
Biocomputational Prediction Approach Targeting FimH by Natural SGLT2 Inhibitors: A Possible Way to Overcome the Uropathogenic Effect of SGLT2 Inhibitor Drugs

 ,
,  ,
, 
 , , ,
, , , 
Abstract
1. Introduction
2. Methodology
2.1. SGLT2 Inhibitors and Target Protein Structure Retrieval
2.2. Physicochemical Properties and Toxicity Potential Prediction
2.3. Molecular Docking
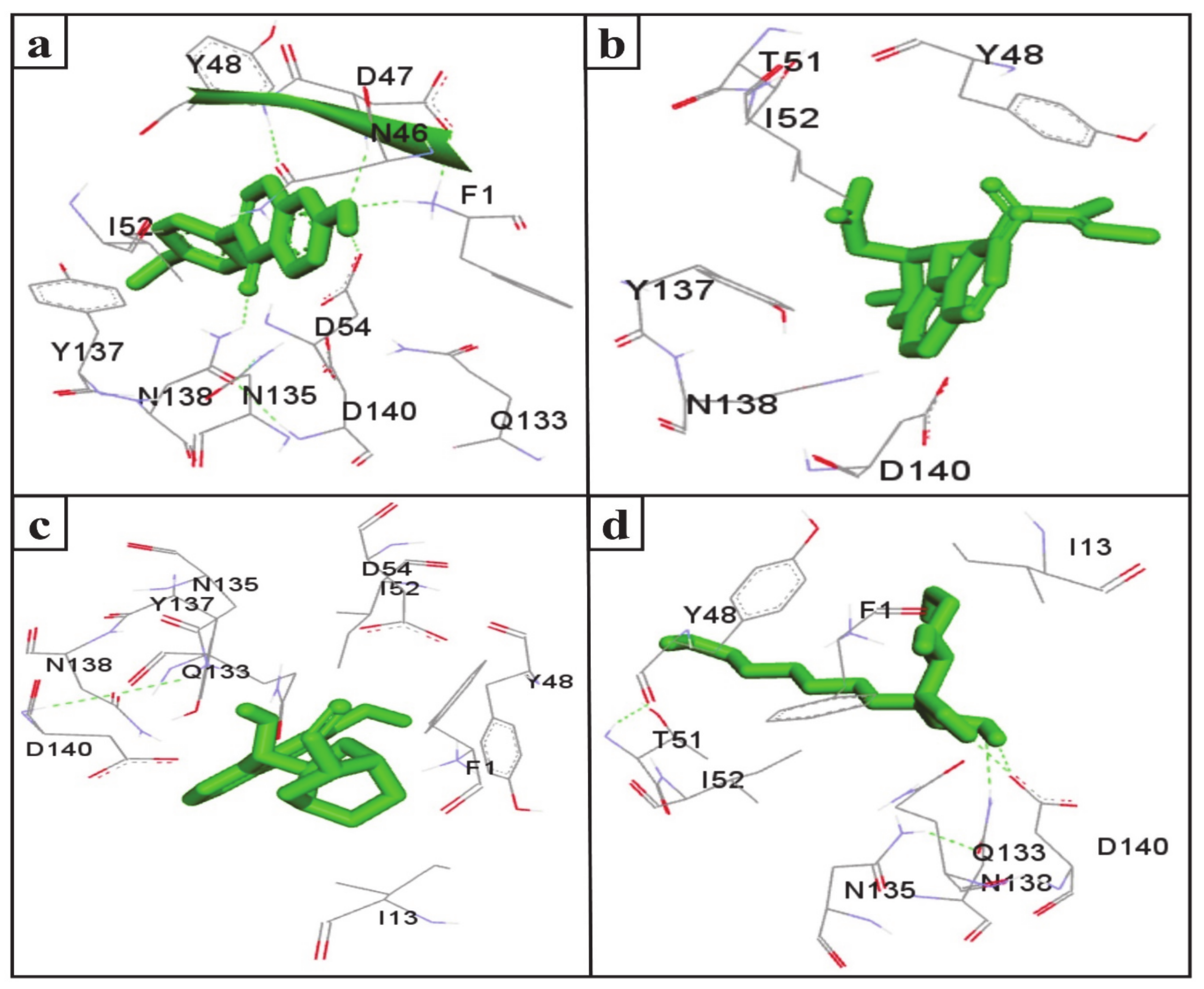
2.4. LIGPLOT+ ANALYSIS
2.5. Molecular Dynamics Simulation
3. Results and Discussion
3.1. Physicochemical Properties and Molecular Docking
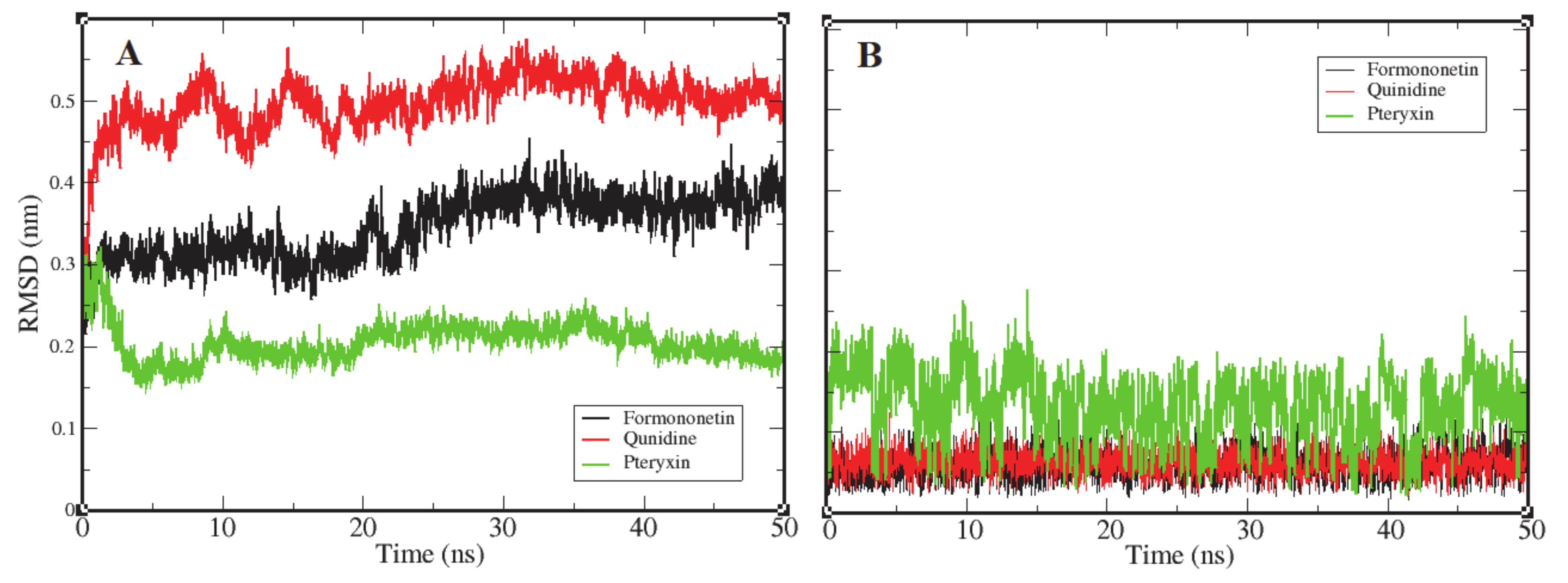
3.2. Root Mean Square Deviation
3.3. Principle Component Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leung, M.Y.; Pollack, L.M.; Colditz, G.A.; Chang, S.H. Life years lost and lifetime health care expenditures associated with diabetes in the U.S., national health interview survey, 1997–2000. Diabetes Care 2015, 38, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.R.; Hux, J.E. Quantifying the risk of infectious diseases for people with diabetes. Diabetes Care 2003, 26, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.W.; Hundborg, H.H.; Lervang, H.H.; Johnsen, S.P.; Schonheyder, H.C.; Sorensen, H.T. Diabetes mellitus as a risk and prognostic factor for community-acquired bacteremia due to enterobacteria: A 10-year, population-based study among adults. Clin. Infect. Dis. 2005, 40, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.W.; Hundborg, H.H.; Lervang, H.H.; Johnsen, S.P.; Sorensen, H.T.; Schonheyder, H.C. Diabetes and outcome of community-acquired pneumococcal bacteremia: A 10-year population based cohort study. Diabetes Care 2004, 27, 70–76. [Google Scholar] [CrossRef]
- Dardi, I.; Kouvatsos, T.; Jabbour, S.A. SGLT2 inhibitors. Biochem. Pharmacol. 2016, 101, 27–39. [Google Scholar] [CrossRef]
- Benfield, T.; Jensen, J.S.; Nordestgaard, B.G. Influence of diabetes and hyperglycaemia on infectious disease hospitalization and outcome. Diabetologia 2007, 50, 549–554. [Google Scholar] [CrossRef]
- Boyko, E.J.; Fihn, S.D.; Scholes, D.; Abraham, L.; Monsey, B. Risk of urinary tract infection and asymptomatic bacteriuria among diabetic and nondiabetic postmenopausal women. Am. J. Epidemiol. 2005, 161, 557–564. [Google Scholar] [CrossRef]
- Geerlings, S.; Fonseca, V.; Castro-Diaz, D.; List, J.; Parikh, S. Genital and urinary tract infections in diabetes: Impact of pharmacologically induced glucosuria. Diabetes Res. Clin. Pract. 2014, 103, 373–381. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Drug Safety Communication: FDA Revises Labels of SGLT2 Inhibitors for Diabetes to Include Warnings About Too Much Acid in the Blood and Serious Urinary Tract Infections. 2015. Available online: http://www.fda.gov/Drugs/DrugSafety/ucm475463.htm (accessed on 10 November 2016).
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Fronzes, R.; Remaut, H.; Waksman, G. Architectures and biogenesis of non-flagellar protein appendages in Gram-negative bacteria. EMBO J. 2008, 27, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Mydock-McGrane, L.K.; Cusumano, Z.T.; Janetka, J.W. Mannose-derived FimH antagonists: A promising anti-virulence therapeutic strategy for urinary tract infections and Crohn’s disease. Expert Opin. Ther. Pat. 2016, 26, 175–197. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, Y.; Akao, Y.; Hirasawa, Y.; Awang, K.; Hadi, A.H.; Sato, S.; Aoyama, C.; Takeo, J.; Shiro, M.; Morita, H. Gneyulins A and B, stilbene trimers, and noidesols A and B, dihydroflavonol-C-glucosides, from the bark of Gnetum gnemonoides. J. Nat. Prod. 2010, 73, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Takeo, J.; Aoyama, C.; Kawahara, H. Na+-glucose cotransporter (SGLT) inhibitory flavonoids from the roots of Sophora flavescens. Bioorg. Med. Chem. 2007, 15, 3445–3449. [Google Scholar] [CrossRef] [PubMed]
- Oranje, P.; Gouka, R.; Burggraaff, L.; Vermeer, M.; Chalet, C.; Duchateau, G.; van der Pijl, P.; Geldof, M.; de Roo, N.; Clauwaert, F.; et al. Novel natural and synthetic inhibitors of solute carriers SGLT1 and SGLT2. Pharmacol. Res. Perspect. 2019, 7, e00504. [Google Scholar] [CrossRef]
- Abgottspon, D.; Rölli, G.; Hosch, L.; Steinhuber, A.; Jiang, X.; Schwardt, O.; Cutting, B.; Smiesko, M.; Jenal, U.; Ernst, B.; et al. Development of an Aggregation Assay to Screen FimH Antagonists. J. Microbiol. Methods 2010, 82, 249–255. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Shakil, S.; Haneef, M.A. simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians. EXCLI J. 2013, 12, 831–857. [Google Scholar]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Toukan, K.; Rahman, A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B 1985, 31, 2643. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Seo, E.; Lee, E.K.; Lee, C.S.; Chun, K.H.; Lee, M.Y.; Jun, H.S. Psoraleacorylifolia L. seed extract ameliorates streptozotocin-induced diabetes in mice by inhibition of oxidative stress. Oxid. Med. Cell Longev. 2014, 2014, 897296. [Google Scholar] [CrossRef]
- Monteiro, M.; Farah, A.; Perrone, D.; Trugo, L.C.; Donangelo, C. Chlorogenic acid compounds from coffee are differentially absorbed and metabolized in humans. J. Nutr. 2007, 137, 2196–2201. [Google Scholar] [CrossRef]
- Mbaze, L.M.; Poumale, H.M.; Wansi, J.D. alpha-Glucosidase inhibitory pentacyclic triterpenes from the stem bark of Fagara tessmannii (Rutaceae). Phytochemistry 2007, 68, 591–595. [Google Scholar] [CrossRef]
- Abdel-Zaher, A.O.; Salim, S.Y.; Assaf, M.H.; Abdel-Hady, R.H. Antidiabetic activity and toxicity of Zizyphus spina-christi leaves. J. Ethnopharmacol. 2005, 101, 129–138. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Udrescu, L.; Bogdan, P.; Chiş, A.; Sîrbu, I.O.; Topîrceanu, A.; Văruţ, R.M.; Udrescu, M. Uncovering New Drug Properties in Target-Based Drug-Drug Similarity Networks. Pharmaceutics 2020, 12, 879. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, R.; Naren, P.; Sudha, V.; Unnikrishnan, M.K.; Pandey, S.; Varghese, M.; Gang, S. Sodium Glucose Co transporter 2 (SGLT2) Inhibitors: A New Sword for the Treatment of Type 2 Diabetes Mellitus. Int. J. Pharma Sci. Res. 2010, 1, 139–147. [Google Scholar]
- Shaikh, S.; Rizvi, S.M.D.; Shakil, S.; Riyaz, S.; Biswas, D.; Jahan, R. Forxiga (Dapagliflozin): Plausible role in the treatment of diabetes associated neurological disorders. Biotechnol. Appl. Biochem. 2016, 63, 145–150. [Google Scholar] [CrossRef]
- Rizvi, S.M.D.; Shakil, S.; Biswas, D.; Shakil, S.; Shaikh, S.; Bagga, P.; Kamal, M.A. Invokana (Canagliflozin) as a Dual Inhibitor of Acetylcholinesterase and Sodium Glucose Co-Transporter 2: Advancement in Alzheimer’s Disease-Diabetes Type 2 Linkage via an Enzoinformatics Study. CNS Neurol. Disord.-Drug Targets 2014, 13, 447–451. [Google Scholar] [CrossRef]
- Kaushal, S.; Singh, H.; Thangaraju, P.; Singh, J. Canagliflozin: A novel SGLT2 inhibitor for Type 2 Diabetes Mellitus. N. Am. J. Med. Sci. 2014, 6, 107–113. [Google Scholar]
- Díez-Sampedro, A.; Barcelona, S. Sugar binding residue affects apparent Na affinity and transport stoichiometry in mouse sodium/glucose cotransporter type 3B. J. Biol. Chem. 2011, 286, 7975–7982. [Google Scholar] [CrossRef]
- Liu, T.; Krofchick, D.; Silverman, M. Effects on conformational states of the rabbit sodium/glucose cotransporter through modulation of polarity and charge at glutamine 457. Biophys. J. 2009, 96, 748–760. [Google Scholar] [CrossRef]
- Wright, E.M.; Turk, E.; Martin, M.G. Molecular basis for glucose galactose malabsorption. Cell Biochem. Biophys. 2002, 36, 115–121. [Google Scholar] [CrossRef]
- Choi, C.I. Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors from Natural Products: Discovery of Next-Generation Antihyperglycemic Agents. Molecules 2016, 21, 1136. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Hosaka, T.; Yano, W.; Itou, T.; Yasumura, M.; Shimizu, Y.; Kondo, T. Metabolic effects of tofogliflozin are efficiently enhanced with appropriate dietary carbohydrate ratio and are distinct from carbohydrate restriction. Physiol. Rep. 2018, 6, e13642. [Google Scholar] [CrossRef] [PubMed]
- Okauchi, S.; Shimoda, M.; Obata, A.; Kimura, T.; Hirukawa, H.; Kohara, K.; Kaneto, H. Protective effects of SGLT2 inhibitor Luseogliflozin on pancreatic β-cells in obese type 2 diabetic db/db mice. Biochem. Biophys. Res. Commun. 2016, 470, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Hirasawa, Y.; Rahman, A.; Kusumawati, I.; Zaini, N.C.; Sato, S.; Aoyama, C.; Takeo, J.; Morita, H. Alstiphyllanines E-H, picraline and ajmaline-type alkaloids from Alstonia macrophylla inhibiting sodium glucose cotransporter. Bioorg. Med. Chem. 2010, 18, 2152–2158. [Google Scholar] [CrossRef]
- Qu, Y.; Chan, J.Y.; Wong, C.W.; Cheng, L.; Xu, C.; Leung, A.W.; Lau, C.B. Antidiabetic Effect of Schisandrae Chinensis Fructus Involves Inhibition of the Sodium Glucose Cotransporter. Drug Dev. Res. 2015, 76, 1–8. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, W.; Yan, X.; Huang, Z.; Zhang, Z.; Wang, P.; Shen, J. Modification on the O-glucoside of Sergliflozin-A: A new strategy for SGLT2 inhibitor design. Bioorg. Med. Chem. Lett. 2016, 26, 2170–2173. [Google Scholar] [CrossRef]
- Wilding, J.P.; Charpentier, G.; Hollander, P.; González-Gálvez, G.; Mathieu, C.; Vercruysse, F.; Usiskin, K.; Law, G.; Black, S.; Canovatchel, W.; et al. Efficacy and safety of canagliflozin in patients with type 2 diabetes mellitus inadequately controlled with metformin and sulphonylurea: A randomized trial. Int. J. Clin. Pract. 2013, 67, 1267–1282. [Google Scholar] [CrossRef]
- Rosenstock, J.; Jelaska, A.; Frappin, G.; Salsali, A.; Kim, G.; Woerle, H.J.; Broedl, U.C.; EMPA-REG MDI Trial Investigators. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care 2014, 37, 1815–1823. [Google Scholar] [CrossRef]
- Jabbour, S.A.; Hardy, E.; Sugg, J.; Parikh, S. Dapagliflozin is effective as add-on therapy to sitagliptin with or without metformin: A 24-Week, multicenter, randomized, double blind, placebo-controlled study. Diabetes Care 2014, 37, 740–750. [Google Scholar] [CrossRef]
- Bode, B.; Stenlof, K.; Harris, S.; Sullivan, D.; Fung, A.; Usiskin, K.; Meininger, G. Long-term efficacy and safety of canagliflozin over 104 weeks in patients aged 55–80 years with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 294–303. [Google Scholar] [CrossRef]
- Haering, H.U.; Merker, L.; Christiansen, A.V.; Roux, F.; Salsali, A.; Kim, G.; Meinicke, T.; Woerle, H.J.; Broedl, U.C.; EMPA-REG EXTEND™ METSU Investigators. Empagliflozin as add-on to metformin plus sulphonylurea in patients with type 2 diabetes. Diabetes Res. Clin. Pract. 2015, 110, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Ronald, A.R.; Nicolle, L.E.; Stamm, E.; Krieger, J.; Warren, J.; Schaeffer, A.; Naber, K.G.; Hooton, T.M.; Johnson, J.; Chambers, S.; et al. Urinary tract infection in adults: Research priorities and strategies. Int. J. Antimicrob. Agents 2001, 17, 343–348. [Google Scholar] [CrossRef]
- Anderson, G.G.; Palermo, J.J.; Schilling, J.D.; Roth, R.; Heuser, J.; Hultgren, S.J. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 2003, 301, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Mydock-McGrane, L.K.; Hannan, T.J.; Janetka, J.W. Rational Design Strategies for FimH Antagonists: New Drugs on the Horizon for Urinary Tract Infection and Crohn’s Disease. Expert Opin. Drug Discov. 2017, 12, 711–731. [Google Scholar] [CrossRef]
- Wellens, A.; Garofalo, C.; Nguyen, H.; Van Gerven, N.; Slättegård, R.; Hernalsteens, J.P.; Wyns, L.; Oscarson, S.; De Greve, H.; Hultgren, S.; et al. Intervening with urinary tract infections using anti-adhesives based on the crystal structure of the FimH-oligomannose-3 complex. PLoS ONE 2008, 3, e2040. [Google Scholar] [CrossRef]
- Hung, C.S.; Bouckaert, J.; Hung, D.; Pinkner, J.; Widberg, C.; DeFusco, A.; Auguste, C.G.; Strouse, R.; Langermann, S.; Waksman, G.; et al. Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection. J. Mol. Microbiol. 2002, 44, 903–915. [Google Scholar] [CrossRef]
- Chen, S.L.; Hung, C.S.; Pinkner, J.S.; Walker, J.N.; Cusumano, C.K.; Li, Z.; Bouckaert, J.; Gordon, J.I.; Hultgren, S. Positive selection identifies an in vivo role for FimH during urinary tract infection in addition to mannose binding. Proc. Natl. Acad. Sci. USA 2009, 106, 22439–22444. [Google Scholar] [CrossRef]
- Mousavifar, L.; Vergoten, G.; Charron, G.; Roy, R. Comparative study of aryl O-, C-, and S-mannopyranosides as potential adhesion inhibitors toward uropathogenic E. coli FimH. Molecules 2019, 24, 3566. [Google Scholar] [CrossRef]
- Mousavifar, L.; Touaibia, M.; Roy, R. Development of mannopyranoside therapeutics against adherent-invasive Escherichia coli infections. Acc. Chem. Res. 2018, 51, 2937–2948. [Google Scholar] [CrossRef]
- Sarshar, M.; Behzadi, P.; Ambrosi, C.; Zagaglia, C.; Palamara, A.T.; Scribano, D. FimH and anti-adhesive therapeutics: A disarming strategy against uropathogens. Antibiotics 2020, 9, 397. [Google Scholar] [CrossRef]
- Ribić, R.; Meštrović, T.; Neuberg, M.; Kozina, G. Effective anti-adhesives of uropathogenic Escherichia coli. Acta Pharm. 2018, 68, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rafsanjany, N.; Senker, J.; Brandt, S.; Dobrindt, U.; Hensel, A. In vivo consumption of cranberry exerts ex vivo antiadhesive activity against FimH-Dominated uropathogenic Escherichia coli: A combined in vivo, ex vivo, and in vitro study of an extract from vaccinium macrocarpon. J. Agric. Food Chem. 2015, 63, 8804–8818. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Shaikh, S.; Shakil, S.; Rizvi, S.M. An enzoinformatics study for prediction of efficacies of three novel penem antibiotics against New Delhi metallo-β-lactamase-1 bacterial enzyme. Interdiscip. Sci. Comput. Life Sci. 2014, 6, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.; Yadav, B.S.; Pandey, P.N.; Tripathi, V. The effect of β-glucan and its potential analog on the structure of Dectin-1 receptor. J. Mol. Graph. Model 2017, 74, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.M.D.; Shakil, S.; Zeeshan, M.; Khan, M.S.; Shaikh, S.; Biswas, D.; Ahmad, A.; Kamal, M.A. An enzoinformatics study targeting polo-like kinases-1 enzyme: Comparative assessment of anticancer potential of compounds isolated from leaves of Ageratum houstonianum. Pharmacogn. Mag. 2014, 10, 14–21. [Google Scholar]
- Verma, A.; Rizvi, S.M.D.; Shaikh, S.; Ansari, M.A.; Shakil, S.; Ghazal, F.; Siddiqui, M.H.; Haneef, M.; Rehman, A. Compounds isolated from Ageratum houstonianum inhibit the activity of matrix metalloproteinases (MMP-2 and MMP-9): An oncoinformatics study. Pharmacogn. Mag. 2014, 10, 18–26. [Google Scholar]
- Erondu, N.; Desai, M.; Ways, K.; Meininger, G. Diabetic ketoacidosis and related events in the canagliflozin type 2 diabetes clinical program. Diabetes Care 2015, 38, 1680–1686. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Compound Name | Physiochemical Parameters | |||||||
|---|---|---|---|---|---|---|---|---|---|
| % of Absorption ** | Topological Polar Surface Area (Å)2 | Molecular Weight | cLogP *** | Hydrogen Bond Donors | Hydrogen Bond Acceptors | Number of Rotatable Bonds | Lipinski’s Violation | ||
| Rule | - | - | <500 | ≤5 | <5 | <10 | ≤10 | ≤1 | |
| 1. | Acerogenin B | 91.85 | 49.69 | 298.38 | 4.50 | 2 | 3 | 0 | 0 |
| 2. | Formononetin | 89.76 | 55.76 | 268.26 | 2.24 | 1 | 4 | 2 | 0 |
| 3. | (−)-kurarinone | 75.80 | 96.22 | 438.51 | 6.11 | 3 | 6 | 7 | 1 |
| 4. | (+)-pteryxin | 78.59 | 88.13 | 386.39 | 3.34 | 0 | 7 | 5 | 0 |
| 5. | Quinidine | 93.27 | 45.59 | 324.42 | 2.61 | 1 | 4 | 4 | 0 |
| 6. | Canagliflozin * | 68.15 | 118.39 | 444.52 | 3.27 | 4 | 5 | 5 | 0 |
| 7. | Heptyl α-d-mannopyranoside * | 74.71 | 99.38 | 278.34 | 0.485 | 4 | 6 | 8 | 0 |
| S.No. | Compound Name | Toxicity Risks | |||
|---|---|---|---|---|---|
| Mutagenic | Tumorigenic | Reproductive Effect | Irritant | ||
| 1. | Acerogenin B | None | None | None | None |
| 2. | Formononetin | None | None | None | None |
| 3. | (−)-kurarinone | None | None | None | None |
| 4. | (+)-pteryxin | None | None | None | High |
| 5. | Quinidine | None | None | None | None |
| 6. | Canagliflozin * | None | None | None | None |
| 7. | Heptyl α-d-mannopyranoside * | None | None | None | None |
| Compounds | SGLT2 | FimH | ||
|---|---|---|---|---|
| Binding Energy (kcal/mol) | Inhibition Constant (µM) | Binding Energy (kcal/mol) | Inhibition Constant (µM) | |
| Canagliflozin * | −7.23 | 5.04 | −3.56 | 2450 |
| Acerogenin B | −6.30 | 24.25 | −4.40 | 598.52 |
| Formononetin | −7.63 | 2.57 | −5.65 | 71.95 |
| (−)-kurarinone | −7.23 | 5.03 | −3.93 | 1310 |
| (+)-pteryxin | −9.01 | 0.248 | −5.50 | 92.97 |
| Quinidine | −8.77 | 0.371 | −5.70 | 66.40 |
| Heptyl α-d-mannopyranoside ** | - | - | −4.46 | 109.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashraqi, M.M.; Chaturvedi, N.; Alam, Q.; Alshamrani, S.; Bahnass, M.M.; Ahmad, K.; Alqosaibi, A.I.; Alnamshan, M.M.; Ahmad, S.S.; Beg, M.M.A.; et al. Biocomputational Prediction Approach Targeting FimH by Natural SGLT2 Inhibitors: A Possible Way to Overcome the Uropathogenic Effect of SGLT2 Inhibitor Drugs. Molecules 2021, 26, 582. https://doi.org/10.3390/molecules26030582
Mashraqi MM, Chaturvedi N, Alam Q, Alshamrani S, Bahnass MM, Ahmad K, Alqosaibi AI, Alnamshan MM, Ahmad SS, Beg MMA, et al. Biocomputational Prediction Approach Targeting FimH by Natural SGLT2 Inhibitors: A Possible Way to Overcome the Uropathogenic Effect of SGLT2 Inhibitor Drugs. Molecules. 2021; 26(3):582. https://doi.org/10.3390/molecules26030582
Chicago/Turabian StyleMashraqi, Mutaib M., Navaneet Chaturvedi, Qamre Alam, Saleh Alshamrani, Mosa M. Bahnass, Khurshid Ahmad, Amany I. Alqosaibi, Mashael M. Alnamshan, Syed Sayeed Ahmad, Mirza Masroor Ali Beg, and et al. 2021. "Biocomputational Prediction Approach Targeting FimH by Natural SGLT2 Inhibitors: A Possible Way to Overcome the Uropathogenic Effect of SGLT2 Inhibitor Drugs" Molecules 26, no. 3: 582. https://doi.org/10.3390/molecules26030582
APA StyleMashraqi, M. M., Chaturvedi, N., Alam, Q., Alshamrani, S., Bahnass, M. M., Ahmad, K., Alqosaibi, A. I., Alnamshan, M. M., Ahmad, S. S., Beg, M. M. A., Mishra, A., Shaikh, S., & Rizvi, S. M. D. (2021). Biocomputational Prediction Approach Targeting FimH by Natural SGLT2 Inhibitors: A Possible Way to Overcome the Uropathogenic Effect of SGLT2 Inhibitor Drugs. Molecules, 26(3), 582. https://doi.org/10.3390/molecules26030582