
Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin

 ,
,  ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
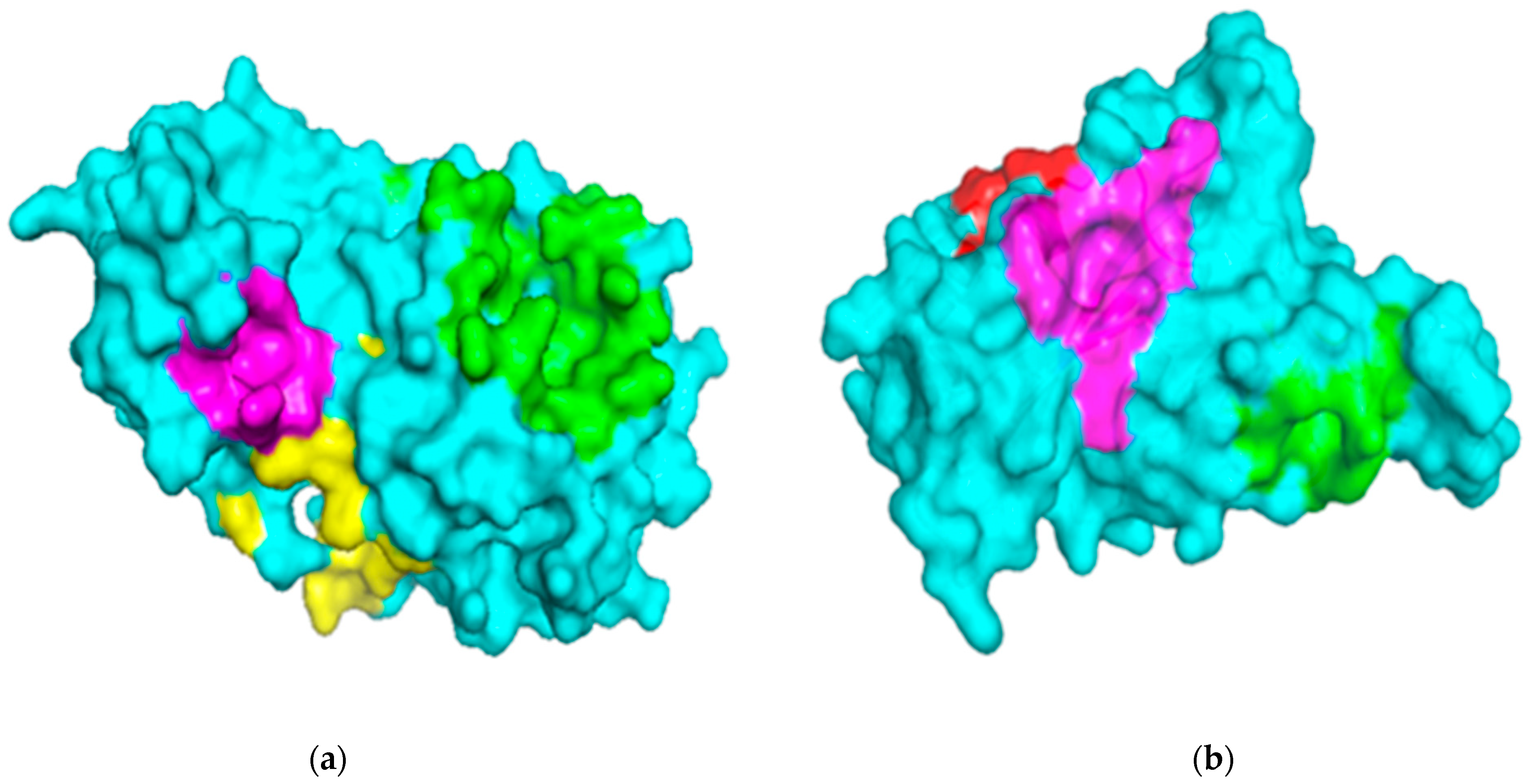
2.1. Description of Binding Sites of Mpro and RBD Structures
2.1.1. Binding Site Analysis of Mpro
2.1.2. Binding Site Analysis of RBD
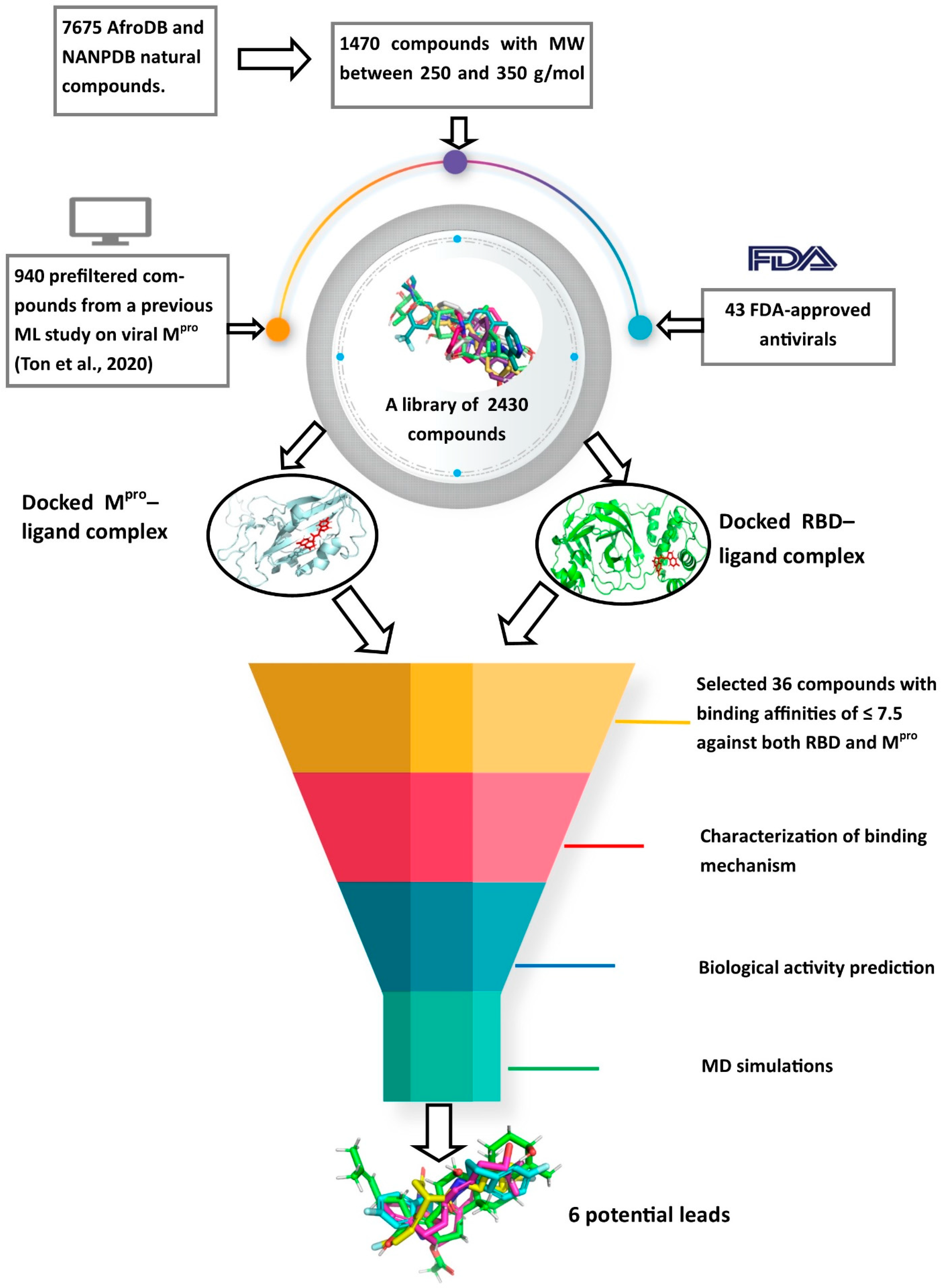
2.2. Virtual Screening Studies
2.2.1. Molecular Docking Studies
Molecular Docking Studies of Mpro
Molecular Docking Studies of RBD
Shortlisted Compounds for Downstream Analysis
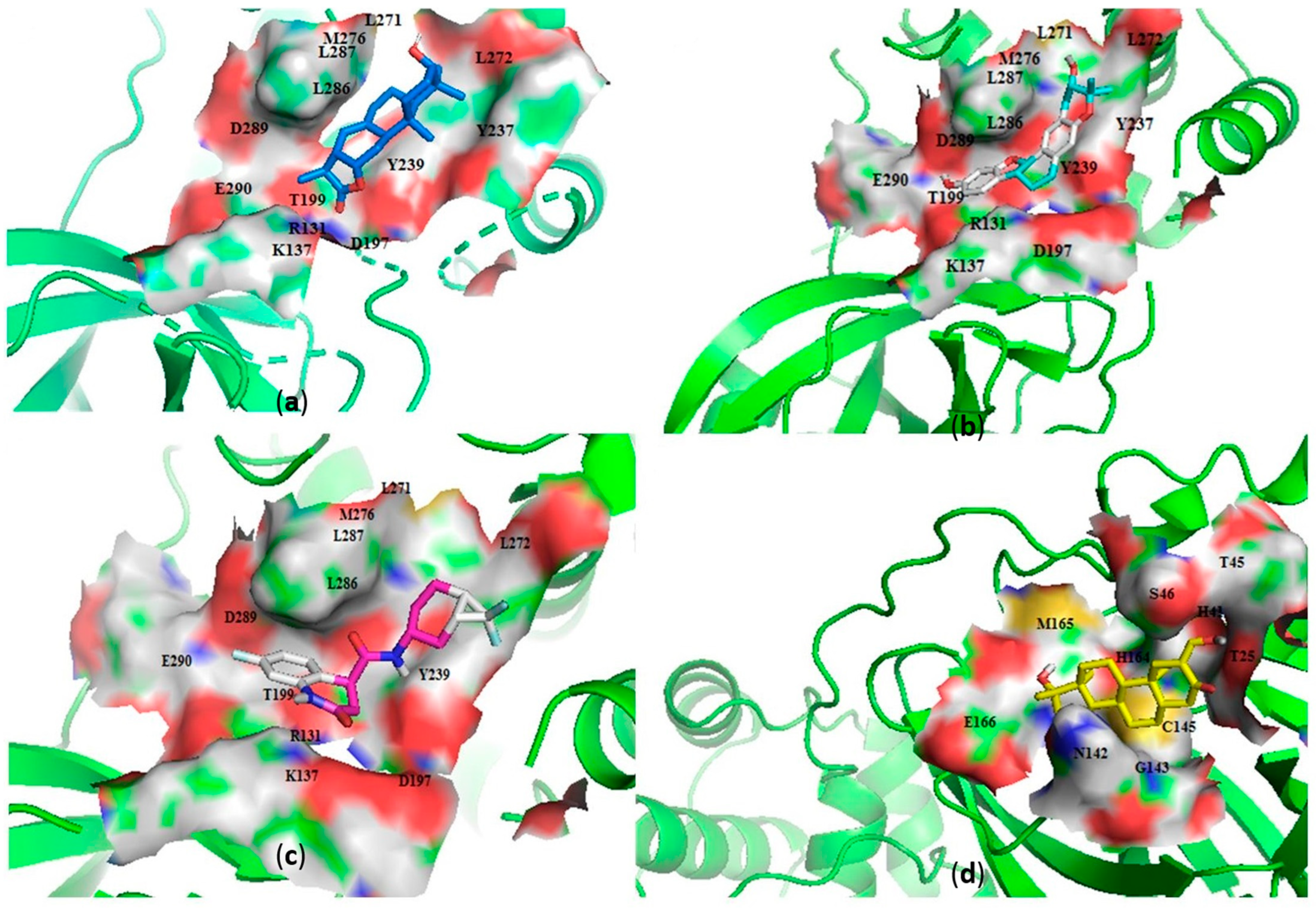
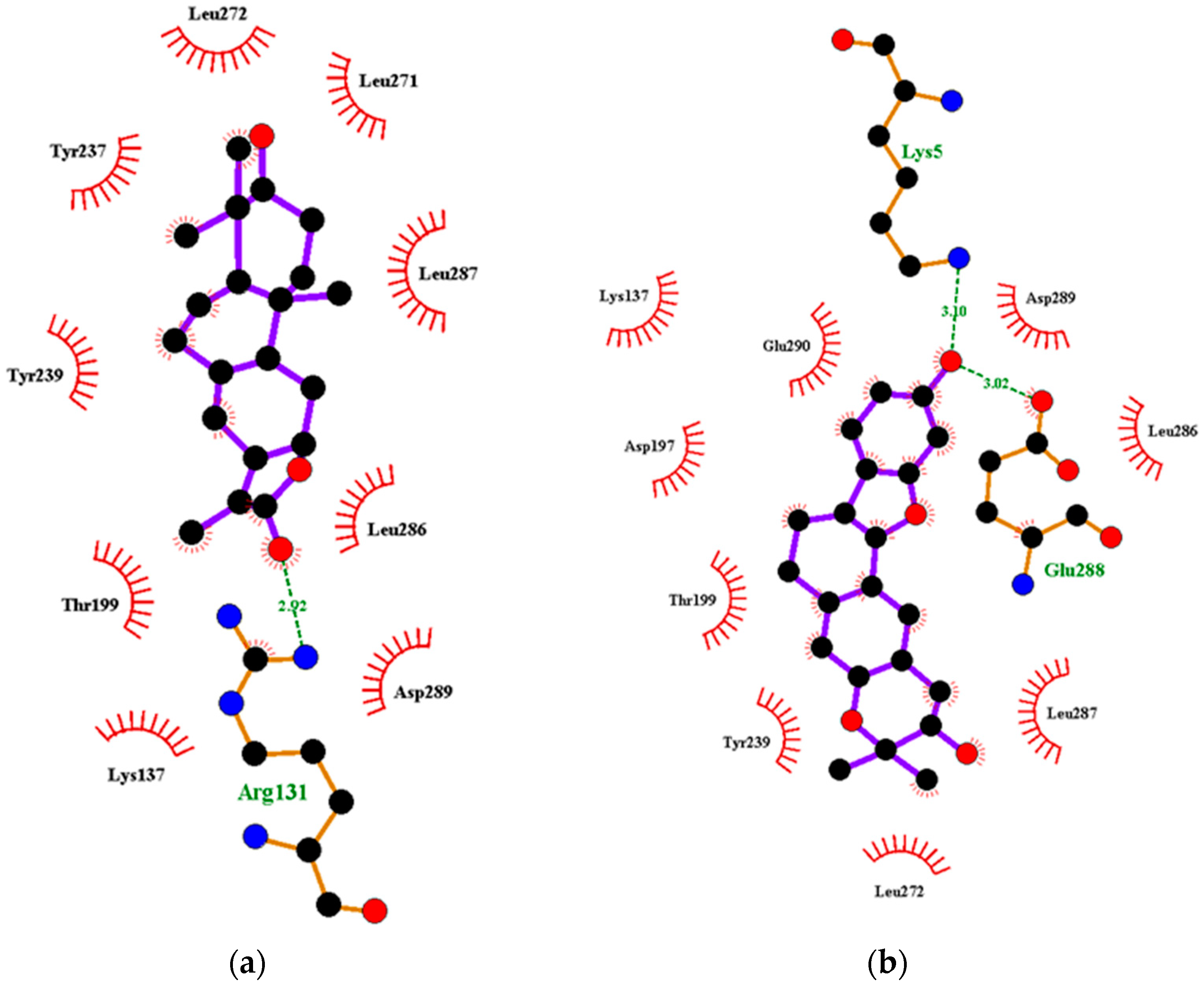
2.2.2. Characterization of the Protein–Ligand Interactions
Characterization of the Mpro–Ligand Interactions
Characterization of the RBD–Ligand Interactions
2.2.3. Predictions of Biological Activities
2.3. Existing Drugs Proposed as Potential Frontline Treatment Options
2.3.1. Similarity Search of Hits
2.3.2. Fusidic Acid and Betulinic Acid as Potential Anti-SARS-CoV-2 Compounds
2.4. Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) Calculations
2.4.1. MM/PBSA-Binding Free Energy Computational Analysis of Mpro
2.4.2. MM/PBSA-Binding Free Energy Computational Analysis of RBD
2.5. Other Contributing Energy Terms
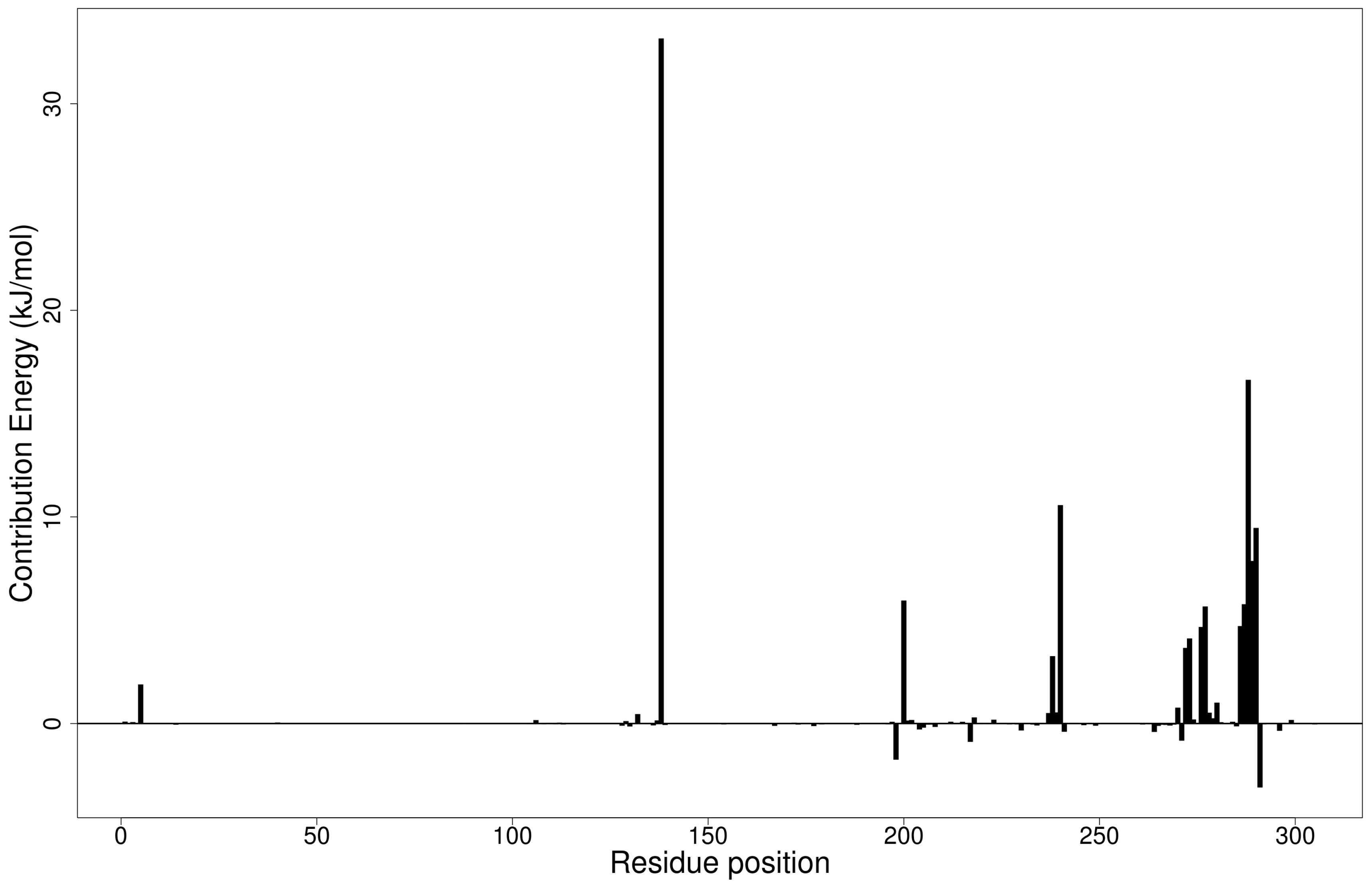
2.5.1. Energy Decomposition per Residue
Per-Residue Energy Decomposition of Mpro–Ligand Complexes
Per-Residue Energy Decomposition of the RBD–Ligand Complexes
2.6. Molecular Dynamics
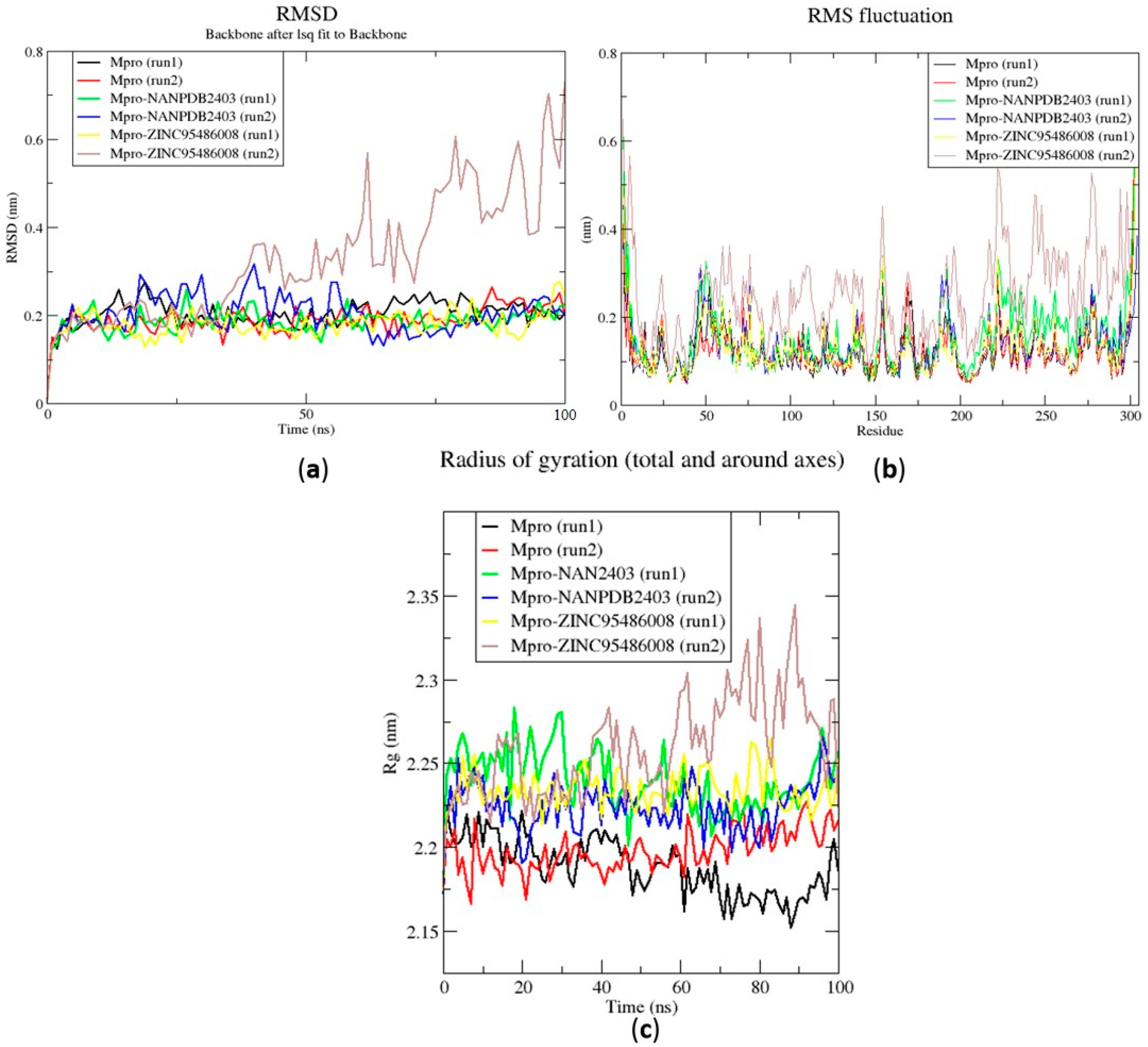
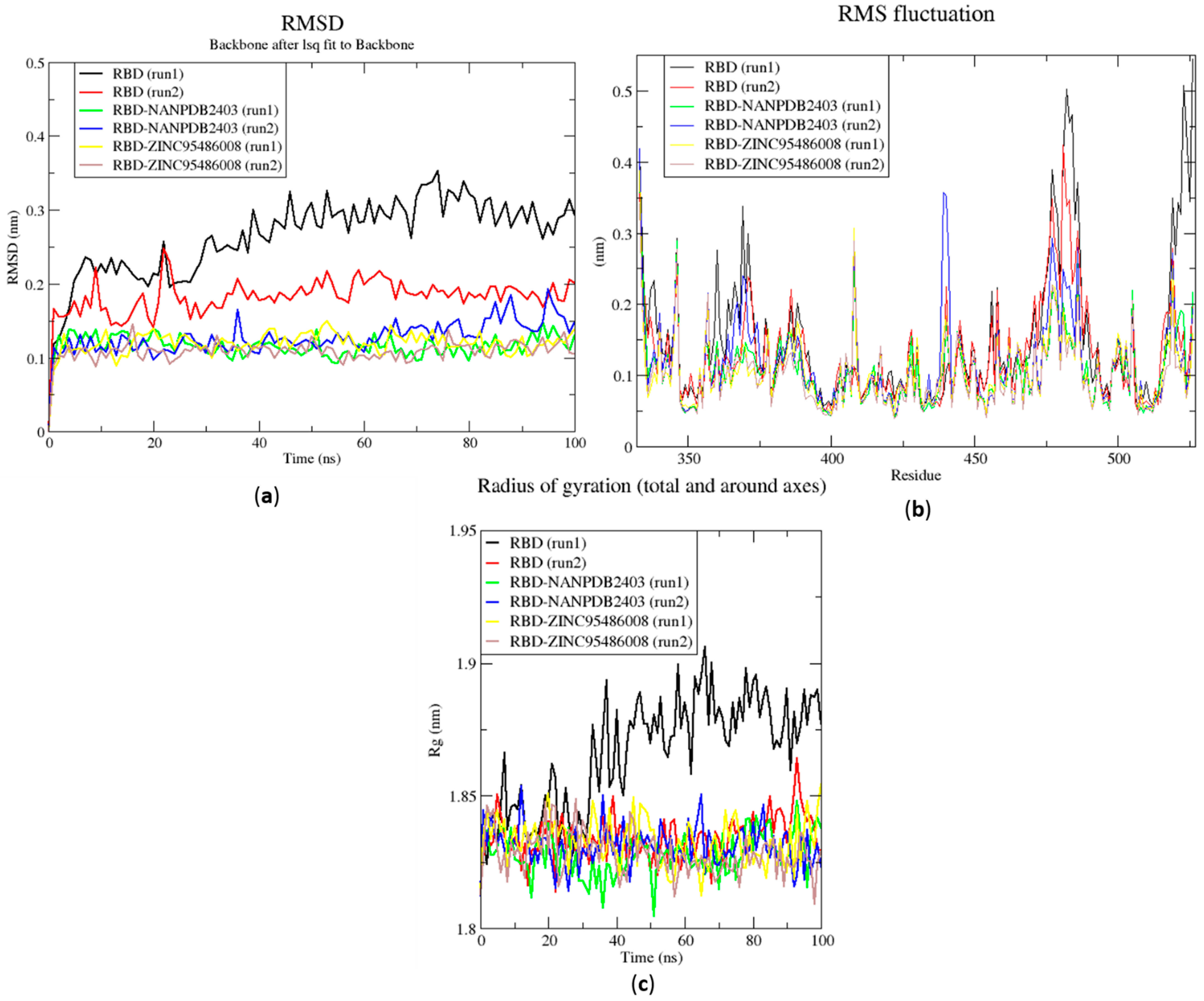
2.6.1. Root Mean Square Deviation of the Complexes for 100 ns MD Simulations
2.6.2. Root Mean Square Fluctuation of the Complexes for 100 ns MD Simulations
2.6.3. Radius of Gyration of the Complexes for 100 ns MD Simulations
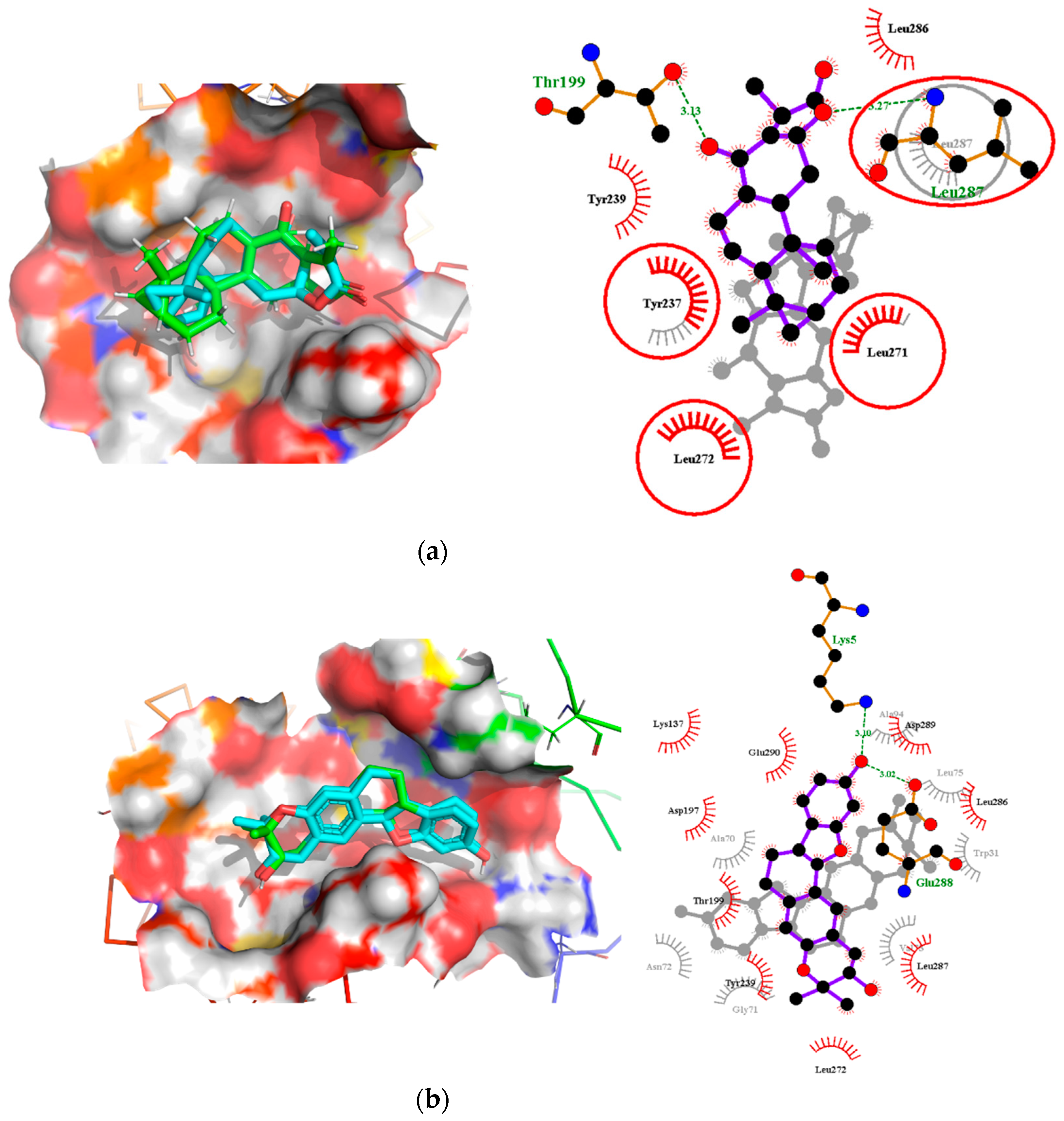
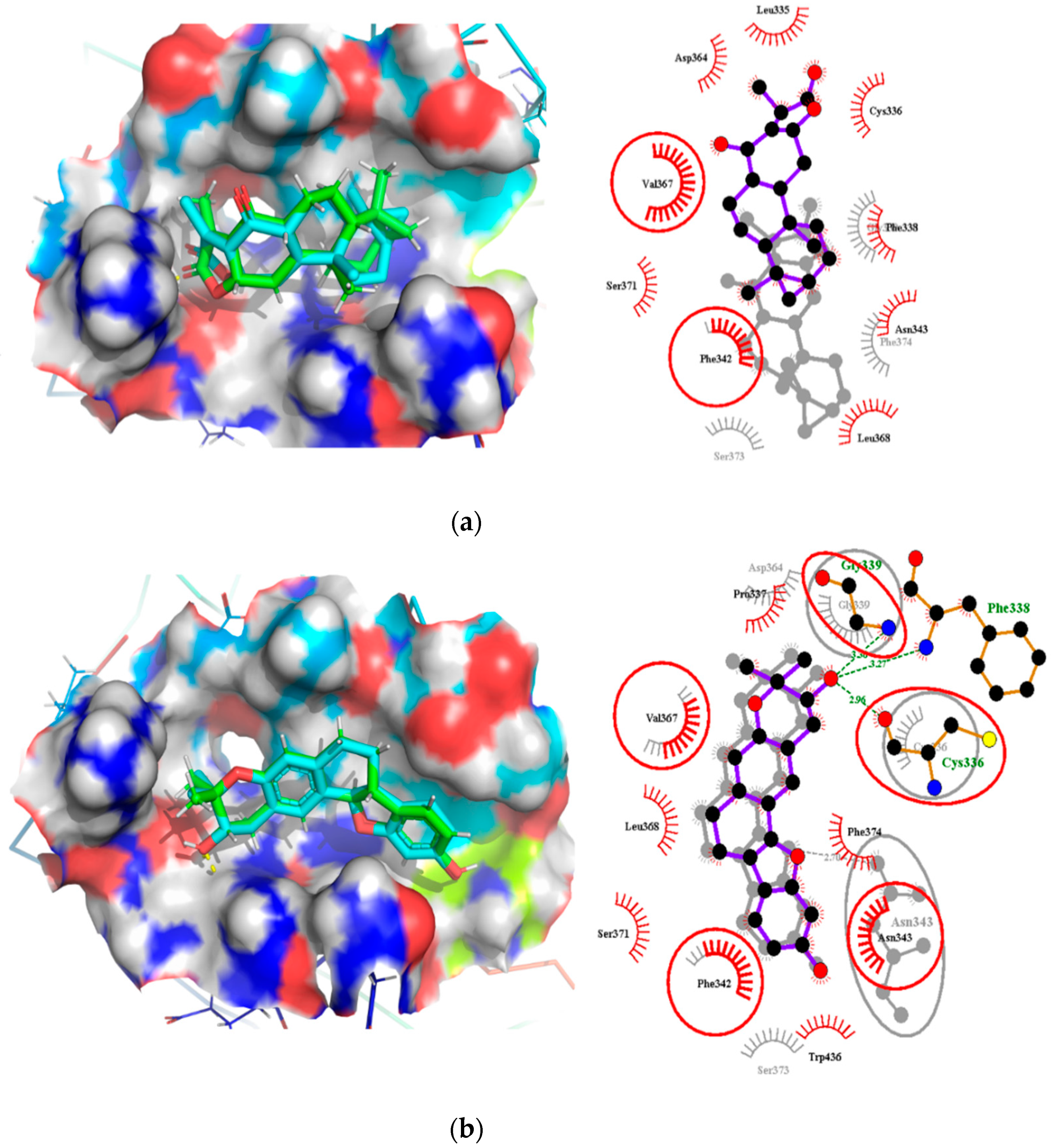
2.7. Comparison of Binding Modes Pre-MD and Post-100 ns MD Simulations
2.7.1. Binding Modes Interactions Analysis between Mpro and Potential Leads
2.7.2. Binding Modes Interactions Analysis between RBD and Potential Leads
2.8. Summary and Implications of the Results
3. Materials and Methods
3.1. Data Sources for SARS-CoV-2 Targets
3.2. The Screening Library
3.3. Preparation of the Protein Structure and Elucidation-Binding Sites
3.4. Virtual Screening of Ligand Library
3.5. Characterization of the Protein–Ligand Interactions
3.6. Prediction of Antiviral Properties of Hit Compounds
3.7. Molecular Dynamics Simulation of Protein–Ligand Complexes
3.8. Analysis of Binding Modes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rabi, A.F.; Al Zoubi, S.M.; Kasasbeh, A.G.; Salameh, M.D.; Al-Nasser, D.A. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Naming the Coronavirus Disease (COVID-19) and the Virus That Causes It. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 3 July 2020).
- WHO. WHO Coronavirus Disease. Available online: https://covid19.who.int/ (accessed on 8 December 2020).
- Aura Vision. Coronavirus (COVID-19) Lockdown Tracker | Aura Vision. Available online: https://auravision.ai/covid19-lockdown-tracker/ (accessed on 18 May 2020).
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19); StatPearls Publishing LLC: Treasure Island, FL, USA, 2020. [Google Scholar]
- Hjelmesæth, J.; Skaare, D. Covid-19 med nedsatt lukte-og smakssans som eneste symptom. Tidsskr. Nor. Laegeforening 2020, 140. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, P.C.; Lopez, M.A.; Mastandrea Bonaviri, G.N.; Garcia-Godoy, F.; D’Addona, A. Taste and smell as chemosensory dysfunctions in COVID-19 infection. Am. J. Dent. 2020, 33, 135–137. [Google Scholar]
- Moein, S.T.; Hashemian, S.M.R.; Mansourafshar, B.; Khorram-Tousi, A.; Tabarsi, P.; Doty, R.L. Smell dysfunction: A biomarker for COVID-19. Int. Forum Allergy Rhinol. 2020, 10, 944–950. [Google Scholar] [CrossRef]
- Yan, C.H.; Faraji, F.; Prajapati, D.P.; Boone, C.E.; DeConde, A.S. Association of chemosensory dysfunction and COVID-19 in patients presenting with influenza-like symptoms. Int. Forum Allergy Rhinol. 2020, 10, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.Y. Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1805–1820. [Google Scholar] [CrossRef]
- Sahin, A.R. 2019 Novel Coronavirus (COVID-19) Outbreak: A Review of the Current Literature. Eurasian J. Med. Oncol. 2020, 4, 1–7. [Google Scholar] [CrossRef]
- Luk, H.K.H.; Li, X.; Fung, J.; Lau, S.K.P.; Woo, P.C.Y. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar]
- Smith, T.; Prosser, T.; Information, C.D.; Solutions, C. COVID-19 Drug Therapy—Potential Options. Elsevier’s Nov. Coronavirus Inf. Cent. 2020, 19–22. Available online: Repository.phb.ac.id/776/1/COVID-19-Drug-Therapy_5.14.2020.pdf (accessed on 18 May 2020).
- Food and Drug Administration COVID-19 Frequently Asked Questions. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-frequently-asked-questions (accessed on 3 July 2020).
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248, 117477. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- FDA. Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment. Press Announc. 2020, 3–5. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment (accessed on 3 July 2020).
- Roberts, M. “Coronavirus: Dexamethasone Proves First Life-Saving Drug—BBC News,” BBC News Online. 2020. Available online: https://www.bbc.com/news/health-53061281 (accessed on 3 July 2020).
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Tang, Q.; Shang, Y.; Liang, S.; Yang, M.; Robinson, N.; Liu, J. Can Chinese Medicine Be Used for Prevention of Corona Virus Disease 2019 (COVID-19)? A Review of Historical Classics, Research Evidence and Current Prevention Programs. Chin. J. Integr. Med. 2020, 26, 243–250. [Google Scholar] [CrossRef]
- MacKenzie, D. The hunt for covid-19 drugs. New Sci. 2020, 245, 10. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Talele, T.; Khedkar, S.; Rigby, A. Successful Applications of Computer Aided Drug Discovery: Moving Drugs from Concept to the Clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, M. The Success of Natural Products in Drug Discovery. Pharmacol. Pharm. 2013, 4, 17–31. [Google Scholar] [CrossRef]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.-C. TCM Database@Taiwan: The World’s Largest Traditional Chinese Medicine Database for Drug Screening In Silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A Select Highly Potent and Diverse Natural Product Library from African Medicinal Plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Hsu, W.C.; Lin, C.C. Antiviral natural products and herbal medicines. J. Tradit. Complement. Med. 2014, 4, 24–35. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Broni, E.; Teye, J.; Quansah, E.; Issah, I.; Wilson, M.D.; Miller, W.A.; Tiburu, E.K.; Bonney, J.H.K. Pharmacoinformatics-based identification of potential bioactive compounds against Ebola virus protein VP24. Comput. Biol. Med. 2019, 113, 103414. [Google Scholar] [CrossRef]
- de Castro, S.; Camarasa, M.J. Polypharmacology in HIV inhibition: Can a drug with simultaneous action against two relevant targets be an alternative to combination therapy? Eur. J. Med. Chem. 2018, 150, 206–227. [Google Scholar] [CrossRef]
- Pizzorno, A.; Padey, B.; Terrier, O.; Rosa-Calatrava, M. Drug repurposing approaches for the treatment of influenza viral infection: Reviving old drugs to fight against a long-lived enemy. Front. Immunol. 2019, 10, 531. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, T.A. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003, 31, 3352–3355. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of Key Interactions Between SARS-CoV-2 Main Protease and Inhibitor Drug Candidates. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Shi, J.; Song, J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Agents 2020, 56, 106012. [Google Scholar] [CrossRef]
- Salman, S.; Shah, F.H.; Idrees, J.; Idrees, F.; Velagala, S.; Ali, J.; Khan, A.A. Virtual screening of immunomodulatory medicinal compounds as promising anti-SARS-CoV-2 inhibitors. Future Virol. 2020, 15, 267–275. [Google Scholar] [CrossRef]
- Maurya, S.K.; Maurya, A.K.; Mishra, N.; Siddique, H.R. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J. Recept. Signal Transduct. 2020, 40, 605–612. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef]
- He, J.; Tao, H.; Yan, Y.; Huang, S.-Y.; Xiao, Y. Molecular Mechanism of Evolution and Human Infection with SARS-CoV-2. Viruses 2020, 12, 428. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, O.V.; Rocha, G.B.; Paluch, A.S.; Costa, L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kiran, G.; Karthik, L.; Shree Devi, M.S.; Sathiyarajeswaran, P.; Kanakavalli, K.; Kumar, K.M.; Ramesh Kumar, D. In Silico computational screening of Kabasura Kudineer—Official Siddha Formulation and JACOM against SARS-CoV-2 spike protein. J. Ayurveda Integr. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods Mol. Biol. 2008, 443, 365–382. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Chang, M.W.; Lindstrom, W.; Olson, A.J.; Belew, R.K. Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries. J. Chem. Inf. Model. 2007, 47, 1258–1262. [Google Scholar] [CrossRef]
- Ray, A.K.; Gupta, P.S.S.; Panda, S.K.; Biswal, S.; Rana, M.K. Repurposing of FDA Approved Drugs for the Identification of Potential Inhibitors of SARS-CoV-2 Main Protease. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Chen, Y.W.; Yiu, C.-P.B.; Wong, K.-Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: Virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research 2020, 9, 129. [Google Scholar] [CrossRef]
- Nejat, R.; Sadr, A.S. Are Losartan and Imatinib Effective Against SARS-CoV2 Pathogenesis? A Pathophysiologic-Based in Silico Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Cheke, R.S. The Molecular Docking Study of Potential Drug Candidates Showing Anti-COVID-19 Activity by Exploring of Therapeutic Targets of SARS-CoV-2. Eurasian J. Med. Oncol. 2020, 4, 185–195. [Google Scholar] [CrossRef]
- Miroshnychenko, K.; Shestopalova, A.V. Combined Use of Amentoflavone and Ledipasvir Could Interfere with Binding of Spike Glycoprotein of SARS-CoV-2 to ACE2: The Results of Molecular Docking Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Chandel, V.; Raj, S.; Rathi, B.; Kumar, D. In silico identification of potent fda approved drugs against coronavirus covid-19 main protease: A drug repurposing approach. Chem. Biol. Lett. 2020, 7, 166–175. [Google Scholar] [CrossRef]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis for receptor recognition by the novel coronavirus from Wuhan. Nature 2020. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Choudhary, S.; Malik, Y.S.; Tomar, S. Identification of SARS-CoV-2 Cell Entry Inhibitors by Drug Repurposing using in silico Structure-based Virtual Screening Approach. Front. Immunol. 2020, 11, 1664. [Google Scholar] [CrossRef]
- Mazzon, M.; Mercer, J. Lipid interactions during virus entry and infection. Cell. Microbiol. 2014, 16, 1493–1502. [Google Scholar] [CrossRef]
- Alen, M.M.F.; Schols, D. Dengue virus entry as target for antiviral therapy. J. Trop. Med. 2012, 2012, 628475. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Kracht, M.; Saul, V.V. The intricate interplay between RNA viruses and NF-κB. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Wu, K.L.; Zhang, X.; Deng, S.; Peng, B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J. Integr. Med. 2020, 18, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Barlough, J.E.; Shacklett, B.L. Antiviral studies of feline infectious peritonitis virus in vitro. Vet. Rec. 1994, 135, 177–179. [Google Scholar] [CrossRef]
- Wen, C.C.; Kuo, Y.H.; Jan, J.T.; Liang, P.H.; Wang, S.Y.; Liu, H.G.; Lee, C.K.; Chang, S.T.; Kuo, C.J.; Lee, S.S.; et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Bailas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.S.; Lee, K.H. Anti-aids agents, 11. betulinic acid and platanic acid as anti-HIV principles from syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994, 57, 243–247. [Google Scholar] [CrossRef]
- Mayaux, J.F.; Bousseau, A.; Pauwels, R.; Huet, T.; Hénin, Y.; Dereu, N.; Evers, M.; Soler, F.; Poujade, C.; De Clercq, E.; et al. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 3564–3568. [Google Scholar] [CrossRef]
- Soler, F.; Poujade, C.; Evers, M.; Carry, J.C.; Hénin, Y.; Bousseau, A.; Huet, T.; Pauwels, R.; De Clercq, E.; Mayaux, J.F.; et al. Betulinic acid derivatives: A new class of specific inhibitors of human immunodeficiency virus type 1 entry. J. Med. Chem. 1996, 39, 1069–1083. [Google Scholar] [CrossRef]
- Zhao, H.; Caflisch, A. Molecular dynamics in drug design. Eur. J. Med. Chem. 2015, 91, 4–14. [Google Scholar] [CrossRef]
- Perez, A.; Morrone, J.A.; Simmerling, C.; Dill, K.A. Advances in free-energy-based simulations of protein folding and ligand binding. Curr. Opin. Struct. Biol. 2016, 36, 25–31. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular dynamics-driven drug discovery: Leaping forward with confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Elmezayen, A.D.; Al-Obaidi, A.; Şahin, A.T.; Yelekçi, K. Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kiel, C.; Case, D.A. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J. Mol. Biol. 2003, 330, 891–913. [Google Scholar] [CrossRef]
- Deng, N.J.; Zhang, P.; Cieplak, P.; Lai, L. Elucidating the energetics of entropically driven protein-ligand association: Calculations of absolute binding free energy and entropy. J. Phys. Chem. B 2011, 115, 11902–11910. [Google Scholar] [CrossRef]
- Campanera, J.M.; Pouplana, R. MMPBSA decomposition of the binding energy throughout a molecular dynamics simulation of amyloid-beta (Aß10-35) aggregation. Molecules 2010, 15, 2730–2748. [Google Scholar] [CrossRef]
- Gupta, A.; Chaudhary, N.; Aparoy, P. MM-PBSA and per-residue decomposition energy studies on 7-Phenyl-imidazoquinolin-4(5H)-one derivatives: Identification of crucial site points at microsomal prostaglandin E synthase-1 (mPGES-1) active site. Int. J. Biol. Macromol. 2018, 119, 352–359. [Google Scholar] [CrossRef]
- Lee, V.S.; Tue-ngeun, P.; Nangola, S.; Kitidee, K.; Jitonnom, J.; Nimmanpipug, P.; Jiranusornkul, S.; Tayapiwatana, C. Pairwise decomposition of residue interaction energies of single chain Fv with HIV-1 p17 epitope variants. Mol. Immunol. 2010, 47, 982–990. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Dankwa, B.; Enninful, K.S.; Adobor, C.; Broni, E.; Ntiamoah, A.; Wilson, M.D. Molecular docking and dynamics simulation studies predict munc18b as a target of mycolactone: A plausible mechanism for granule exocytosis impairment in Buruli Ulcer Pathogenesis. Toxins 2019, 11, 181. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Doiphode, S.; Vyas, R.; Swamy, K.V. Molecular docking and simulation studies on SARS-CoV-2 Mpro reveals Mitoxantrone, Leucovorin, Birinapant, and Dynasore as potent drugs against COVID-19. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Joshi, T.; Pundir, H.; Mathpal, S.; Chandra, S. Structure-based screening of novel lichen compounds against SARS Coronavirus main protease (Mpro) as potentials inhibitors of COVID-19. Mol. Divers. 2020. [Google Scholar] [CrossRef] [PubMed]
- Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.H.; Tahir ul Qamar, M.; Li, G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: Integrated computational approach. J. Mol. Liq. 2020, 320, 114493. [Google Scholar] [CrossRef] [PubMed]
- Razzaghi-Asl, N.; Ebadi, A.; Shahabipour, S.; Gholamin, D. Identification of a potential SARS-CoV2 inhibitor via molecular dynamics simulations and amino acid decomposition analysis. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.J.; Froufe, H.J.C.; Costa, A.F.T.; Santos, A.F.; Oliveira, L.G.; Osório, S.R.M.; Abreu, R.M.V.; Pintado, M.; Ferreira, I.C.F.R. Docking studies in target proteins involved in antibacterial action mechanisms: Extending the knowledge on standard antibiotics to antimicrobial mushroom compounds. Molecules 2014, 19, 1672–1684. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2017; Volume 1607, pp. 627–641. [Google Scholar]
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J. Enzyme Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Harrison, C. Coronavirus puts drug repurposing on the fast track. Nat. Biotechnol. 2020, 38, 379–381. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Gajula, M.; Kumar, A.; Ijaq, J. Protocol for Molecular Dynamics Simulations of Proteins. Bio-Protocol 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Vaught, A. Graphing with Gnuplot and Xmgr: Two graphing packages available under Linux. Linux J. 1996, 1996, 7. [Google Scholar]
- Parasuraman, S. Prediction of activity spectra for substances. J. Pharmacol. Pharmacother. 2011, 2, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Dankwa, B.; Odame, E.A.; Agamah, F.E.; Doe, L.; Teye, J.; Agyapong, O.; Miller, W.A.; Mosi, L.; Wilson, M.D. In Silico Screening of Isocitrate Lyase for Novel Anti-Buruli Ulcer Natural Products Originating from Africa. Molecules 2018, 23, 1550. [Google Scholar] [CrossRef] [PubMed]
- Jamkhande, P.; Barde, S. Evaluation of anthelmintic activity and in silico PASS assisted prediction of Cordia dichotoma (Forst.) root extract. Anc. Sci. Life 2014, 34, 39. [Google Scholar] [CrossRef] [PubMed]
- Stasevich, M.V.; Zvarich, V.I.; Novikov, V.P.; Zagorodnyaya, S.D.; Povnitsa, O.Y.; Chaika, M.A.; Nesterkina, M.V.; Kravchenko, I.A.; Druzhilovskii, D.S.; Poroikov, V.V. 9,10-Anthraquinone Dithiocarbamates as Potential Pharmaceutical Substances with Pleiotropic Actions: Computerized Prediction of Biological Activity and Experimental Validation. Pharm. Chem. J. 2020, 53, 905–913. [Google Scholar] [CrossRef]
- Cheng, X.; Ivanov, I. Molecular dynamics. Methods Mol. Biol. 2012, 929, 243–285. [Google Scholar]
- Dong, Y.W.; Liao, M.L.; Meng, X.L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef]
- Sinha, S.; Wang, S.M. Classification of VUS and unclassified variants in BRCA1 BRCT repeats by molecular dynamics simulation. Comput. Struct. Biotechnol. J. 2020, 18, 723–736. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pocket Area (Å2) | Volume (Å3) | Residues Lining Pockets | |
|---|---|---|---|
| Mpro | |||
| 1 | 557.1 | 920.6 | Thr24, Thr25, Thr26, Leu27, His41, Cys44, Thr45, Ser46, Met49, Leu50, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, Asp187, Arg188, Gln189, Thr190, Gln192 |
| 2 | 380.0 | 465.1 | Met6, Ala7, Phe8, Pro9, Gly11, Lys12, Val13, Gln127, Phe150, Ile152, Asp153, Tyr154, Val157, Phe291, Asp295, Arg298, Gln299, Val303, Thr304 |
| 3 | 107.5 | 151.3 | Phe3, Arg4, Lys5, Trp207, Leu282, Ser284, Glu288, Phe291 |
| 4 | 193.5 | 225.2 | Pro108, Gly109, Gln110, Pro132, Ile200, Thr201, Val202, Asn203, Glu240, His246, Ile249, Thr292, Pro293, Phe294 |
| 5 | 86.6 | 131.4 | Glu14, Gly15, Met17, Val18, Trp31, Ala70, Gly71, Val73, Asn95, Lys97 |
| RBD | |||
| A | 148.3 | 169.2 | Arg454, Phe456, Arg457, Lys458, Asp467, Ser469, Glu471, Ile472, Tyr473, Pro491 |
| B | 52.3 | 90.3 | Phe342, Asn343, Leu368, Ser371, Ser373, Phe374 |
| C | 146.8 | 160.6 | Glu340, Val341, Ala344, Arg346, Phe347, Ala348, Asn354, Arg355, Lys356, Ala397, Ser399, Val511 |
| Compound | Source | Binding Energy (kcal/mol) | Hydrogen Bonds [Bond Length (Ȧ)] | Hydrophobic Bonds | |||
|---|---|---|---|---|---|---|---|
| Mpro | RBD | Mpro | RBD | Mpro | RBD | ||
| Selected Hits | |||||||
| NANPDB2403 | ANC | −8.1 | −7.8 | Leu287 (3.22) | - | Thr199, Tyr237, Tyr239, Leu271, Leu272, Leu286. | Leu335, Cys336, Phe338, Phe342, Asn343, Asp364, Val367, Leu368, Ser371, |
| NANPDB2245 | ANC | −8.0 | −7.7 | Arg131 (2.92) | Asn343 (2.96) | Lys137, Thr199, Tyr237, Tyr239, Leu271, Leu272, Leu286, Leu287, Asp289 | Leu335, Cyc336, Phe338, Gly339, Asp364, Val367, Leu368, Ser371, Phe374 |
| ZINC000055656943 | ML | −8.0 | −8.0 | Asp197 (2.80) | - | Arg131, Thr198, Thr199, Tyr237, Tyr239, Leu272, Leu287 | Leu335, Cys336, Phe338, Phe342, Asp364, Val367, Leu368, Ser371, Phe374, |
| ZINC000095486008 | ANC | −8.2 | −7.8 | Lys5 (3.1), Glu288 (3.02) | Cys336 (2.96), Phe338 (3.26), Gly339 (3.3) | Lys137, Asp197, Thr199, Tyr239, Leu272, Leu286, Leu287, Asp289, Glu290 | Pro337, Phe342, Asn343, Val367, Leu368, Ser371, Phe374, Trp436 |
| ZINC001645993538 | ML | −7.7 | −7.5 | Thr199 (314) | - | Lys137, Asp197, Tyr239, Leu272, Leu286, Leu287, Glu288, Asp289 | Cys336, Phe338, Asp364, Val367, Leu368, Ser371, Phe374 |
| Known Antivirals and Experimental Drugs | |||||||
| Oxymetholone | −7.8 | −7.7 | Thr25 (2.81), Glu166 (2.9, 3.00) | Cys336 (3.0), Asn343 (3.09) | His41, Ser46, Thr45, Asn142, Gly143, Cys145, His164, Met165 | Leu335, Phe338, Gly339, Phe342, Asp364, Val367, Leu368, Ser371, Phe374 | |
| Dexamethasone | −7.6 | −6.7 | Asp197 (2.9, 3.24), Met276 (3.01), Leu287 (3.29, 3.32) | Arg355 (2.99, 3.01), Thr430 (3.18), Glu516 (2.76) | Lys137, Thr198, Thr199, Tyr239, Leu271, Gly275, Leu286, Leu287, Asp289 | Pro426, Phe429, Pro463, Phe464, Phe515 | |
| Remdesivir | −6.8 | −6.3 | Lys137 (3.19), Thr199 (2.82, 3.05), Leu287 (3.09), Asp289 (2.84) | Gly496 (2.84, 2.96), Asn501 (2.9) | Arg131, Asp197, Thr198, Tyr237, Asn238, Tyr239, Leu271, Leu272, Asn274, Gly275, Met276, Leu286 | Arg403, Tyr453, Leu455, Ser494, Tyr495, Phe497, Tyr505 | |
| Hydroxychloroquine | −5.9 | −5.5 | Asp197 (3.05, 3.22), Thr199 (3.26) | Thr345 (2.97), Asn354 (3.04), Ala397 (2.72), Ser399 (2.99), | Arg131, Thr198, Tyr237, Tyr239, Leu272, Met276, Ala285, Leu286, Leu287, Asp289 | Glu340, Val341, Ala344, Arg346, Phe347, Ala348, Arg355, Lys356, Asp398 | |
| Chloroquine | −5.5 | −4.9 | Tyr239 (3.2) | - | Arg131, Asp197, Thr198, Thr199, Tyr237, Leu272, Leu286, Leu287, Asp289, | Arg403, Tyr449, Tyr453, Ser494, Tyr495, Gly496, Phe497, Asn501, Tyr505 | |
| Compound | van der Waals Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | SASA Energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| Mpro | |||||
| NANPDB2245 | −85.61 +/− 11.970 | −6.274 +/− 7.537 | 46.495 +/− 10.814 | −10.829 +/− 1.110 | −56.223 +/− 11.988 |
| NANPDB2403 | −77.965 +/− 12.063 | −6.624 +/− 7.992 | 36.397 +/− 13.775 | −9.939 +/− 1.139 | −58.132 +/− 13.000 |
| ZINC000095486008 | −98.620 +/− 15.067 | −20.464 +/− 14.240 | 84.718 +/− 29.042 | −12.692 +/− 1.538 | −47.058 +/− 20.877 |
| ZINC000055656943 | −18.966 +/− 26.649 | −2.907 +/− 8.983 | 7.468 +/− 57.684 | −2.692 +/− 4.061 | −17.097 +/− 45.262 |
| ZINC001645993538 | −84.952 +/− 12.296 | −20.470 +/− 13.867 | 62.338 +/− 24.852 | −10.702 +/− 1.140 | −53.785 +/− 18.652 |
| Oxymetholone | −60.820 +/− 13.039 | −3.207 +/− 5.288 | 27.787 +/− 20.226 | −8.485 +/− 1.967 | −44.724 +/− 17.562 |
| Remdesivir | −114.276 +/− 18.798 | −19.410 +/− 12.604 | 89.056 +/− 41.414 | −13.726 +/− 2.248 | −58.356 +/− 31.051 |
| RBD | |||||
| NANPDB2245 | −30.310 +/− 43.669 | −2.337 +/− 4.496 | 14.435 +/− 40.707 | −3.930 +/− 5.769 | −22.142 +/− 39.775 |
| NANPDB2403 | −79.080 +/− 14.764 | −2.714 +/− 7.624 | 39.552 +/− 18.265 | −10.898 +/− 1.698 | −53.140 +/− 20.905 |
| ZINC000095486008 | −119.217 +/− 10.410 | −8.227 +/− 7.728 | 77.567 +/− 12.472 | −15.298 +/− 1.031 | −65.174 +/− 10.495 |
| ZINC000055656943 | −58.972 +/− 54.205 | −11.991 +/− 12.656 | 34.870 +/− 57.870 | −7.003 +/− 6.417 | −43.096 +/− 39.685 |
| ZINC001645993538 | −109.967 +/− 10.090 | −0.990 +/− 6.308 | 63.10 +/− 8.655 | −13.921 +/− 0.760 | −61.778 +/− 9.594 |
| Oxymetholone | −109.874 +/− 9.028 | −15.240 +/− 7.816 | 74.123 +/− 15.363 | −13.752 +/− 0.876 | −64.742 +/− 14.235 |
| Remdesivir | −100.708 +/− 18.622 | −11.616 +/− 11.476 | 80.060 +/− 24.762 | −12.206 +/− 1.981 | −44.471 +/− 19.222 |
| Mpro | ||||
|---|---|---|---|---|
| Pre-MD Interactions | Post-MD Interactions (100 ns) | |||
| Compound Name | H-Bond Residues | Hydrophobic Bond Residues | H-Bond Residues | Hydrophobic Bond Residues |
| NANPDB2403 | Thr199, Leu287 | Tyr237, Tyr239, Leu271, Leu272, Leu286 | - | Tyr237, Leu271, Leu272, Leu287 |
| ZINC95486008 | Gly5, Glu288 | Lys137, Asp197, Thr199, Tyr239, Leu272, Leu286, Leu287, Asp289, Glu290 | - | Trp31, Ala70, Gly71, Asn72, Val73, Leu75, Ala94 |
| RBD | ||||
|---|---|---|---|---|
| Pre-MD Interactions | Post-MD Interactions (100 ns) | |||
| Compound Name | H-Bond Residues | Hydrophobic Bond Residues | H-Bond Residues | Hydrophobic Bond Residues |
| NANPDB2403 | - | Leu335, Cys336, Phe338, Phe342, Asn343, Asp364, Val367, Leu368, Ser371 | - | Gly339, Phe342, Val367, Ser373, Phe374 |
| ZINC95486008 | Cys336, Phe338,Gly339 | Pro337, Phe342, Asn343, Val367, Leu368, Ser371, Phe374, Trp436 | Asn343 | Cys336, Gly339, Phe342, Asp364, Val367, Ser373 |
| Ligand ID | Common/IUPAC Name | 2D Structure |
|---|---|---|
| NANPDB2245 | Helioscopinolide B |  |
| NANPDB2403 | Retusolide B |  |
| Fusidic acid | Fusidic acid |  |
| ZINC000095486008 | (2R,10R,18S)-17,17-dimethyl-3,16-dioxapentacyclo (1 1.8.0.02,10.04,9.015,20) henicosa-1(13),4,6,8,14,20-hexaene-6,18-diol |  |
| ZINC000055656943 | (4S)-7-fluoro-N-((1S)-5-fluoro-2,3-dihydro-1H-inden-1-yl)-2-oxo-1,2,3,4-tetrahydroquinoline-4-carboxamide |  |
| ZINC001645993538 | (4S)-N-((1R,3R,6R)-7,7-difluorobicyclo (4.1.0) heptan-3-yl)-7-fluoro-2-oxo-1,2,3,4-tetrahydroquinoline-4-carboxamide |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwofie, S.K.; Broni, E.; Asiedu, S.O.; Kwarko, G.B.; Dankwa, B.; Enninful, K.S.; Tiburu, E.K.; Wilson, M.D. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 2021, 26, 406. https://doi.org/10.3390/molecules26020406
Kwofie SK, Broni E, Asiedu SO, Kwarko GB, Dankwa B, Enninful KS, Tiburu EK, Wilson MD. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules. 2021; 26(2):406. https://doi.org/10.3390/molecules26020406
Chicago/Turabian StyleKwofie, Samuel K., Emmanuel Broni, Seth O. Asiedu, Gabriel B. Kwarko, Bismark Dankwa, Kweku S. Enninful, Elvis K. Tiburu, and Michael D. Wilson. 2021. "Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin" Molecules 26, no. 2: 406. https://doi.org/10.3390/molecules26020406
APA StyleKwofie, S. K., Broni, E., Asiedu, S. O., Kwarko, G. B., Dankwa, B., Enninful, K. S., Tiburu, E. K., & Wilson, M. D. (2021). Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules, 26(2), 406. https://doi.org/10.3390/molecules26020406