; Mencarelli, M.; Di Blasio, A.M.; Bernardinelli, L.; Zangheri, G.; Vignali, M.; Gentilini, D. Epigenome Wide Association and Stochastic Epigenetic Mutation Analysis on Cord Blood of Preterm Birth. Int. J. Mol. Sci. 2020, 21, 5044. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. Identification and characterization of human ppfia4 gene in silico. Int. J. Mol. Med. 2003, 12, 1009–1014. [Google Scholar] [CrossRef]
- Siebold, C.; Yamashita, T.; Monnier, P.P.; Mueller, B.K.; Pasterkamp, R.J. RGMs: Structural Insights, Molecular Regulation, and Downstream Signaling. Trends Cell Biol. 2017, 27, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Kameda, Y. Hoxa3 and signaling molecules involved in aortic arch patterning and remodeling. Cell Tissue Res. 2009, 336, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nat. Cell Biol. 1991, 350, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Nishimaki, T.; Takeichi, M.; Chisaka, O. Homeobox Gene Hoxa3 Is Essential for the Formation of the Carotid Body in the Mouse Embryos. Dev. Biol. 2002, 247, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manley, N.R.; Capecchi, M.R. The role of Hoxa-3 in mouse thymus and thyroid development. Development 1995, 121, 1989–2003. [Google Scholar] [PubMed]
- Kessel, M. Respecification of vertebral identities by retinoic acid. Development 1992, 115, 487–501. [Google Scholar] [PubMed]
- Marshall, H.; Nonchev, S.; Sham, M.H.; Muchamore, I.; Lumsden, A.; Krumlauf, R. Retinoic acid alters hindbrain hox code and induces transformation of rhombomeres 2/3 into a 4/5 identity. Nature 1992, 360, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Roux, M.; Ryckebüsch, L.; Niederreither, K.; Dollé, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar] [CrossRef]
- Nassif, A.; Senussi, I.; Meary, F.; Loiodice, S.; Hotton, D.; Robert, B.; Bensidhoum, M.; Berdal, A.; Babajko, S. Msx1 role in craniofacial bone morphogenesis. Bone 2014, 66, 96–104. [Google Scholar] [CrossRef]
- Liang, J.; Hoff, J.V.D.; Lange, J.; Ren, Y.; Fan, M.; Kuijpers-Jagtman, A. MSX1 mutations and associated disease phenotypes: Genotype-phenotype relations. Eur. J. Hum. Genet. 2016, 24, 1663–1670. [Google Scholar] [CrossRef] [Green Version]
- Satokata, I.; Maas, R.L. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat. Genet. 1994, 6, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Catron, K.M.; Abate-Shen, C. A role for the Msx-1 homeodomain in transcriptional regulation: Residues in the N-terminal arm mediate TATA binding protein interaction and transcriptional repression. Proc. Natl. Acad. Sci. USA 1996, 93, 1764–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillet, P.; Oulad-Abdelghani, M.; Vicaire, S.; Garnier, J.-M.; Schuhbaur, B.; Dollé, P.; Chambon, P. Efficient Cloning of cDNAs of Retinoic Acid-Responsive Genes in P19 Embryonal Carcinoma Cells and Characterization of a Novel Mouse Gene, Stra1 (Mouse LERK-2/Eplg2). Dev. Biol. 1995, 170, 420–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, N.; Ragge, N.; Kariminejad, A.; Buffet, A.; Ghaderi-Sohi, S.; Martinovic, J.; Calvas, P. Mutation analysis of the STRA6 gene in isolated and non-isolated anophthalmia/microphthalmia. Clin. Genet. 2013, 83, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Turan, S.; Bastepe, M. GNAS Spectrum of Disorders. Curr. Osteoporos. Rep. 2015, 13, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldred, M.A.; Trembath, R.C. Activating and inactivating mutations in the human gnas1 gene. Hum. Mutat. 2000, 16, 183–189. [Google Scholar] [CrossRef]
- Le Norcy, E.; Reggio-Paquet, C.; De Kerdanet, M.; Mignot, B.; Rothenbuhler, A.; Chaussain, C.; Linglart, A. Dental and craniofacial features associated with GNAS loss of function mutations. Eur. J. Orthod. 2020, 42, 525–533. [Google Scholar] [CrossRef]
- Krebs, L.T.; Iwai, N.; Nonaka, S.; Welsh, I.C.; Lan, Y.; Jiang, R.; Saijoh, Y.; O’Brien, T.P.; Hamada, H.; Gridley, T. Notch signaling regulates left-right asymmetry determination by inducing nodal expression. Genes Dev. 2003, 17, 1207–1212. [Google Scholar] [CrossRef] [Green Version]
- Maksimovic, J.; Gordon, L.; Oshlack, A. Swan: Subset-quantile within array normalization for illumina infinium humanmethylation450 beadchips. Genome Biol. 2012, 13, R44. [Google Scholar] [CrossRef] [Green Version]
- Lalevée, S.; Anno, Y.N.; Chatagnon, A.; Samarut, E.; Poch, O.; Laudet, V.; Benoit, G.; Lecompte, O.; Rochette-Egly, C. Genome-wide in Silico Identification of New Conserved and Functional Retinoic Acid Receptor Response Elements (Direct Repeats Separated by 5 bp). J. Biol. Chem. 2011, 286, 33322–33334. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Storey, J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2006, 23, 257–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2016, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.; Bäcklin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.-L.; Flaegstad, T.; Forestier, E.; Frost, B.-M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OAVS cohort | N = 41 | |
| Sex | 19 females/22 males | |
| Age (years) | Median = 6 (first quartile = 3—third quartile = 14) | |
| Main anomalies | Our cohort | |
| Craniofacial | Hemifacial microsomia | 39/41 (95%) |
| Mandibular hypoplasia | 37/41 (90%) | |
| Cleft lip/palate | 8/41 (20%) | |
| Ear | Microtia/anotia | 41/41 (100%) |
| Asymmetric ears | ||
| Atresia of the external auditory canal | ||
| Dysplastic ears | ||
| Ocular | Microphthalmos | 24/41 (58%) |
| Coloboma upper eyelid | ||
| Epibulbar dermoid | ||
| Other eye anomalies | ||
| Vertebral | Hemivertebrae | 12/41 (29%) |
| Fusion | ||
| Scoliosis | ||
| Kyphosis | ||
| Other vertebral anomalies | ||
| Cardiovascular | 15/41 (37%) | |
| Genitourinary | 7/41 (17%) | |
| Brain | 7/41 (17%) | |
| Developmental delay | 9/41 (22%) | |
| Radial defects | 3/41 (7.3%) | |
| Other organs/systems | 8/41(19.5%) | |
| Locus | Number of Cases | OAVS Code (N° SEVs) |
|---|---|---|
| PPFIA4 | 6 | OAVS02 (5), OAVS08 (5), OAVS12 (5), OAVS23 (4), OAVS41 (4), OAVS45 (4) |
| ISOC2 | 3 | OAVS09 (5), OAVS34 (6), OAVS35 (6) |
| LLPH | 3 | OAVS04 (6), OAVS20 (5), OAVS26 (6) |
| RGMA | 3 | OAVS16 (10), OAVS27 (10), OAVS42 (6) |
| ADGB;LOC101928661 | 2 | OAVS14 (7), OAVS32 (4) |
| CATSPERE | 2 | OAVS33 (4), OAVS35 (3) |
| CCDC71L | 2 | OAVS14 (2), OAVS30 (2) |
| DDX60 | 2 | OAVS01 (6), OAVS19 (4) |
| FRG1;FRG2 | 2 | OAVS20 (7), OAVS28 (5) |
| GGCT | 2 | OAVS26 (6), OAVS33 (3) |
| LCLAT1 | 2 | OAVS02 (3), OAVS26 (2) |
| LINC01166 | 2 | OAVS26 (6), OAVS02 (3) |
| LINC01558 | 2 | OAVS30 (5), OAVS32 (4) |
| MIR596 | 2 | OAVS28 (6), OAVS03 (2) |
| NPY | 2 | OAVS30 (5), OAVS16 (3) |
| OXGR1 | 2 | OAVS40 (4), OAVS43 (3) |
| PLSCR1 | 2 | OAVS33 (8), OAVS02 (3) |
| RASSF6 | 2 | OAVS12 (3), OAVS24 (3) |
| SIX3;SIX2 | 2 | OAVS32 (5), OAVS40 (4) |
| TNFRSF9;PARK7 | 2 | OAVS16 (4), OAVS43 (4) |
| VMO1 | 2 | OAVS05 (9), OAVS20 (3) |
| ZNF814 | 2 | OAVS35 (7), OAVS21 (4) |
| Locus | Number of Cases | OAVS Code (N° SEVs) |
|---|---|---|
| UPP1 | 2 | OAVS27 (5), OAVS42 (5) |
| HOXA3 | 2 | OAVS16 (4), OAVS27 (4) |
| LOC100507547 | 2 | OAVS27 (6), OAVS16 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guida, V.; Calzari, L.; Fadda, M.T.; Piceci-Sparascio, F.; Digilio, M.C.; Bernardini, L.; Brancati, F.; Mattina, T.; Melis, D.; Forzano, F.; et al. Genome-Wide DNA Methylation Analysis of a Cohort of 41 Patients Affected by Oculo-Auriculo-Vertebral Spectrum (OAVS). Int. J. Mol. Sci. 2021, 22, 1190. https://doi.org/10.3390/ijms22031190
Guida V, Calzari L, Fadda MT, Piceci-Sparascio F, Digilio MC, Bernardini L, Brancati F, Mattina T, Melis D, Forzano F, et al. Genome-Wide DNA Methylation Analysis of a Cohort of 41 Patients Affected by Oculo-Auriculo-Vertebral Spectrum (OAVS). International Journal of Molecular Sciences. 2021; 22(3):1190. https://doi.org/10.3390/ijms22031190
Chicago/Turabian StyleGuida, Valentina, Luciano Calzari, Maria Teresa Fadda, Francesca Piceci-Sparascio, Maria Cristina Digilio, Laura Bernardini, Francesco Brancati, Teresa Mattina, Daniela Melis, Francesca Forzano, and et al. 2021. "Genome-Wide DNA Methylation Analysis of a Cohort of 41 Patients Affected by Oculo-Auriculo-Vertebral Spectrum (OAVS)" International Journal of Molecular Sciences 22, no. 3: 1190. https://doi.org/10.3390/ijms22031190