1. Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer deaths worldwide. According to the 2018 statistical report of global cancer burden (GLOBOCAN), HCC is the sixth for incidence and the fourth for mortality cancer, accounting for 841,080 new cases and 781,631 deaths per year worldwide [
1]. HCC lesions originate from chronic liver fibrosis and cirrhosis, which arise from repeated cycles of injury and repair. Tissue injuries originate from several sources including chronic viral hepatitis (hepatitis B and C), excessive alcohol intake, non-alcoholic fatty liver disease, aflatoxin exposure, tobacco smoking and diabetes [
2,
3].
With its consistent immune/inflammatory pathogenesis, HCC appears as a strong candidate for the application of immune-based therapies; however, this strategy has been shown to be only partially successful [
4]. Currently, the survival of HCC patients that are not eligible for curative therapy (i.e., resection, local ablation and liver transplantation) depends on their response to the less efficient systemic chemotherapy [
5,
6]. The refractoriness of HCC, not only to classical chemotherapy but also to targeted therapy, is still poorly understood; HCC progression and resistance can be affected by multiple biological processes such as epigenetic modulation [
7], immune microenvironment in the tumor site [
8] and mechanisms of chemoresistance (MOC) [
9]. Therefore, HCC patient stratification into homogeneous progression groups is critical for the identification of potential biological processes involved in cancer progression, which then form the bases for the selection of the most appropriate treatment or possibly shed new light on novel druggable biological targets.
Despite the extensive advancement in earlier diagnosis, therapy decision-making and interdisciplinary evaluation, the prognosis of HCC patients remains poor. Ongoing prognostic models integrate tumor node metastasis (TNM) staging, liver function, comorbidities and other parameters to predict HCC progression and prognosis [
10]; however, since HCC is a very heterogeneous disease, the prognostic performance of classical methods is still not satisfactory. Nowadays taking into account large scale omics data is becoming fundamental when establishing novel prognostic and predictive tools that can better represent a broader HCC scenario.
The use of whole gene expression and methylation analysis of tumors have proved that it is possible to highlight patterns and signatures related to prognosis, tumor classification and response to treatment [
11,
12]. Meanwhile, Machine Learning methods have been trained and applied into genomic data to discover new molecular signatures, interpret complex biological mechanisms and predict clinical outcomes from biomedical datasets [
13,
14]. Therefore, besides traditional clinical-pathological risk factor models, an efficient predictive model that can classify patients in different cancer progression groups is highly desirable. Moreover, the features used by this model for making predictions could be the bricks of an optimized prognostic model capable of stratifying patients in relation to cancer progression, eventually providing a more oriented therapy decision and an improved clinical management.
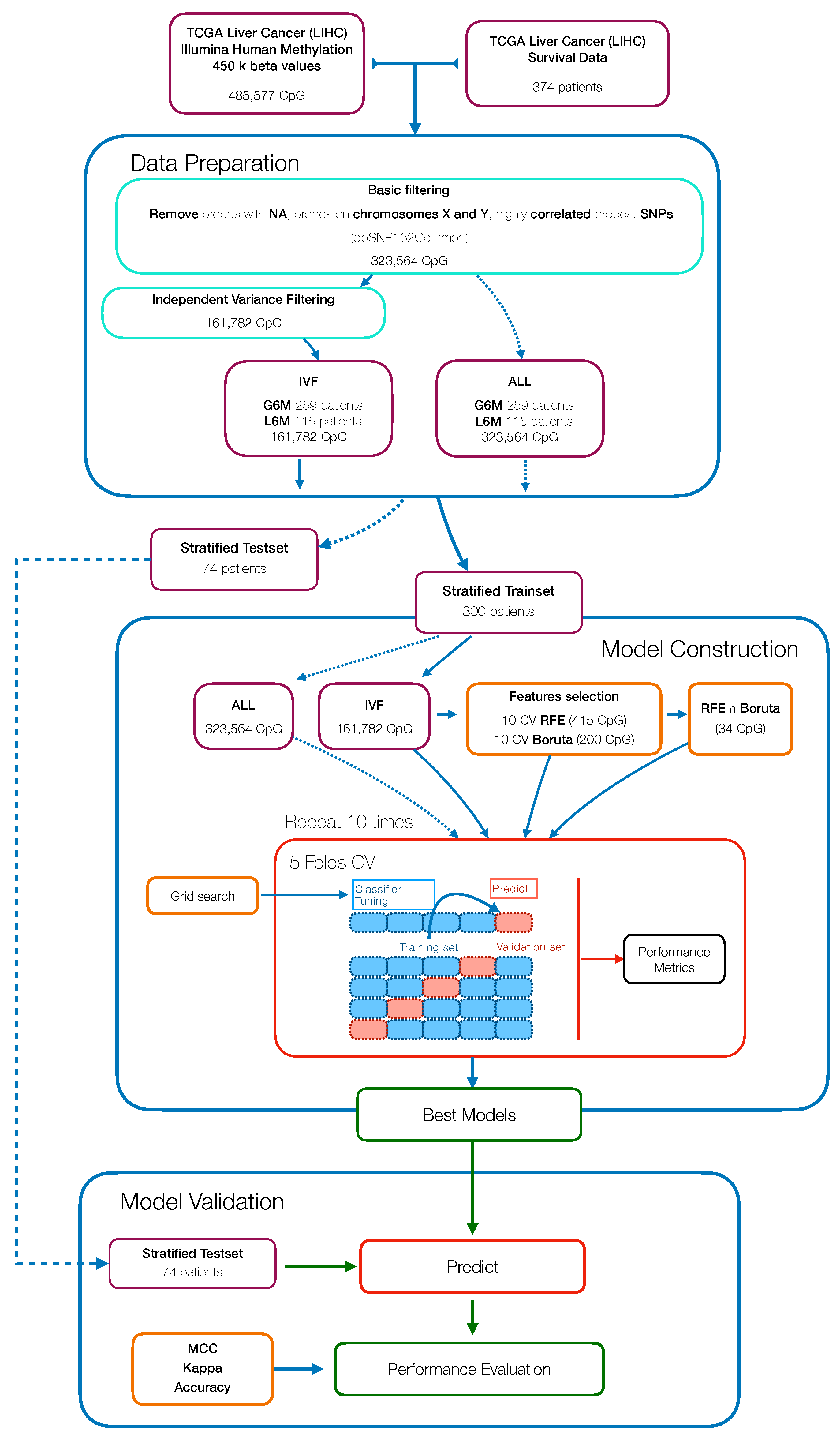
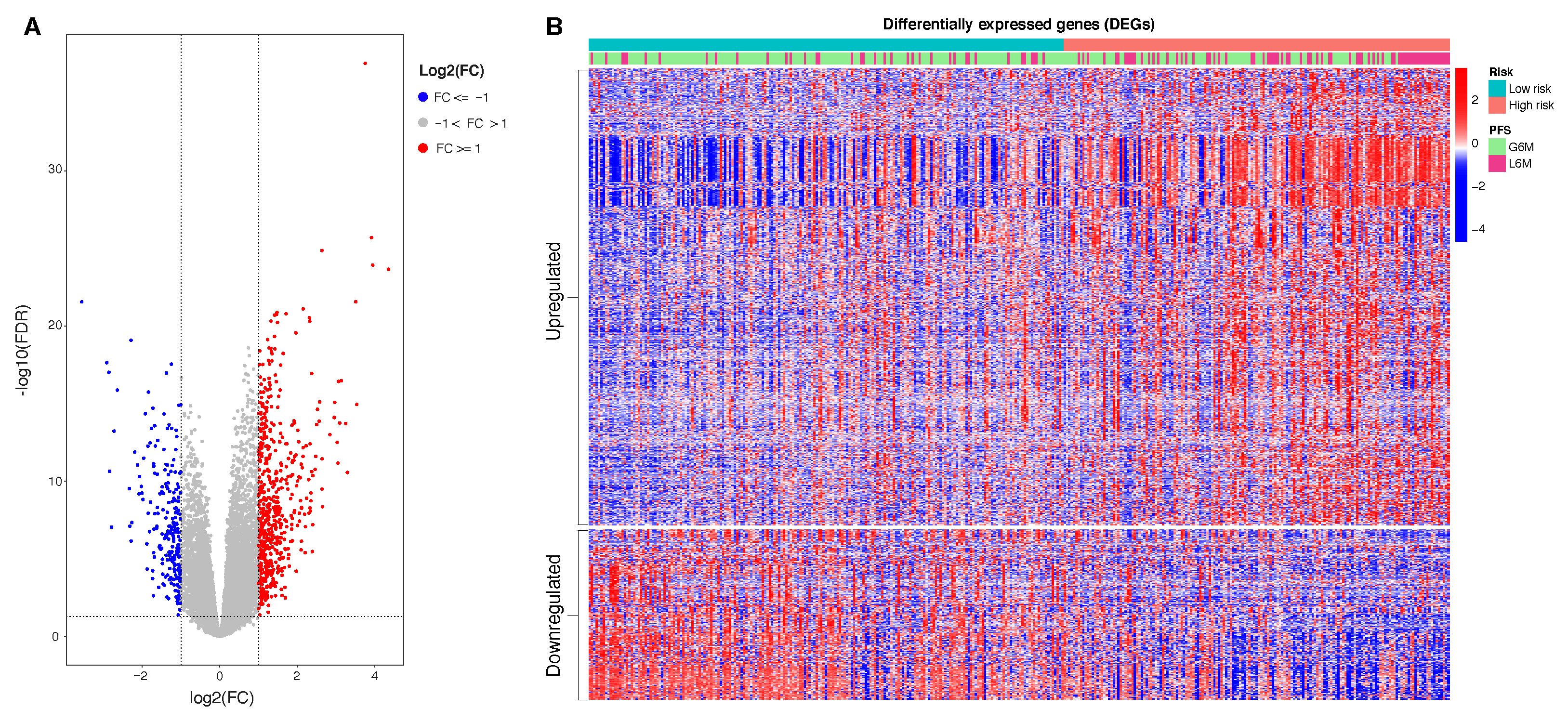
In this study, we aimed to build and evaluate a predictive model able to classify HCC patients with a progression-free survival (PFS) time greater or less than six months by using their methylation profiles. HCC patients were from a large dataset within the Liver Hepatocellular Carcinoma (LIHC) project of The Cancer Genome Atlas (TCGA). We initially used a Machine Learning algorithm (Random Forest) combined with different features selection algorithms to select the best prediction subset of methylation probes on cytosine-phosphate-guanine dinucleotides (CpGs), resulting in a final 34 CpGs-based model for PFS prediction. Then, starting from these final 34 markers, we performed a univariate Cox regression analysis to select PFS relevant CpG probes followed by the construction of a CpGs-based prognostic signature using a stepwise model selection. A four-CpGs-based risk model was successfully built, validated and used to stratify the patients in high risk and low risk for an early cancer progression. Finally, we conducted an analysis of differentially expressed genes (DEGs) followed by a functional enrichment analysis to gain more insights into the biological differences between high risk and low risk patients as well as into the processes involved. This prognostic signature could be useful in the HCC patient administration, by providing a stratification system that reliably separates patients with respect to the progression prognosis, ultimately impacting both therapy and clinical decision-making.
4. Discussion
HCC remains one of the most frequent malignancies and a leading challenge for public health worldwide. Even after the application of curative treatments, such as resection, local ablation and liver transplantation, and the extensive advancement in earlier diagnosis, staging systems and therapy decision-making, the long-term prognosis of HCC remains poor. The evaluation of conventional parameters such as proper liver function, vessel invasion, tumor staging and biomarker levels are commonly used in HCC prognosis prediction; nevertheless, their performance is still not satisfactory and this could be due to the high degree of heterogeneity among HCC cases. Therefore, the recognition of novel prognostic biomarkers from large scale omics data and the establishment of more accurate prognostic models could dynamically recapitulate the biological progression of HCC and may have a superior predictive accuracy than conventional ongoing parameters. Recently, numerous studies with gene expression, microRNAs and methylation profiling have shown great potential in prognosis prediction and staging systems of HCC [
50,
51,
52,
53,
54]. The main goal of this strategy is to stratify patients into homogeneous prognosis clusters, which then can provide the bases for the option of most appropriate interventions. Although several studies have been conducted, many perspectives still remain unexplored. A prediction model that can reliably classify HCC patients into homogeneous groups with respect to the PFS time can be of particular importance in the context of HCC where the disease progression is still not entirely definable by conventional prognosis parameters in the attempt to recapitulate prognosis and treatment response.
The present study aimed to identify efficient prognostic markers to stratify HCC patients according to their epigenetic features involved in tumor pathogenesis and progression. We applied a combination of Machine Learning algorithms that performed a supervised features selection (i.e., Boruta, RFE) on methylation data to increase the chances of catching CpG markers related to the PFS [
55,
56]. The subsets of probes obtained were used to train RF models to select the best predictive model resulting in a final 34 CpGs-based model.
One of the advantages of RF is the built-in variable importance measure that ranks the features with respect to their relevance for prediction. This is performed calculating the Gini Importance (Mean Decrease in Impurity—MDI) [
57] or the Permutation Importance (Mean Decrease in Accuracy—MDA) that can correct the Gini importance bias [
58]. However, techniques that estimate the variable importance are not able to capture patterns of dependency between features and response; they only represent the strength of this dependency as a single number, and the obtained results can be difficult to interpret [
59].
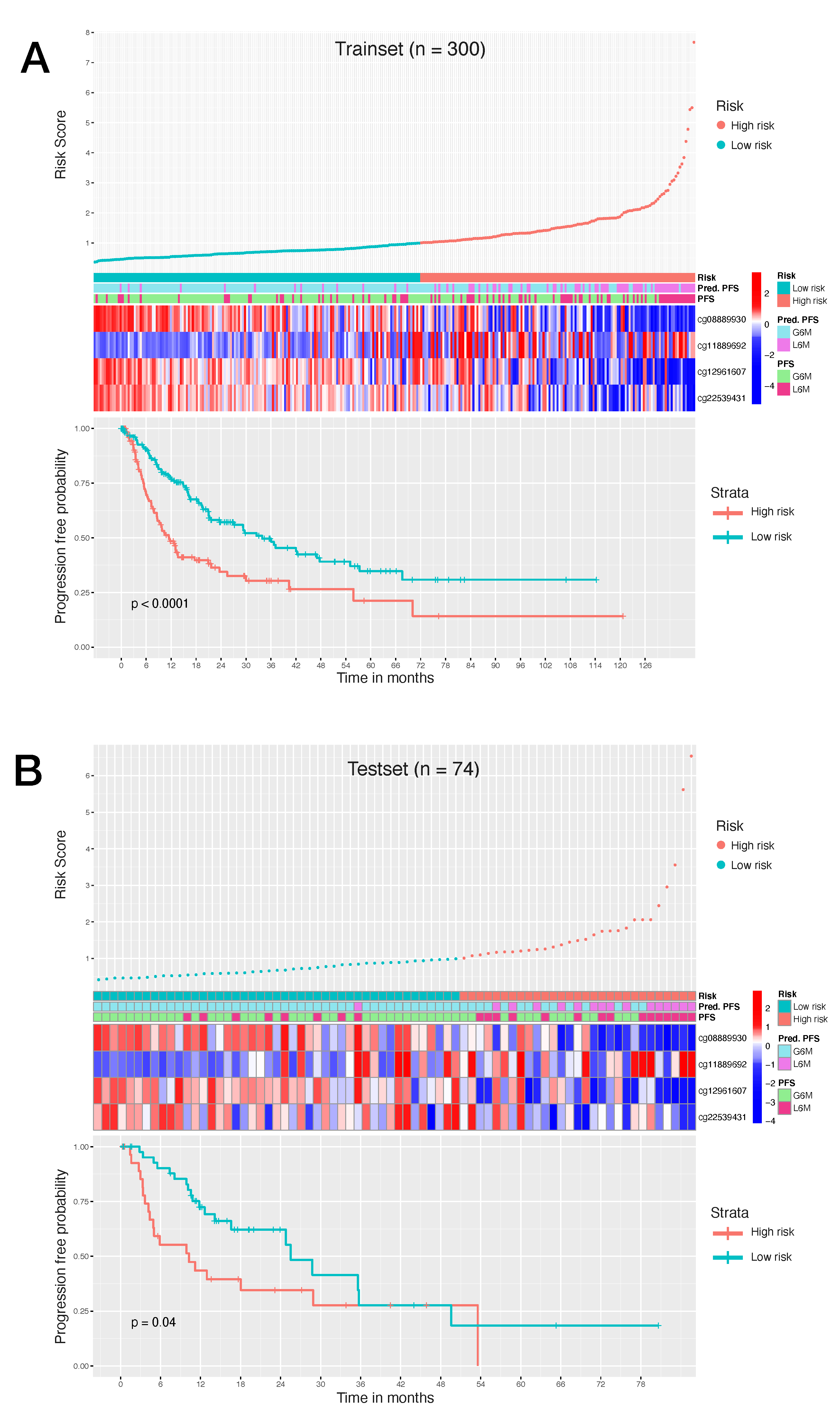
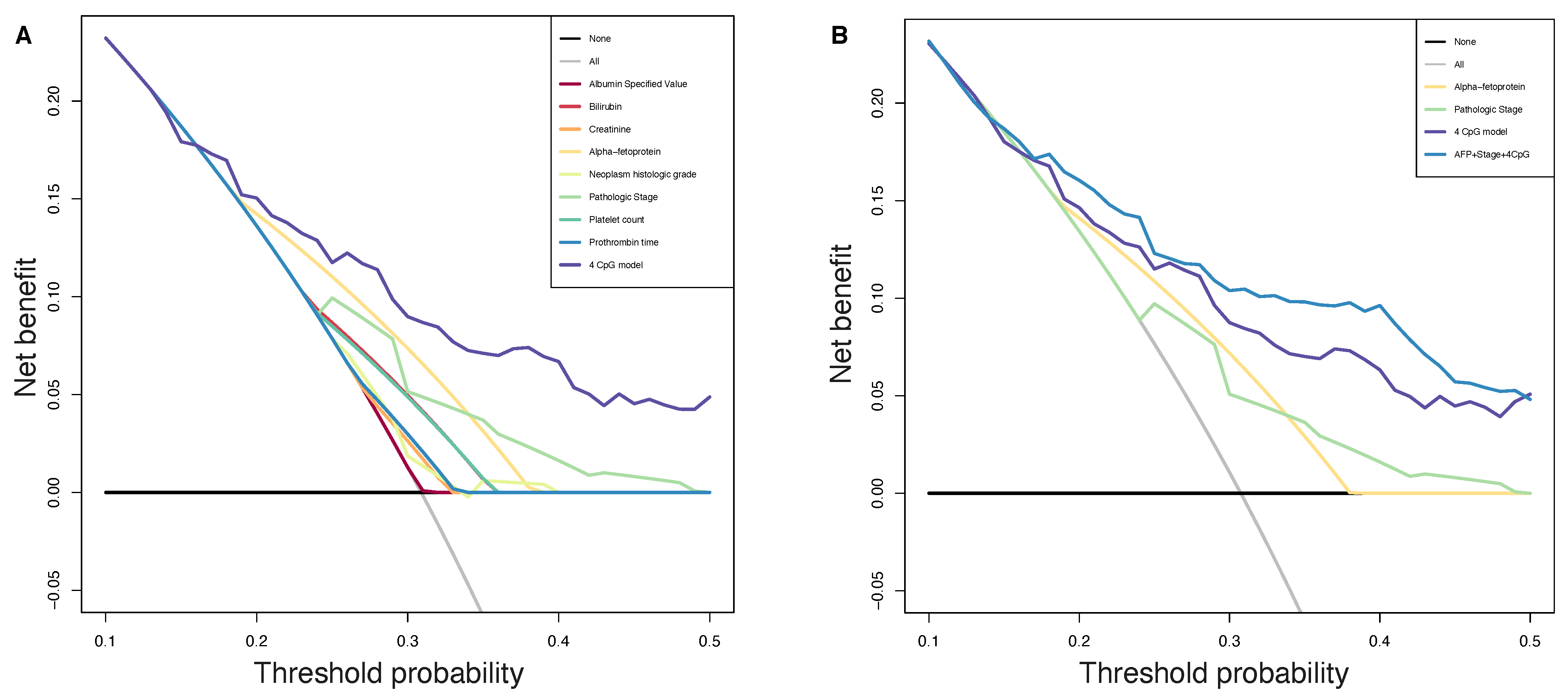
Consequently, to evaluate the size and direction of the relationship between the 34 CpG features and the PFS, but also to investigate their prognostic impact, we firstly performed the univariate Cox regression analysis of the final 34 markers and then, we established a 4-CpG-based prognostic model for HCC. The signature was validated using the internal left-out testing set, indicating the stability of this model in terms of ability to stratify HCC patients into high and low risk groups for early cancer progression. To assess the adequacy and the clinical advantages of our signature over currently used parameters, we performed a decision curve analysis proving that our method showed a significantly improved performance when compared to in-use conventional clinical parameters (i.e., preoperative AFP levels and AJCC pathologic stage), thus indicating a more powerful and dynamic reflection of HCC heterogeneity. This was true either if the 4 CpG signature was considered alone or in combination with well-recognized prognosis parameters. The dataset considered in this analysis is the most comprehensive collection of multi-omic data from HCC cases [
53]. Unfortunately, we were not able to find larger datasets to integrate our analysis. This could be considered as a limitation even if in the attempt to address the lack of an independent validation set, we followed the recommendations described in Shi et al. [
60]. The approach presented here relies on the analysis of methylation data. This design has several advantages: the DNA methylation level can be easily accessible in circulating tumor DNA (ctDNA) allowing a noninvasive ‘liquid biopsy’; unlike methods based on somatic alterations analysis in ctDNA, methylation methods do not need the identification of somatic mutations in the tumor; the collection of peripheral blood to obtain cfDNA is less invasive compared with tumor biopsy. However, further studies are needed to validate the feasibility of using the defined CpG sites from ctDNA to guide a personalized approach for HCC patients.
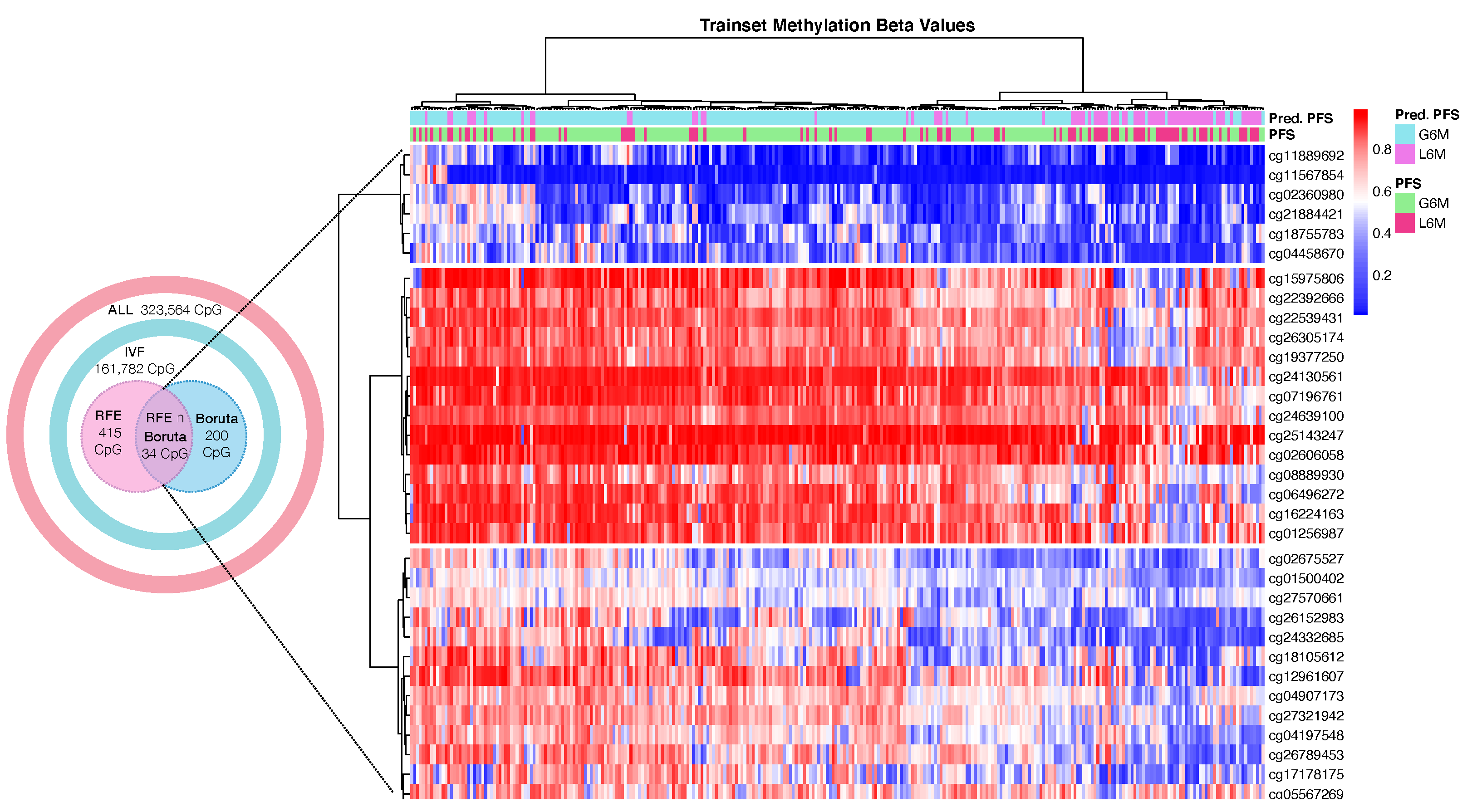
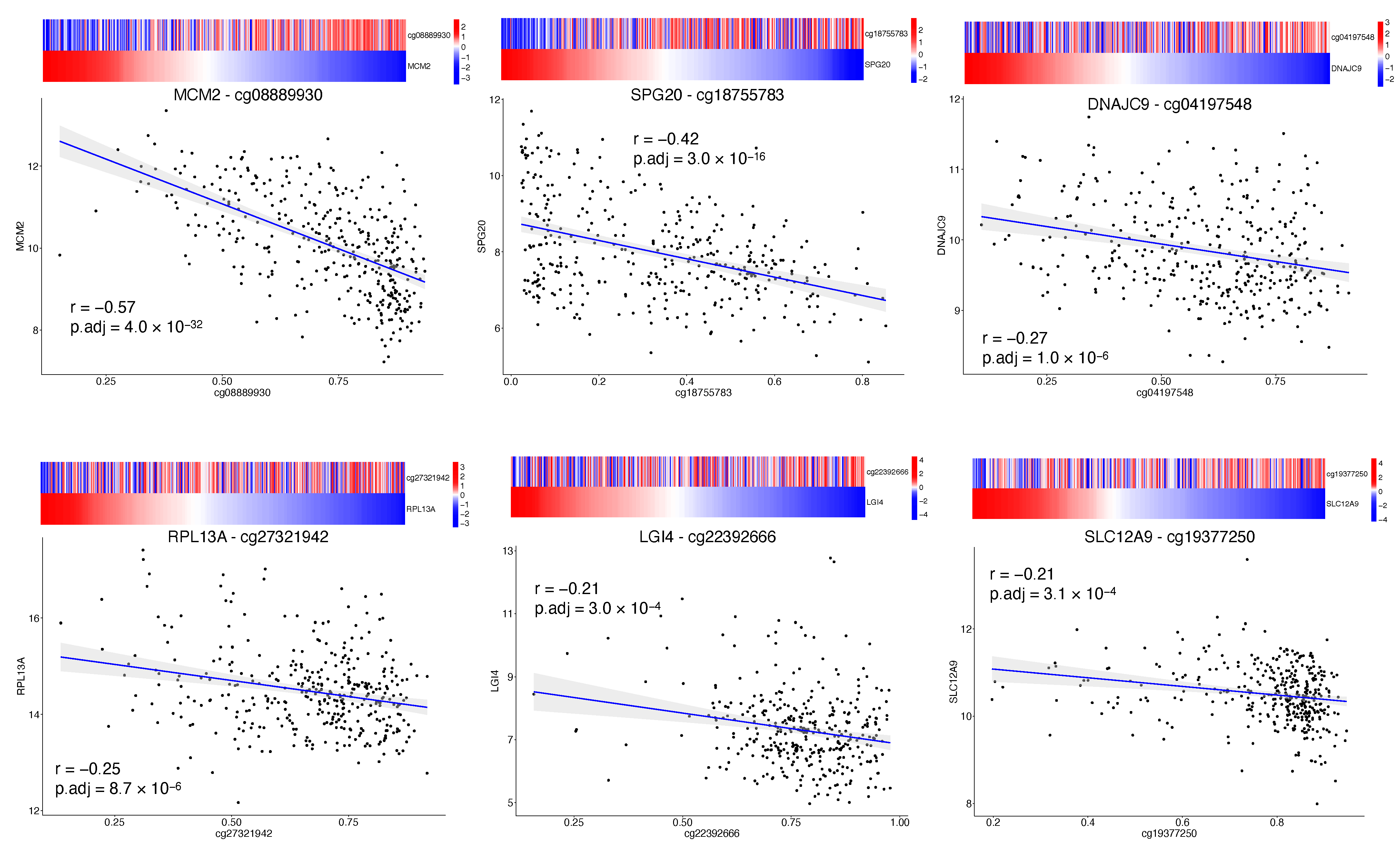
Our CpGs signature comprises the methylation level of 4 CpG sites (
Table 2), of which the cg08889930 was also correlated with
MCM2 expression (
Figure 3); this site likely represents the methylation status of
MCM2 gene and so an indicator of
MCM2 expression. High risk for progression is driven by a lower level of cg08889930 methylation that results in an overexpression of
MCM2, which in turn is associated with cancer progression and poor prognosis [
43]. Aberrant DNA methylated-differentially expressed genes and pathways in HCC have been previously reported [
61,
62]. Our epigenetic Machine Learning model better discriminated among previously reported methylated genes, pointing out MCM2 methylation as a key point to define risk of progression for HCC patients.
MCM2 belongs to the minichromosome maintenance (MCM) protein complex which is involved in the initiation of DNA replication and DNA unwinding [
42].
MCM2 is a subunit/component of the hexameric protein complex that consists of
MCM2-7 and directly interacts with
MCM5 [
63]. In vitro studies indicate that
MCM2 silencing inhibits cell proliferation by affecting the G1/S transition and conversely the overexpression of
MCM2 promotes cell proliferation in lung cancer cells [
64]; moreover,
MCM2 knockdown inhibits cell migration in lung cancer cells. Notably, during the last few years, several studies have evaluated the action of in use treatments against
MCM2. In particular,
MCM2 is a therapeutic target of Trichostatin A in colon cancer cells [
65],
MCM2 has been proposed as therapeutic target of lovastatin in human non-small cell lung carcinomas [
66], the combination of
MCM2 silencing and carboplatin treatment may represent a novel therapeutic strategy to treat ovarian cancer [
67]. Here we propose that the CpG site cg08889930 could represent the methylation status of the
MCM2 gene and consequently the expression level of
MCM2. Its mehylation level could serve not only as prognostic marker to evaluate HCC patient progression but also as predictive marker to evaluate the efficacy of a therapeutic intervention. Another important implication is that the MCM complex offers a fascinating target for drug development in HCC, since it is an essential replication factor that couples DNA replication to both cell cycle progression and checkpoint regulation [
68].
Information relative to the potential implication for the development of novel therapies [
11,
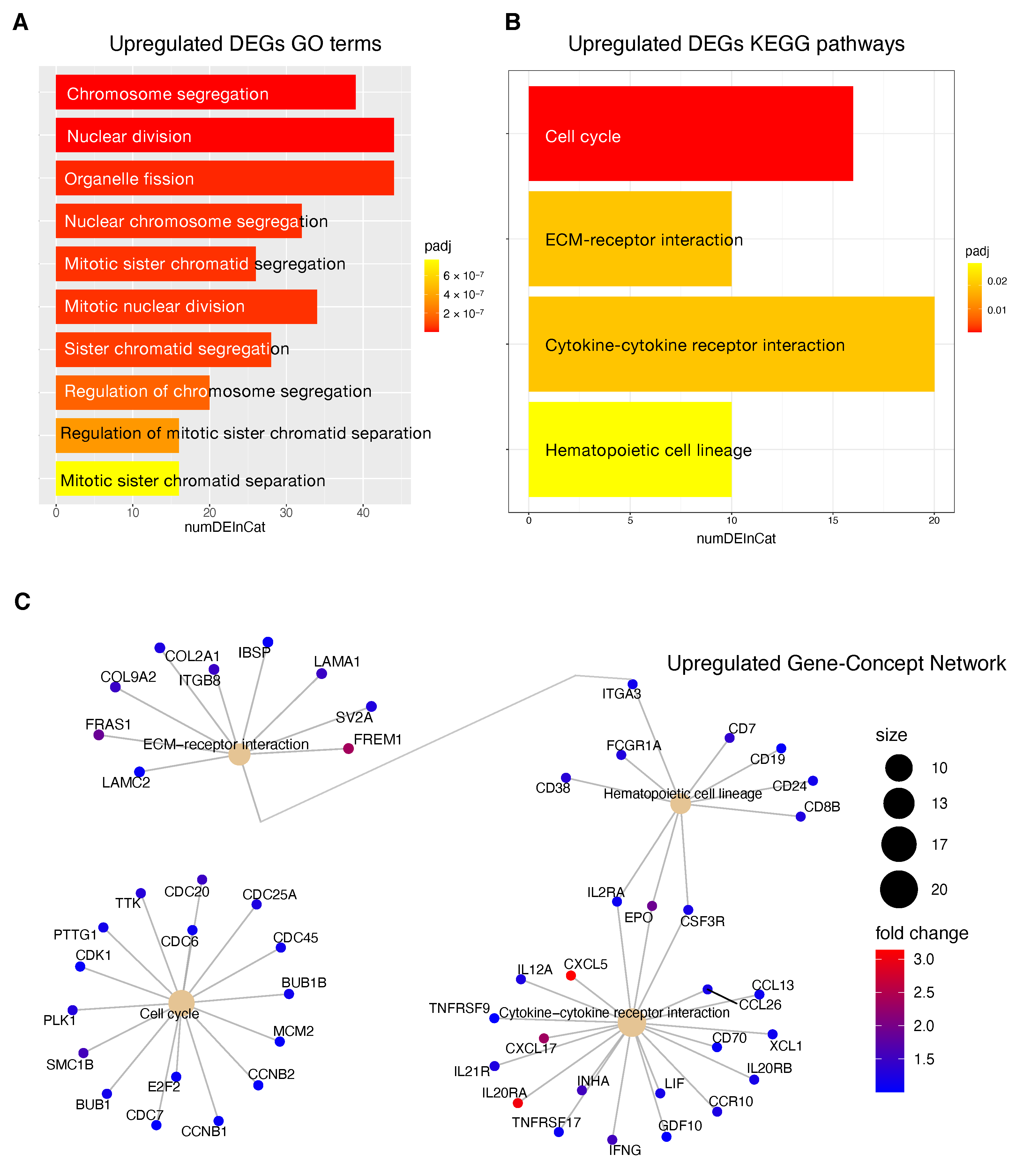
69], was also highlighted by the transcriptome analysis for the HCC patients stratified according to the proposed 4-CpG signature. In particular, from the functional enrichment analysis, we found that upregulated genes in the context of the high risk HCC patient group are implicated in important pathways
Figure 8C) such as extracellular matrix (ECM) receptor interaction, cell cycle, cytokine receptor interaction and hematopoietic cell lineage. Of note, we also found that
MCM2 is upregulated in patients belonging to the high risk group and this could be at least in part recapitulated by the enrichment of genes involved in the control of DNA replication, cell division and cell cycle (
Figure 8). One of the most common features of cancer is cell cycle deregulation which leads to unscheduled proliferation and genomic instability. In humans, the cell cycle is controlled by a subfamily of cyclin-dependent kinases (
CDKs) and several modulators [
70]. Results indicate that
CDK1 is the only CDK that is essential for cell cycle progression [
71]. Here, the high risk HCC patient group has been found to be characterized by the upregulation of several effectors that play a role within the
CDK1 network and that are usually upregulated in cancer (
Figure 8). Overexpression of
CDK1,
MCM2,
E2F2,
PLK1,
CCNB1/2,
BUB1,
BUB1B,
CDC25 has been associated with aberrant proliferation in many cancer types [
72] including HCC [
73]. This could be of interest for the designing of inhibitors of cell cycle protein pathways to be used as anticancer drugs.
One of the major constituents of the environment is the extracellular matrix (ECM). The ECM supervises crucial processes like intratumoral signaling, transport mechanisms, metabolisms and immunogenicity. For its activity, the ECM has been associated with tumor establishment, disease progression and therapy resistance in several tumors [
74]. The ECM aberration plays also a role in the carcinogenesis and progression of HCC [
75]. In this context, the overexpression of ECM-receptor interactor accelerates liver cancer cell metastasis in vessel and settlement in metastatic sites [
76]. In the present study, we found a high expression of some upregulated ECM interactors (
Figure 8C) that have been correlated to migration and invasion (
LAMC2,
FRAS1) [
77,
78], drug resistance (
ITGB8) [
79] and proliferation (
LAMA1) [
80].
Tumor progression is promoted by the crosstalk of different cells populations within the tumor microenvironment (TME) and this communication is guaranteed by the release of key mediators such as cytokines and chemokines. These signaling molecules and their receptors affect multiple processes including tumor cell proliferation, invasion and metastasis, tumor immune response and angiogenesis [
81]. In this context, in the high risk HCC patient group, the most upregulated genes within the cytokines-chemokines network were
CXCL5,
CXCL17 and
IL20RA (
Figure 8C).
CXCL5 overexpression promotes HCC cell proliferation, invasion and intratumoral neutrophil infiltration [
82]. Moreover, a high
CXCL17 expression and a higher rate of tumor-infiltrating CXCL17-expressing cells have been found to be associated with unfavorable prognosis in HCC patients [
83].
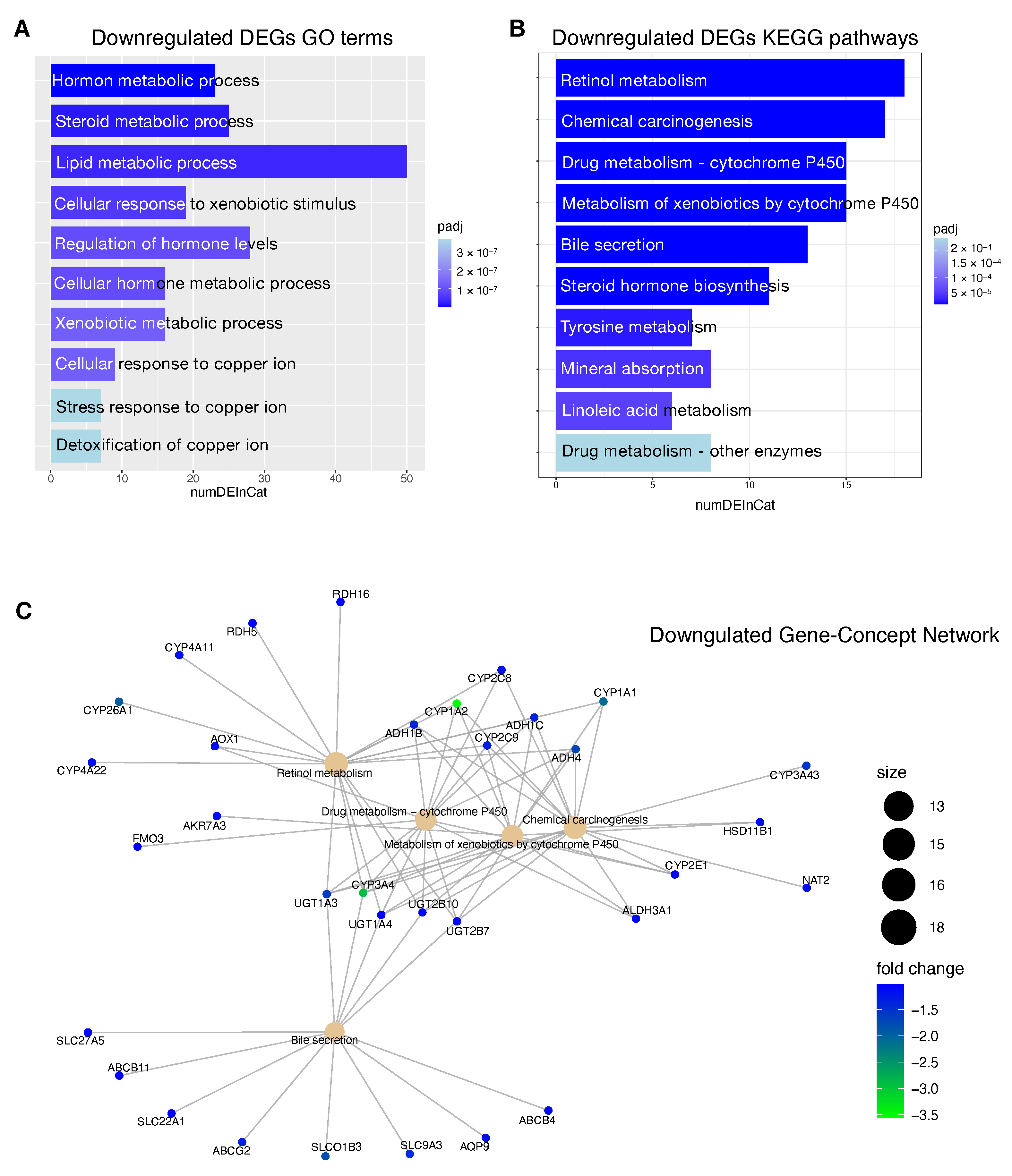
The downregulated genes in high risk HCC patients include several metabolic genes, drug metabolism genes, transporters and carriers genes (
Figure 7). This scenario has important implications for the choice of an appropriate chemotherapy or an appropriate adjuvant chemotherapy. Until 2007, no effective therapies were available for HCC patients that failed to be treated with locoregional approaches. Between 2007 and 2016, sorafenib was the only systemic drug approved for advanced HCC. Currently, palliative treatment strategies in patients with advanced HCC comprise new pharmacological therapies based on inhibitors of tyrosine kinases (TKIs) like sorafenib and regorafenib, but also classical chemotherapeutic agents and novel immunotherapy strategies [
84]. However, several mechanisms of chemoresistance (MOC) [
9] can significantly affect the response of HCC patients to the currently used pharmacological treatments. We found that patients at high risk of progression are characterized by the deregulation of some genes involved in mechanisms of chemoresistance (
SLC22A1,
SLCO1B3,
ABCG2,
CYP3A4).
SLC22A1 is a member of the solute carriers (SLC) family, a collection of proteins that play an essential role in the uptake of anticancer drugs. Loss of
SLC22A1 in the plasma membrane of tumor cells results in a reduction of sorafenib uptake, that has been correlated with less favorable prognosis of HCC patients treated with this drug [
85]. Furthermore, restoring
SLC22A1 expression results in an improved receptivity of sorafenib in HCC cells [
86]. Another dowregulated gene is
SLCO1B3, a component of a family of genes that plays a role in the transport of TKIs drugs.
SLCO1B3 accomplishes the uptake of cabozantinib [
87], clears sorafenib glucuronidated metabolites [
88] and its downregulation in HCC patients contributes to chemoresistance [
89]. The ATP-binding cassette
ABCG2 plays a crucial role in sorafenib efflux and higher
ABCG2 expression has also been correlated with chemoresistance in HCC and reduced overall survival in HCC patients [
90]. In our study, high risk HCC patients are characterized by the downregulation of the gene
ABCG2, so in this case the higher expression level could mainly affect the sorafenib behavior in low risk HCC patients. Several CYPs genes are deregulated in high risk patients including crucial genes such as
CYP3A4,
CYP2C9,
CYP1A2 (
Figure 7C). Deregulated expression of these genes involved in drug and xenobiotics metabolism may affect prodrug activation or drug inactivation, both leading to a lower bioavailability of the functional drug. Sorafenib, regorafenib, cabozantinib and lenvatinib are metabolized by
CYP3A4 through an oxidation process [
91,
92,
93]. Downregulation of
CYP3A4 in HCC microsomes leads to a significantly altered sorafenib metabolism in the liver tumor tissue of HCC patients [
94].

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








