Apoptosis in the Extraosseous Calcification Process


Abstract
1. Extraosseous Calcification
1.1. Membrane Vesicles
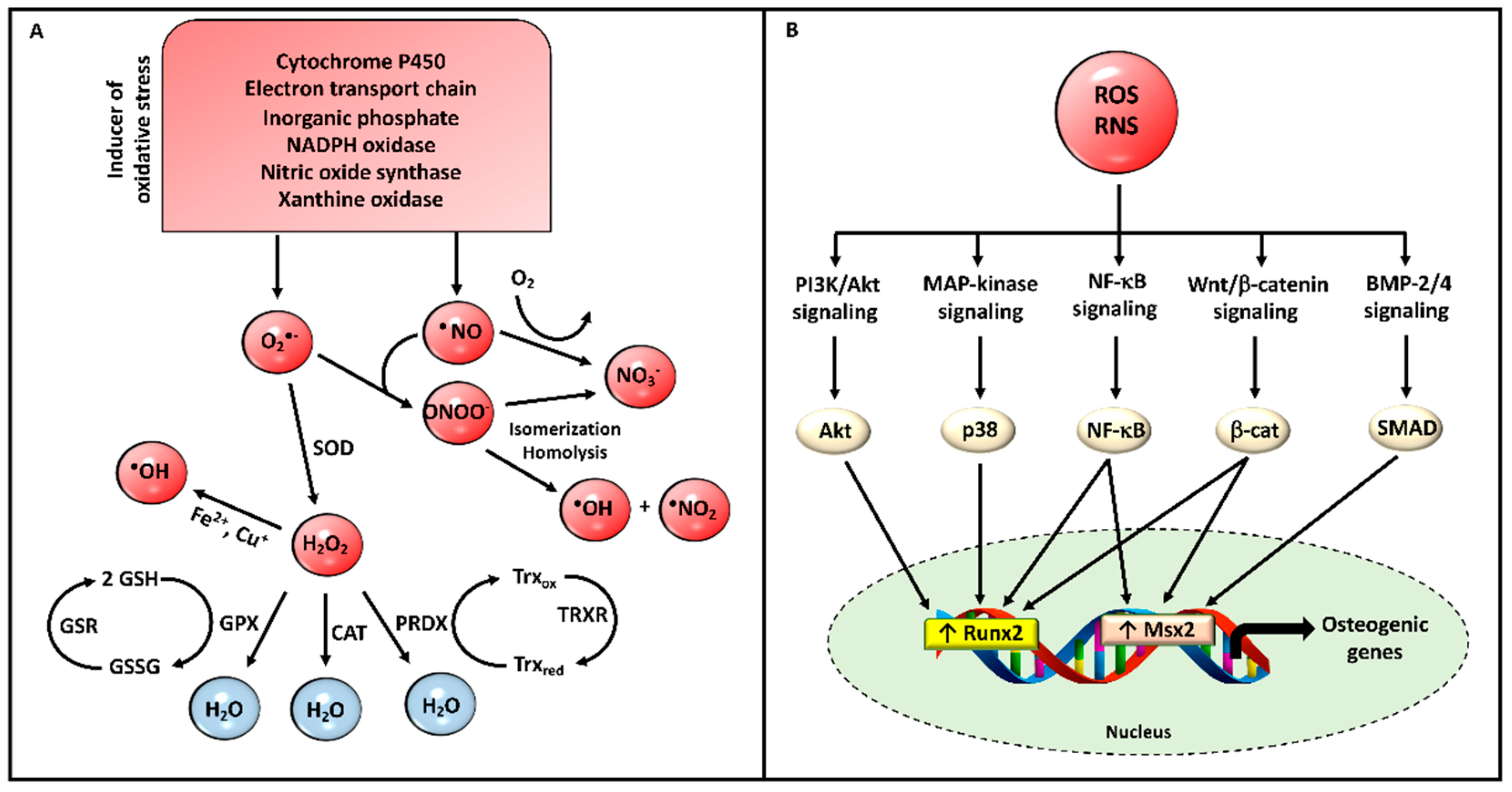
1.2. Mitochondria and Oxidative Stress
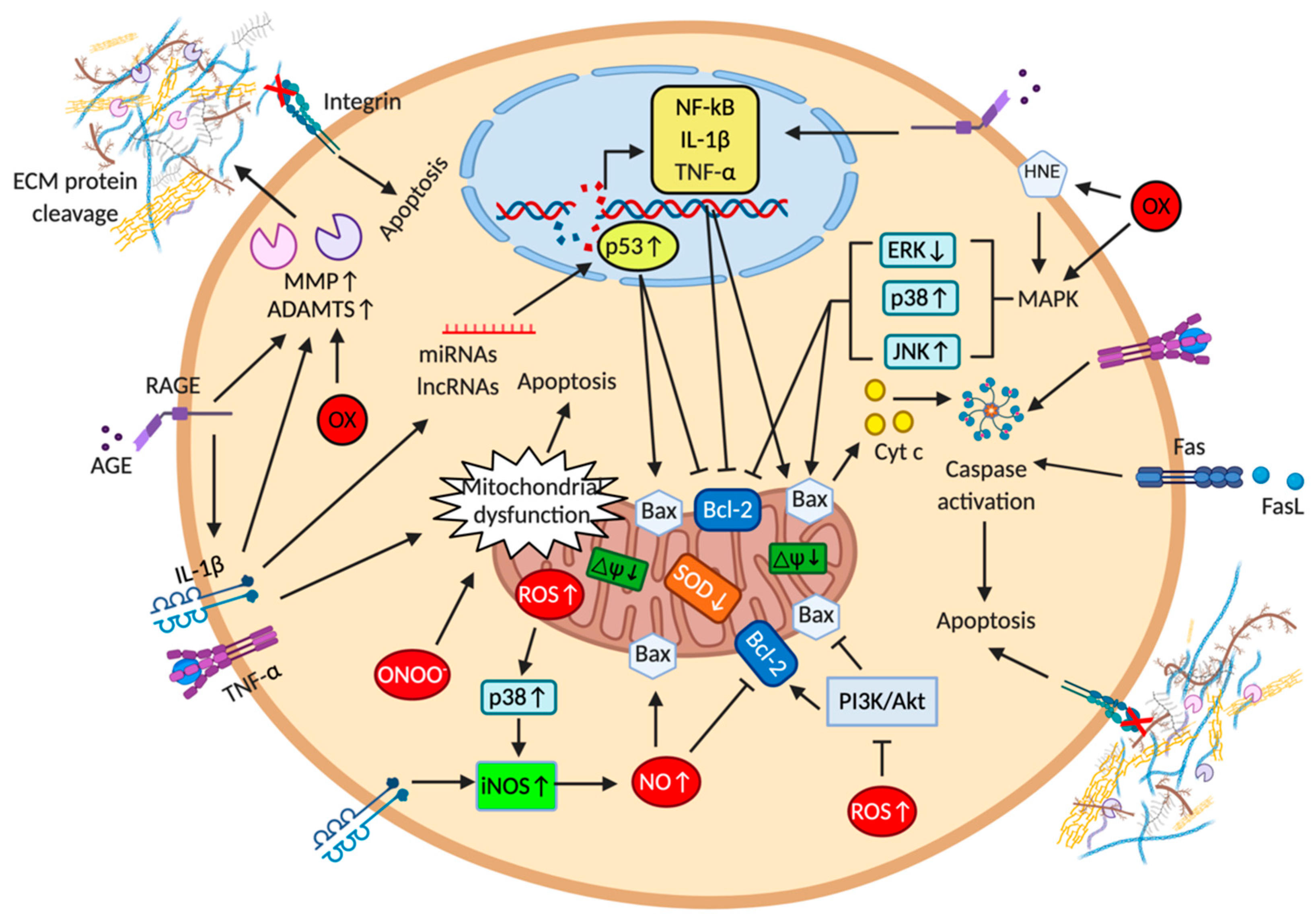
2. Apoptosis and Cartilage Calcification
2.1. Extracellular Matrix Components in the Context of Cartilage Apoptosis
2.2. Death Receptors and Cytokines in the Context of Cartilage Apoptosis
2.3. Mitochondria and Reactive Oxygen and Nitrogen Species in the Context of Cartilage Apoptosis
2.4. Microribonucleic Acids and Long Non-Coding RNA in the Context of Cartilage Apoptosis
3. Apoptosis and Vascular Calcification
3.1. Changes in Matrix Components in the Context of Vascular Apoptosis
3.2. Death Receptors, Cytokines and Growth Factors in the Context of Vascular Apoptosis
3.3. Mitochondria and Reactive Oxygen and Nitrogen Species in the Context of Vascular Apoptosis
3.4. Microribonucleic Acids and Long Non-Coding RNA in the Context of Vascular Apoptosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M. Apoptosis and Calcification. Scanning Microsc. 1995, 9, 1137–1175. [Google Scholar] [PubMed]
- Sharma, H.P.; Jain, P.; Amit, P.; Sinha, A. Apoptosis (Programmed Cell Death)—A review. World J. Pharm. Res. 2014, 3, 1854–1872. [Google Scholar]
- Fuchs, Y.; Steller, H. Live to Die another Way: Modes of Programmed Cell Death and the Signals Emanating from Dying Cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Fung, M.C.; Hardwick, J.M. In Vivo CaspaseTracker Biosensor System for Detecting Anastasis and Non-Apoptotic Caspase Activity. Sci. Rep. 2015, 5, 9015. [Google Scholar] [CrossRef] [PubMed]
- Zakharov, I.I.; Savitskaya, M.A.; Onishchenko, G.E. The Problem of Apoptotic Processes Reversibility. Biochem. Mosc. 2020, 85, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B. Cartilage Histomorphometry. Methods Mol. Med. 2007, 135, 147–166. [Google Scholar] [CrossRef]
- Clarke, B. Normal Bone Anatomy and Physiology. Clin. J. Am. Soc. Nephrol. 2008, 3, S131–S139. [Google Scholar] [CrossRef]
- Li, Q.; Uitto, J. Mineralization/Anti-Mineralization Networks in the Skin and Vascular Connective Tissues. Am. J. Pathol. 2013, 183, 10–18. [Google Scholar] [CrossRef]
- Boraldi, F.; Lofaro, F.D.; Costa, S.; Moscarelli, P.; Quaglino, D. Rare Co-Occurrence of Beta-Thalassemia and Pseudoxanthoma Elasticum: Novel Biomolecular Findings. Front. Med. 2020, 6, 322. [Google Scholar] [CrossRef]
- Giachelli, C.M. Ectopic Calcification: Gathering Hard Facts about Soft Tissue Mineralization. Am. J. Pathol. 1999, 154, 671–675. [Google Scholar] [CrossRef]
- Mackey, R.H.; Venkitachalam, L.; Sutton-Tyrrell, K. Calcifications, Arterial Stiffness and Atherosclerosis. Atheroscler. Large Arter. Cardiovasc. Risk 2007, 44, 234–244. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, Q.; Uitto, J. Ectopic Mineralization Disorders of the Extracellular Matrix of Connective Tissue: Molecular Genetics and Pathomechanisms of Aberrant Calcification. Matrix Biol. 2014, 33, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, D.; Boraldi, F.; Lofaro, F.D. The Biology of Vascular Calcification. Int. Rev. Cell Mol. Biol. 2020, 354, 261–353. [Google Scholar] [CrossRef]
- Kirsch, T. Biomineralization—An Active or Passive Process? Connect. Tissue Res. 2012, 53, 438–445. [Google Scholar] [CrossRef]
- Kirsch, T. Determinants of Pathological Mineralization. Curr. Opin. Rheumatol. 2006, 18, 174–180. [Google Scholar] [CrossRef]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous Calcification Inhibitors in the Prevention of Vascular Calcification: A Consensus Statement From the COST Action EuroSoftCalcNet. Front. Cardiovas. Med. 2018, 5. [Google Scholar] [CrossRef]
- Black, A.; Kanat, I. A Review of Soft Tissue Calcifications. J. Foot Surg. 1985, 24, 243–250. [Google Scholar]
- Meyers, C.; Lisiecki, J.; Miller, S.; Levin, A.; Fayad, L.; Ding, C.; Sono, T.; McCarthy, E.; Levi, B.; James, A.W. Heterotopic Ossification: A Comprehensive Review. JBMR Plus 2019, 3. [Google Scholar] [CrossRef]
- Seethapathy, H.; Noureddine, L. Calciphylaxis: Approach to Diagnosis and Management. Adv. Chronic Kidney Dis. 2019, 26, 484–490. [Google Scholar] [CrossRef]
- Kim, K.M.; Trump, B.F. Amorphous Calcium Precipitations in Human Aortic Valve. Calcfied Tissue Res. 1975, 18, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Michotte, Y.; Smeyers-Verbeke, J.; Ebinger, G.; Maurus, R.; Pelsmaekers, J.; Lowenthal, A.; Massart, D.L. Brain Calcification in a Case of Acute Lymphoblastic Leukaemia. J. Neurol. Sci. 1975, 25, 145–152. [Google Scholar] [CrossRef]
- Prieto, R.M.; Gomila, I.; Söhnel, O.; Costa-Bauza, A.; Bonnin, O.; Grases, F. Study on the Structure and Composition of Aortic Valve Calcific Deposits. Etiological Aspects. JBPC 2011, 02, 19–25. [Google Scholar] [CrossRef][Green Version]
- Mikroulis, D.; Mavrilas, D.; Kapolos, J.; Koutsoukos, P.G.; Lolas, C. Physicochemical and Microscopical Study of Calcific Deposits from Natural and Bioprosthetic Heart Valves. Comparison and Implications for Mineralization Mechanism. J. Mater. Sci. Mater. Med. 2002, 13, 885–889. [Google Scholar] [CrossRef]
- Evan, A.P.; Coe, F.L.; Lingeman, J.E.; Worcester, E. Insights on the Pathology of Kidney Stone Formation. Urol. Res. 2005, 33, 383–389. [Google Scholar] [CrossRef]
- Riley, G.P.; Harrall, R.L.; Constant, C.R.; Cawston, T.E.; Hazleman, B.L. Prevalence and Possible Pathological Significance of Calcium Phosphate Salt Accumulation in Tendon Matrix Degeneration. Ann. Rheum. Dis. 1996, 55, 109–115. [Google Scholar] [CrossRef]
- Ivanovski, O.; Drüeke, T.B. A New Era in the Treatment of Calcium Oxalate Stones? Kidney Int. 2013, 83, 998–1000. [Google Scholar] [CrossRef]
- Perrotta, I.; Perri, E. Ultrastructural, Elemental and Mineralogical Analysis of Vascular Calcification in Atherosclerosis. Microsc. Microanal. 2017, 23, 1030–1039. [Google Scholar] [CrossRef]
- Yang, B.Y.; Sartoris, D.J.; Resnick, D.; Clopton, P. Calcium Pyrophosphate Dihydrate Crystal Deposition Disease: Frequency of Tendon Calcification about the Knee. J. Rheumatol. 1996, 23, 883–888. [Google Scholar]
- Fuerst, M.; Bertrand, J.; Lammers, L.; Dreier, R.; Echtermeyer, F.; Nitschke, Y.; Rutsch, F.; Schäfer, F.K.W.; Niggemeyer, O.; Steinhagen, J.; et al. Calcification of Articular Cartilage in Human Osteoarthritis. Arthritis Rheum. 2009, 60, 2694–2703. [Google Scholar] [CrossRef]
- Ryu, K.; Iriuchishima, T.; Oshida, M.; Kato, Y.; Saito, A.; Imada, M.; Aizawa, S.; Tokuhashi, Y.; Ryu, J. The Prevalence of and Factors Related to Calcium Pyrophosphate Dihydrate Crystal Deposition in the Knee Joint. Osteoarthr. Cartil. 2014, 22, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, H.; Ogawa, R.; Okuma, K.; Harato, K.; Niki, Y.; Kobayashi, S.; Nagura, T. Detection of Calcium Pyrophosphate Dihydrate Crystals in Knee Meniscus by Dual-Energy Computed Tomography. J. Orthop. Surg. Res. 2018, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Yasui, M.; Yase, Y.; Kihira, T.; Adachi, K.; Suzuki, Y. Magnesium and Calcium Contents in CNS Tissues of Amyotrophic Lateral Sclerosis Patients from the Kii Peninsula, Japan. Eur. Neurol. 1992, 32, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.M.; McCord, G.C.; Kumar, R. Hydroxyapatite Crystal Deposition Disease. Semin. Musculoskelet. Radiol. 2003, 7, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Tonelli, M.; Gheduzzi, D.; Ronchetti, I.P.; Quaglino, D. Identification of Mineralized Elastic Fibers on Wet Samples by SEM. Microsc. Res. Tech. 2005, 67, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Bertazzo, S.; Gentleman, E.; Cloyd, K.L.; Chester, A.H.; Yacoub, M.H.; Stevens, M.M. Nano-Analytical Electron Microscopy Reveals Fundamental Insights into Human Cardiovascular Tissue Calcification. Nat. Mater. 2013, 12, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Coe, F.L.; Worcester, E.M.; Evan, A.P. Idiopathic Hypercalciuria and Formation of Calcium Renal Stones. Nat. Rev. Nephrol. 2016, 12, 519–533. [Google Scholar] [CrossRef]
- Sammel, L.M.; Claus, J.R. Calcium Chloride and Tricalcium Phosphate Effects on the Pink Color Defect in Cooked Ground and Intact Turkey Breast. Meat Sci. 2007, 77, 492–498. [Google Scholar] [CrossRef]
- McCarty, D.J. Crystals and Arthritis. Disease-a-Month 1994, 40, 258–299. [Google Scholar] [CrossRef]
- Delogne, C.; Lawford, P.V.; Habesch, S.M.; Carolan, V.A. Characterization of the Calcification of Cardiac Valve Bioprostheses by Environmental Scanning Electron Microscopy and Vibrational Spectroscopy. J. Microsc. 2007, 228, 62–77. [Google Scholar] [CrossRef]
- Burstein, L.S.; Boskey, A.L.; Tannenbaum, P.J.; Posner, A.S.; Mandel, I.D. The Crystal Chemistry of Submandibular and Parotid Salivary Gland Stones. J. Oral. Pathol. Med. 1979, 8, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.Y. Apatite-Type Crystal Deposition in Arthritic Cartilage. Scan. Electron. Microsc. 1985, 4, 1555–1566. [Google Scholar]
- Reid, J.D.; Andersen, M.E. Medial Calcification (Whitlockite) in the Aorta. Atherosclerosis 1993, 101, 213–224. [Google Scholar] [CrossRef]
- Lagier, R.; Baud, C.A. Magnesium Whitlockite, a Calcium Phosphate Crystal of Special Interest in Pathology. Pathol. Res. Pract. 2003, 199, 329–335. [Google Scholar] [CrossRef]
- Kodaka, T.; Hirayama, A.; Sano, T.; Debari, K.; Mayahara, M.; Nakamura, M. Fine Structure and Mineral Components of Primary Calculi in Some Human Prostates. J. Electron. Microsc. 2008, 57, 133–141. [Google Scholar] [CrossRef]
- Scott, R.; Kendall, C.; Stone, N.; Rogers, K. Elemental vs. Phase Composition of Breast Calcifications. Sci. Rep. 2017, 7, 136. [Google Scholar] [CrossRef]
- Karwowski, W.; Naumnik, B.; Szczepański, M.; Myśliwiec, M. The Mechanism of Vascular Calcification—A Systematic Review. Med. Sci. Monit. 2012, 18, RA1–RA11. [Google Scholar] [CrossRef]
- Boraldi, F.; Bartolomeo, A.; De Biasi, S.; Orlando, S.; Costa, S.; Cossarizza, A.; Quaglino, D. Innovative Flow Cytometry Allows Accurate Identification of Rare Circulating Cells Involved in Endothelial Dysfunction. PLoS ONE 2016, 11, e0160153. [Google Scholar] [CrossRef]
- Shira, G.; Ziegler, S.; Gahl, W.; Ferreira, C. Disorders and Mechanisms of Ectopic Calcification. In Genetics of Bone Biology and Skeletal Disease, 2nd ed.; Academic Press: San Diego, CA, USA, 2018; pp. 571–595. ISBN 978-0-12-804182-6. [Google Scholar]
- Opdebeeck, B.; Orriss, I.R.; Neven, E.; D’Haese, P.C.; Verhulst, A. Extracellular Nucleotides Regulate Arterial Calcification by Activating Both Independent and Dependent Purinergic Receptor Signaling Pathways. Int. J. Mol. Sci. 2020, 21, 7636. [Google Scholar] [CrossRef]
- Rilla, K.; Mustonen, A.-M.; Arasu, U.T.; Härkönen, K.; Matilainen, J.; Nieminen, P. Extracellular Vesicles Are Integral and Functional Components of the Extracellular Matrix. Matrix Biol. 2019, 75–76, 201–219. [Google Scholar] [CrossRef]
- Azoidis, I.; Cox, S.C.; Davies, O.G. The Role of Extracellular Vesicles in Biomineralisation: Current Perspective and Application in Regenerative Medicine. J. Tissue Eng. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular Smooth Muscle Cell Calcification Is Mediated by Regulated Exosome Secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Majeska, R.J.; Wuthier, R.E. Studies on Matrix Vesicles Isolated from Chick Epiphyseal Cartilage. Association of Pyrophosphatase and ATPase Activities with Alkaline Phosphatase. Biochim. Biophys. Acta 1975, 391, 51–60. [Google Scholar] [CrossRef]
- Fedde, K.N. Human Osteosarcoma Cells Spontaneously Release Matrix-Vesicle-like Structures with the Capacity to Mineralize. Bone Miner. 1992, 17, 145–151. [Google Scholar] [CrossRef]
- Anderson, H.C. Matrix Vesicles and Calcification. Curr. Rheumatol. Rep. 2003, 5, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Houston, D.A.; Farquharson, C.; MacRae, V.E. Characterisation of Matrix Vesicles in Skeletal and Soft Tissue Mineralisation. Bone 2016, 87, 147–158. [Google Scholar] [CrossRef]
- Hsu, H.H.; Camacho, N.P. Isolation of Calcifiable Vesicles from Human Atherosclerotic Aortas. Atherosclerosis 1999, 143, 353–362. [Google Scholar] [CrossRef]
- Shao, J.-S.; Cai, J.; Towler, D.A. Molecular Mechanisms of Vascular Calcification: Lessons Learned from the Aorta. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1423–1430. [Google Scholar] [CrossRef]
- Golub, E.E. Biomineralization and Matrix Vesicles in Biology and Pathology. Semin. Immunopathol. 2011, 33, 409–417. [Google Scholar] [CrossRef]
- Golub, E. Role of Matrix Vesicles in Biomineralization. Biochim. Biophys. Acta 2009, 1790, 1592–1598. [Google Scholar] [CrossRef]
- Gurley, K.A.; Reimer, R.J.; Kingsley, D.M. Biochemical and Genetic Analysis of ANK in Arthritis and Bone Disease. Am. J. Hum. Genet. 2006, 79, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the Mouse Ank Gene in Control of Tissue Calcification and Arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.C.; Rosenthal, A.K.; Kurup, I.V.; Masuda, I.; Medhora, M.; Ryan, L.M. Parallel Regulation of Extracellular ATP and Inorganic Pyrophosphate: Roles of Growth Factors, Transduction Modulators, and ANK. Connect. Tissue Res. 2011, 52, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Szeri, F.; Lundkvist, S.; Donnelly, S.; Engelke, U.F.H.; Rhee, K.; Williams, C.J.; Sundberg, J.P.; Wevers, R.A.; Tomlinson, R.E.; Jansen, R.S.; et al. The Membrane Protein ANKH Is Crucial for Bone Mechanical Performance by Mediating Cellular Export of Citrate and ATP. PLoS Genet. 2020, 16, e1008884. [Google Scholar] [CrossRef] [PubMed]
- Boskey, A.L.; Maresca, M.; Ullrich, W.; Doty, S.B.; Butler, W.T.; Prince, C.W. Osteopontin-Hydroxyapatite Interactions in Vitro: Inhibition of Hydroxyapatite Formation and Growth in a Gelatin-Gel. Bone Miner. 1993, 22, 147–159. [Google Scholar] [CrossRef]
- Roberts, S.; Narisawa, S.; Harmey, D.; Millán, J.L.; Farquharson, C. Functional Involvement of PHOSPHO1 in Matrix Vesicle–Mediated Skeletal Mineralization. J. Bone Miner. Res. 2007, 22, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Yamamoto, T.; Tsuchiya, E.; Hongo, H.; Tsuboi, K.; Kudo, A.; Abe, M.; Yoshida, T.; Nagai, T.; Khadiza, N.; et al. Ultrastructural and Biochemical Aspects of Matrix Vesicle-Mediated Mineralization. Jpn. Dent. Sci. Rev. 2017, 53, 34–45. [Google Scholar] [CrossRef]
- Orimo, H. The Mechanism of Mineralization and the Role of Alkaline Phosphatase in Health and Disease. J. Nippon Med. Sch. 2010, 77, 4–12. [Google Scholar] [CrossRef]
- Kirsch, T. Annexins—Their Role in Cartilage Mineralization. Front. Biosci. 2005, 10, 576–581. [Google Scholar] [CrossRef]
- Kockx, M.M.; Muhring, J.; Bortier, H.; De Meyer, G.R.; Jacob, W. Biotin- or Digoxigenin-Conjugated Nucleotides Bind to Matrix Vesicles in Atherosclerotic Plaques. Am. J. Pathol. 1996, 148, 1771–1777. [Google Scholar]
- Hashimoto, S.; Ochs, R.L.; Rosen, F.; Quach, J.; McCabe, G.; Solan, J.; Seegmiller, J.E.; Terkeltaub, R.; Lotz, M. Chondrocyte-Derived Apoptotic Bodies and Calcification of Articular Cartilage. Proc. Natl. Acad. Sci. USA 1998, 95, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification in Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Wang, W.; Pfander, D. Functional Differences Between Growth Plate Apoptotic Bodies and Matrix Vesicles. J. Bone Miner. Res. 2003, 18, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, N.; Wu, B.; Wu, S.; Zhang, Y.; Sun, Y. The Role of Mitochondria in Vascular Calcification. J. Transl. Int. Med. 2020, 8, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as Playmakers of Apoptosis, Autophagy and Senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Bonucci, E.; Derenzini, M.; Marinozzi, V. The Organic-Inorganic Relationship in Calcified Mitochondria. J. Cell Biol. 1973, 59, 185. [Google Scholar] [CrossRef]
- Weinbach, E.C.; Von Brand, T. Formation, Isolation and Composition of Dense Granules from Mitochondria. Biochim. Biophys. Acta 1967, 148, 256–266. [Google Scholar] [CrossRef]
- Halstead, L.B. Are Mitochondria Directly Involved in Biological Mineralisation? The Mitochondrion and the Origin of Bone. Calcified Tissue Res. 1969, 3, 103–105. [Google Scholar] [CrossRef]
- Martin, J.H.; Matthews, J.L. Mitochondrial Granules in Chondrocytes, Osteoblasts and Osteocytes. An Ultrastructural and Microincineration Study. Clin. Orthop. Relat. Res. 1970, 68, 273–278. [Google Scholar] [CrossRef]
- Holtrop, M.E. The Ultrastructure of the Epiphyseal Plate. I. The Flattened Chondrocyte. Calcif. Tissue Res. 1972, 9, 131–139. [Google Scholar] [CrossRef]
- Brighton, C.T.; Hunt, R.M. Mitochondrial Calcium and Its Role in Calcification. Histochemical Localization of Calcium in Electron Micrographs of the Epiphyseal Growth Plate with K-Pyroantimonate. Clin. Orthop. Relat. Res. 1974, 100, 406–416. [Google Scholar] [CrossRef]
- Anderson, H.C. Mechanisms of Pathologic Calcification. Rheum. Dis. Clin. N. Am. 1988, 14, 303–319. [Google Scholar]
- Omelon, S.; Ariganello, M.; Bonucci, E.; Grynpas, M.; Nanci, A. A Review of Phosphate Mineral Nucleation in Biology and Geobiology. Calcif. Tissue Int. 2013, 93, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Fenton, A.R.; Jongens, T.A.; Holzbaur, E.L.F. Mitochondrial Dynamics: Shaping and Remodeling an Organelle Network. Curr. Opin. Cell Biol. 2020, 68, 28–36. [Google Scholar] [CrossRef]
- Wolf, C.; López Del Amo, V.; Arndt, S.; Bueno, D.; Tenzer, S.; Hanschmann, E.M.; Berndt, C.; Methner, A. Redox Modifications of Proteins of the Mitochondrial Fusion and Fission Machinery. Cells 2020, 9, 815. [Google Scholar] [CrossRef]
- Chen, W.R.; Zhou, Y.J.; Sha, Y.; Wu, X.P.; Yang, J.Q.; Liu, F. Melatonin Attenuates Vascular Calcification by Inhibiting Mitochondria Fission via an AMPK/Drp1 Signalling Pathway. J. Cell Mol. Med. 2020, 24, 6043–6054. [Google Scholar] [CrossRef]
- Forrester Steven, J.; Kikuchi Daniel, S.; Hernandes Marina, S.; Xu, Q.; Griendling Kathy, K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Liaudet, L.; Vassalli, G.; Pacher, P. Role of Peroxynitrite in the Redox Regulation of Cell Signal Transduction Pathways. Front. Biosci. 2009, 14, 4809–4814. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS Signalling: Adaptive Redox Switches through Oxidative/Nitrosative Protein Modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Ljubuncic, P.; Bar-Shai, M.; Reznick, A. The Role of Reactive Nitrogen Species (RNS) in the Activation of Nuclear Factor Kappa B (NFkB) and Its Implications for Biological Systems: The Question of Balance. In Oxidants in Biology: A Question of Balance; Springer: Dordrecht, The Netherlands, 2008; pp. 67–109. ISBN 978-1-4020-8398-3. [Google Scholar]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81. [Google Scholar] [CrossRef]
- Zhao, G.; Xu, M.; Zhao, M.; Dai, X.; Kong, W.; Wilson, G.; Guan, Y.; Wang, C.; Wang, X. Activation of Nuclear Factor-Kappa B Accelerates Vascular Calcification by Inhibiting Ankylosis Protein Homolog Expression. Kidney Int. 2012, 82, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The Antioxidant Tempol Ameliorates Arterial Medial Calcification in Uremic Rats: Important Role of Oxidative Stress in the Pathogenesis of Vascular Calcification in Chronic Kidney Disease. J. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yamashita, M.; Horimai, C.; Hayashi, M. Smooth Muscle-Selective Nuclear Factor-ΚB Inhibition Reduces Phosphate-Induced Arterial Medial Calcification in Mice with Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e007248. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Miki, H. Redox Regulation of Wnt Signalling via Nucleoredoxin. Free Radic. Res. 2010, 44, 379–388. [Google Scholar] [CrossRef]
- Bandara, N.; Gurusinghe, S.; Lim, S.Y.; Chen, H.; Chen, S.; Wang, D.; Hilbert, B.; Wang, L.-X.; Strappe, P. Molecular Control of Nitric Oxide Synthesis through ENOS and Caveolin-1 Interaction Regulates Osteogenic Differentiation of Adipose-Derived Stem Cells by Modulation of Wnt/β-Catenin Signaling. Stem. Cell Res. Ther. 2016, 7, 182. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-ΚB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Shao, J.-S.; Lai, C.-F.; Huang, E.; Cai, J.; Behrmann, A.; Cheng, S.-L.; Towler, D.A. Aortic Msx2-Wnt Calcification Cascade Is Regulated by TNF-Alpha-Dependent Signals in Diabetic Ldlr-/- Mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2589–2596. [Google Scholar] [CrossRef]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef]
- Byon, C.H.; Heath, J.M.; Chen, Y. Redox Signaling in Cardiovascular Pathophysiology: A Focus on Hydrogen Peroxide and Vascular Smooth Muscle Cells. Redox Biol. 2016, 9, 244–253. [Google Scholar] [CrossRef]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef]
- Johnson, K.A.; Polewski, M.; Terkeltaub, R.A. Transglutaminase 2 Is Central to Induction of the Arterial Calcification Program by Smooth Muscle Cells. Circ. Res. 2008, 102, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Harris, S.E.; Ghosh-Choudhury, G.; Ghosh-Choudhury, N. Reactive Oxygen Species Derived from Nox4 Mediate BMP2 Gene Transcription and Osteoblast Differentiation. Biochem. J. 2011, 433, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.L.; Shao, J.S.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. MSX2 Promotes Osteogenesis and Suppresses Adipogenic Differentiation of Multipotent Mesenchymal Progenitors. J. Biol. Chem. 2003, 278, 45969–45977. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Bone Development and Extracellular Matrix Protein Genes by RUNX2. Cell Tissue Res. 2010, 339, 189–195. [Google Scholar] [CrossRef]
- Roughley, P.J. Articular Cartilage and Changes in Arthritis: Noncollagenous Proteins and Proteoglycans in the Extracellular Matrix of Cartilage. Arthritis Res. 2001, 3, 342–347. [Google Scholar] [CrossRef]
- Hunziker, E.B. Mechanism of Longitudinal Bone Growth and Its Regulation by Growth Plate Chondrocytes. Microsc. Res. Tech. 1994, 28, 505–519. [Google Scholar] [CrossRef]
- Szuwart, T.; Kierdorf, H.; Kierdorf, U.; Clemen, G. Ultrastructural Aspects of Cartilage Formation, Mineralization, and Degeneration during Primary Antler Growth in Fallow Deer (Dama Dama). Ann. Anat. 1998, 180, 501–510. [Google Scholar] [CrossRef]
- Messner, K. Postnatal Development of the Cruciate Ligament Insertions in the Rat Knee. Morphological Evaluation and Immunohistochemical Study of Collagens Types I and II. Cells Tissues Organs 1997, 160, 261–268. [Google Scholar] [CrossRef]
- Sun, M.M.G.; Beier, F. Chondrocyte Hypertrophy in Skeletal Development, Growth, and Disease. Birth Defects Res. C Embryo Today Rev. 2014, 102, 74–82. [Google Scholar] [CrossRef]
- Lefebvre, V.; Smits, P. Transcriptional Control of Chondrocyte Fate and Differentiation. Birth Defects Res. C Embryo Today Rev. 2005, 75, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.S.; Shapiro, I.M. The Fate of the Terminally Differentiated Chondrocyte: Evidence for Microenvironmental Regulation of Chondrocyte Apoptosis. Crit. Rev. Oral Biol. Med. 2002, 13, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Adams, C.S.; Freeman, T.; Srinivas, V. Fate of the Hypertrophic Chondrocyte: Microenvironmental Perspectives on Apoptosis and Survival in the Epiphyseal Growth Plate. Birth Defects Res. C Embryo Today Rev. 2005, 75, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, J.; Moore, D.C.; Zuo, C.; Wu, Q.; Xie, L.; von der Mark, K.; Yuan, X.; Chen, D.; Warman, M.L.; et al. SHP2 Regulates the Osteogenic Fate of Growth Plate Hypertrophic Chondrocytes. Sci. Rep. 2017, 7, 12699. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Chondrocyte Hypertrophy and Osteoarthritis: Role in Initiation and Progression of Cartilage Degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R.; Terkeltaub, R. Emerging Regulators of the Inflammatory Process in Osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-ΚB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Guitian, R.; Vázquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis Chondrocytes Die by Apoptosis. A Possible Pathway for Osteoarthritis Pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Sharif, M.; Whitehouse, A.; Sharman, P.; Perry, M.; Adams, M. Increased Apoptosis in Human Osteoarthritic Cartilage Corresponds to Reduced Cell Density and Expression of Caspase-3. Arthritis Rheum. 2004, 50, 507–515. [Google Scholar] [CrossRef]
- Huppertz, B.; Frank, H.G.; Kaufmann, P. The Apoptosis Cascade--Morphological and Immunohistochemical Methods for Its Visualization. Anat. Embryol. 1999, 200, 1–18. [Google Scholar] [CrossRef]
- Musumeci, G.; Loreto, C.; Carnazza, M.L.; Martinez, G. Characterization of Apoptosis in Articular Cartilage Derived from the Knee Joints of Patients with Osteoarthritis. Knee Surg. Sports Traumatol. Arthrosc. 2011, 19, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte Apoptosis: A Cause or Consequence of Osteoarthritis? Int. J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.T.; Burton-Wurster, N.; Borden, C.; Hueffer, K.; Bloom, S.E.; Lust, G. Chondrocyte Necrosis and Apoptosis in Impact Damaged Articular Cartilage. J. Orthop. Res. 2001, 19, 703–711. [Google Scholar] [CrossRef]
- Kourí, J.B.; Aguilera, J.M.; Reyes, J.; Lozoya, K.A.; González, S. Apoptotic Chondrocytes from Osteoarthrotic Human Articular Cartilage and Abnormal Calcification of Subchondral Bone. J. Rheumatol. 2000, 27, 1005–1019. [Google Scholar]
- Thomas, C.M.; Fuller, C.J.; Whittles, C.E.; Sharif, M. Chondrocyte Death by Apoptosis Is Associated with Cartilage Matrix Degradation. Osteoarthr. Cartil. 2007, 15, 27–34. [Google Scholar] [CrossRef]
- Roach, H.I.; Aigner, T.; Kouri, J.B. Chondroptosis: A Variant of Apoptotic Cell Death in Chondrocytes? Apoptosis 2004, 9, 265–277. [Google Scholar] [CrossRef]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell Death of Chondrocytes Is a Combination between Apoptosis and Autophagy during the Pathogenesis of Osteoarthritis within an Experimental Model. Apoptosis 2010, 15, 631–638. [Google Scholar] [CrossRef]
- Johnson, K.; Pritzker, K.; Goding, J.; Terkeltaub, R. The Nucleoside Triphosphate Pyrophosphohydrolase Isozyme PC-1 Directly Promotes Cartilage Calcification through Chondrocyte Apoptosis and Increased Calcium Precipitation by Mineralizing Vesicles. J. Rheumatol. 2001, 28, 2681–2691. [Google Scholar]
- Hashimoto, S.; Ochs, R.L.; Komiya, S.; Lotz, M. Linkage of Chondrocyte Apoptosis and Cartilage Degradation in Human Osteoarthritis. Arthritis Rheum. 1998, 41, 1632–1638. [Google Scholar] [CrossRef]
- Jiao, K.; Zhang, J.; Zhang, M.; Wei, Y.; Wu, Y.; Qiu, Z.Y.; He, J.; Cao, Y.; Hu, J.; Zhu, H.; et al. The Identification of CD163 Expressing Phagocytic Chondrocytes in Joint Cartilage and Its Novel Scavenger Role in Cartilage Degradation. PLoS ONE 2013, 8, e53312. [Google Scholar] [CrossRef]
- Kirsch, T.; Swoboda, B.; Nah, H. Activation of Annexin II and V Expression, Terminal Differentiation, Mineralization and Apoptosis in Human Osteoarthritic Cartilage. Osteoarthr. Cartil. 2000, 8, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyama, H.; Healey, R.M.; Terkeltaub, R.A.; Coutts, R.D.; Amiel, D. Calcification of Human Articular Knee Cartilage Is Primarily an Effect of Aging Rather than Osteoarthritis. Osteoarthr. Cartil. 2007, 15, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Ochs, R.L.; Schwarz, H.; Lotz, M. Chondrocyte Apoptosis Induced by Nitric Oxide. Am. J. Pathol. 1995, 146, 75–85. [Google Scholar] [PubMed]
- Hashimoto, S.; Setareh, M.; Ochs, R.L.; Lotz, M. Fas/Fas Ligand Expression and Induction of Apoptosis in Chondrocytes. Arthritis Rheum. 1997, 40, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Bartolomeo, A.; Di Bari, C.; Cocconi, A.; Quaglino, D. Donor’s Age and Replicative Senescence Favour the in-Vitro Mineralization Potential of Human Fibroblasts. Exp. Gerontol. 2015, 72, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Burns, J.S.; Bartolomeo, A.; Dominici, M.; Quaglino, D. Mineralization by Mesenchymal Stromal Cells Is Variously Modulated Depending on Commercial Platelet Lysate Preparations. Cytotherapy 2018, 20, 335–342. [Google Scholar] [CrossRef]
- Boraldi, F.; Moscarelli, P.; Lofaro, F.D.; Sabia, C.; Quaglino, D. The Mineralization Process of Insoluble Elastin Fibrillar Structures: Ionic Environment vs Degradation. Int. J. Biol. Macromol. 2020, 149, 693–706. [Google Scholar] [CrossRef]
- Fujita, T.; Meguro, T.; Izumo, N.; Yasutomi, C.; Fukuyama, R.; Nakamuta, H.; Koida, M. Phosphate Stimulates Differentiation and Mineralization of the Chondroprogenitor Clone ATDC5. Jnp. J. Pharmacol. 2001, 85, 278–281. [Google Scholar] [CrossRef][Green Version]
- Magne, D.; Bluteau, G.; Faucheux, C.; Palmer, G.; Vignes-Colombeix, C.; Pilet, P.; Rouillon, T.; Caverzasio, J.; Weiss, P.; Daculsi, G.; et al. Phosphate Is a Specific Signal for ATDC5 Chondrocyte Maturation and Apoptosis-Associated Mineralization: Possible Implication of Apoptosis in the Regulation of Endochondral Ossification. J. Bone Miner. Res. 2003, 18, 1430–1442. [Google Scholar] [CrossRef]
- Yuan, F.L.; Xu, R.S.; Ye, J.X.; Zhao, M.D.; Ren, L.J.; Li, X. Apoptotic Bodies from Endplate Chondrocytes Enhance the Oxidative Stress-Induced Mineralization by Regulating PPi Metabolism. J. Cell Mol. Med. 2019, 23, 3665–3675. [Google Scholar] [CrossRef]
- Ea, H.K.; Uzan, B.; Rey, C.; Lioté, F. Octacalcium Phosphate Crystals Directly Stimulate Expression of Inducible Nitric Oxide Synthase through P38 and JNK Mitogen-Activated Protein Kinases in Articular Chondrocytes. Arthritis Res. Ther. 2005, 7, R915–R926. [Google Scholar] [CrossRef] [PubMed]
- Ea, H.K.; Monceau, V.; Camors, E.; Cohen-Solal, M.; Charlemagne, D.; Lioté, F. Annexin 5 Overexpression Increased Articular Chondrocyte Apoptosis Induced by Basic Calcium Phosphate Crystals. Ann. Rheum. Dis. 2008, 67, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.; Lieberherr, M.; Bordat, C.; Velard, F.; Côme, D.; Lioté, F.; Ea, H.-K. Intracellular Calcium Oscillations in Articular Chondrocytes Induced by Basic Calcium Phosphate Crystals Lead to Cartilage Degradation. Osteoarthr. Cartil. 2012, 20, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, J.; Kirsch, T. Annexin-Mediated Ca2+ Influx Regulates Growth Plate Chondrocyte Maturation and Apoptosis. J. Biol. Chem. 2003, 278, 3762–3769. [Google Scholar] [CrossRef]
- Ea, H.-K.; Chobaz, V.; Nguyen, C.; Nasi, S.; van Lent, P.; Daudon, M.; Dessombz, A.; Bazin, D.; McCarthy, G.; Jolles-Haeberli, B.; et al. Pathogenic Role of Basic Calcium Phosphate Crystals in Destructive Arthropathies. PLoS ONE 2013, 8, e57352. [Google Scholar] [CrossRef]
- McCarthy, G.M.; Dunne, A. Calcium Crystal Deposition Diseases—Beyond Gout. Nat. Rev. Rheumatol. 2018, 14, 592–602. [Google Scholar] [CrossRef]
- Blasioli, D.J.; Kaplan, D.L. The Roles of Catabolic Factors in the Development of Osteoarthritis. Tissue Eng. Part B Rev. 2014, 20, 355–363. [Google Scholar] [CrossRef]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Heinegård, D.; Saxne, T. The Role of the Cartilage Matrix in Osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 50–56. [Google Scholar] [CrossRef]
- Homandberg, G.A.; Hui, F.; Wen, C.; Kuettner, K.E.; Williams, J.M. Hyaluronic Acid Suppresses Fibronectin Fragment Mediated Cartilage Chondrolysis: I. In Vitro. Osteoarthr. Cartil. 1997, 5, 309–319. [Google Scholar] [CrossRef]
- Lark, M.W.; Bayne, E.K.; Flanagan, J.; Harper, C.F.; Hoerrner, L.A.; Hutchinson, N.I.; Singer, I.I.; Donatelli, S.A.; Weidner, J.R.; Williams, H.R.; et al. Aggrecan Degradation in Human Cartilage. Evidence for Both Matrix Metalloproteinase and Aggrecanase Activity in Normal, Osteoarthritic, and Rheumatoid Joints. J. Clin. Investig. 1997, 100, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Tchetina, E.V.; Kobayashi, M.; Yasuda, T.; Meijers, T.; Pidoux, I.; Poole, A.R. Chondrocyte Hypertrophy Can Be Induced by a Cryptic Sequence of Type II Collagen and Is Accompanied by the Induction of MMP-13 and Collagenase Activity: Implications for Development and Arthritis. Matrix Biol. 2007, 26, 247–258. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, J. The AGE of the Matrix: Chemistry, Consequence and Cure. Curr. Opin. Pharmacol. 2004, 4, 301–305. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, J.; Verzijl, N.; Bank, R.A.; Lafeber, F.P.; Bijlsma, J.W.; TeKoppele, J.M. Age-Related Decrease in Proteoglycan Synthesis of Human Articular Chondrocytes: The Role of Nonenzymatic Glycation. Arthritis Rheum. 1999, 42, 1003–1009. [Google Scholar] [CrossRef]
- Loeser, R.F.; Yammani, R.R.; Carlson, C.S.; Chen, H.; Cole, A.; Im, H.-J.; Bursch, L.S.; Yan, S.D. Articular Chondrocytes Express the Receptor for Advanced Glycation End Products: Potential Role in Osteoarthritis. Arthritis Rheum. 2005, 52, 2376–2385. [Google Scholar] [CrossRef]
- Kim, H.A.; Suh, D.I.; Song, Y.W. Relationship between Chondrocyte Apoptosis and Matrix Depletion in Human Articular Cartilage. J. Rheumatol. 2001, 28, 2038–2045. [Google Scholar]
- Loeser, R.F. Integrins and Chondrocyte–Matrix Interactions in Articular Cartilage. Matrix Biol. 2014, 39, 11–16. [Google Scholar] [CrossRef]
- Pulai, J.I.; Del Carlo, M.; Loeser, R.F. The Alpha5beta1 Integrin Provides Matrix Survival Signals for Normal and Osteoarthritic Human Articular Chondrocytes in Vitro. Arthritis Rheum. 2002, 46, 1528–1535. [Google Scholar] [CrossRef]
- Almonte-Becerril, M.; Costell, M.; Kouri, J.B. Changes in the Integrins Expression Are Related with the Osteoarthritis Severity in an Experimental Animal Model in Rats. J. Orthop. Res. 2014, 32, 1161–1166. [Google Scholar] [CrossRef]
- Zemmyo, M.; Meharra, E.J.; Kühn, K.; Creighton-Achermann, L.; Lotz, M. Accelerated, Aging-Dependent Development of Osteoarthritis in Α1 Integrin–Deficient Mice. Arthritis Rheum. 2003, 48, 2873–2880. [Google Scholar] [CrossRef]
- Pennock, A.T.; Robertson, C.M.; Emmerson, B.C.; Harwood, F.L.; Amiel, D. Role of Apoptotic and Matrix-Degrading Genes in Articular Cartilage and Meniscus of Mature and Aged Rabbits during Development of Osteoarthritis. Arthritis Rheum. 2007, 56, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Sun, X.; Wang, Z.; Chen, Q. CD95-Induced Osteoarthritic Chondrocyte Apoptosis and Necrosis: Dependency on P38 Mitogen-Activated Protein Kinase. Arthritis Res. Ther. 2006, 8, R37. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Lee, Y.J.; Seong, S.C.; Choe, K.W.; Song, Y.W. Apoptotic Chondrocyte Death in Human Osteoarthritis. J. Rheumatol. 2000, 27, 455–462. [Google Scholar] [PubMed]
- Ryu, J.-H.; Shin, Y.; Huh, Y.H.; Yang, S.; Chun, C.-H.; Chun, J.-S. Hypoxia-Inducible Factor-2 α Regulates Fas-Mediated Chondrocyte Apoptosis during Osteoarthritic Cartilage Destruction. Cell Death Differ. 2012, 19, 440–450. [Google Scholar] [CrossRef]
- Yang, S.; Kim, J.; Ryu, J.-H.; Oh, H.; Chun, C.-H.; Kim, B.J.; Min, B.H.; Chun, J.-S. Hypoxia-Inducible Factor-2α Is a Catabolic Regulator of Osteoarthritic Cartilage Destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. The Role of Cytokines in Cartilage Matrix Degeneration in Osteoarthritis. Clin. Orthop. Relat. Res. 2004, S27–S36. [Google Scholar] [CrossRef]
- Mengshol, J.A.; Vincenti, M.P.; Coon, C.I.; Barchowsky, A.; Brinckerhoff, C.E. Interleukin-1 Induction of Collagenase 3 (Matrix Metalloproteinase 13) Gene Expression in Chondrocytes Requires P38, c-Jun N-Terminal Kinase, and Nuclear Factor KappaB: Differential Regulation of Collagenase 1 and Collagenase 3. Arthritis Rheum. 2000, 43, 801–811. [Google Scholar] [CrossRef]
- Verma, P.; Dalal, K. ADAMTS-4 and ADAMTS-5: Key Enzymes in Osteoarthritis. J. Cell. Biochem. 2011, 112, 3507–3514. [Google Scholar] [CrossRef]
- Séguin, C.A.; Bernier, S.M. TNFalpha Suppresses Link Protein and Type II Collagen Expression in Chondrocytes: Role of MEK1/2 and NF-KappaB Signaling Pathways. J. Cell. Physiol. 2003, 197, 356–369. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Pelletier, J.-P.; Fahmi, H. Cyclooxygenase-2 and Prostaglandins in Articular Tissues. Semin. Arthritis Rheum. 2003, 33, 155–167. [Google Scholar] [CrossRef]
- Attur, M.G.; Patel, I.R.; Patel, R.N.; Abramson, S.B.; Amin, A.R. Autocrine Production of IL-1 Beta by Human Osteoarthritis-Affected Cartilage and Differential Regulation of Endogenous Nitric Oxide, IL-6, Prostaglandin E2, and IL-8. Proc. Assoc. Am. Physicians 1998, 110, 65–72. [Google Scholar] [PubMed]
- Davies, C.M.; Guilak, F.; Weinberg, J.B.; Fermor, B. Reactive Nitrogen and Oxygen Species in Interleukin-1-Mediated DNA Damage Associated with Osteoarthritis. Osteoarthr. Cartil. 2008, 16, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Xu, M.; Xo, R.; Mates, A.; Wilson, G.L.; Pearsall, A.W.; Grishko, V. Mitochondrial DNA Damage Is Involved in Apoptosis Caused by Pro-Inflammatory Cytokines in Human OA Chondrocytes. Osteoarthr. Cartil. 2010, 18, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Kojima, T.; Kato, D.; Matsubara, H.; Takigawa, M.; Ishiguro, N. A Selective Estrogen Receptor Modulator Inhibits Tumor Necrosis Factor-α-Induced Apoptosis through the ERK1/2 Signaling Pathway in Human Chondrocytes. Biochem. Biophys. Res Commun 2012, 421, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP Kinase Signaling Pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Maneiro, E.; Martín, M.A.; de Andres, M.C.; López-Armada, M.J.; Fernández-Sueiro, J.L.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Mitochondrial Respiratory Activity Is Altered in Osteoarthritic Human Articular Chondrocytes. Arthritis Rheum. 2003, 48, 700–708. [Google Scholar] [CrossRef]
- Petit, P.X.; Zamzami, N.; Vayssière, J.L.; Mignotte, B.; Kroemer, G.; Castedo, M. Implication of Mitochondria in Apoptosis. Mol. Cell. Biochem. 1997, 174, 185–188. [Google Scholar] [CrossRef]
- Huser, C.A.M.; Davies, M.E. Calcium Signaling Leads to Mitochondrial Depolarization in Impact-Induced Chondrocyte Death in Equine Articular Cartilage Explants. Arthritis Rheum. 2007, 56, 2322–2334. [Google Scholar] [CrossRef]
- Ruiz-Romero, C.; Calamia, V.; Mateos, J.; Carreira, V.; Martínez-Gomariz, M.; Fernández, M.; Blanco, F.J. Mitochondrial Dysregulation of Osteoarthritic Human Articular Chondrocytes Analyzed by Proteomics: A Decrease in Mitochondrial Superoxide Dismutase Points to a Redox Imbalance. Mol. Cell Proteom. 2009, 8, 172–189. [Google Scholar] [CrossRef]
- Liu, H.; Li, Z.; Cao, Y.; Cui, Y.; Yang, X.; Meng, Z.; Wang, R. Effect of Chondrocyte Mitochondrial Dysfunction on Cartilage Degeneration: A Possible Pathway for Osteoarthritis Pathology at the Subcellular Level. Mol. Med. Rep. 2019, 20, 3308–3316. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Transient Receptor Potential A1 Is a Sensory Receptor for Multiple Products of Oxidative Stress. J. Neurosci. 2008, 28, 2485–2494. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, L.J.; Hämäläinen, M.; Nummenmaa, E.; Ilmarinen, P.; Vuolteenaho, K.; Nieminen, R.M.; Lehtimäki, L.; Moilanen, E. Monosodium Iodoacetate-Induced Inflammation and Joint Pain Are Reduced in TRPA1 Deficient Mice—Potential Role of TRPA1 in Osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2017–2026. [Google Scholar] [CrossRef]
- Nummenmaa, E.; Hämäläinen, M.; Moilanen, L.J.; Paukkeri, E.-L.; Nieminen, R.M.; Moilanen, T.; Vuolteenaho, K.; Moilanen, E. Transient Receptor Potential Ankyrin 1 (TRPA1) Is Functionally Expressed in Primary Human Osteoarthritic Chondrocytes. Arthritis Res. Ther. 2016, 18, 185. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Zhang, L.; Ding, L.; Huang, Z.; Xu, B.; Li, X.; Wang, P.; Mao, J. Transient Receptor Potential Ankyrin 1 (Trpa1) Mediates Il-1β-Induced Apoptosis in Rat Chondrocytes via Calcium Overload and Mitochondrial Dysfunction. J. Inflamm. 2018, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic Cell Death Is Not a Widespread Phenomenon in Normal Aging and Osteoarthritis Human Articular Knee Cartilage: A Study of Proliferation, Programmed Cell Death (Apoptosis), and Viability of Chondrocytes in Normal and Osteoarthritic Human Knee Cartilage. Arthritis Rheum. 2001, 44, 1304–1312. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Yan, M.; Yang, M.; Wang, S.; Pan, J.; Li, L.; Tan, J. Oxidative Damage Induces Apoptosis and Promotes Calcification in Disc Cartilage Endplate Cell through ROS/MAPK/NF-ΚB Pathway: Implications for Disc Degeneration. Biochem. Biophys. Res. Commun. 2019, 516, 1026–1032. [Google Scholar] [CrossRef]
- Vaillancourt, F.; Fahmi, H.; Shi, Q.; Lavigne, P.; Ranger, P.; Fernandes, J.C.; Benderdour, M. 4-Hydroxynonenal Induces Apoptosis in Human Osteoarthritic Chondrocytes: The Protective Role of Glutathione-S-Transferase. Arthritis Res. Ther. 2008, 10, R107. [Google Scholar] [CrossRef]
- Li, D.; Ni, S.; Miao, K.-S.; Zhuang, C. PI3K/Akt and Caspase Pathways Mediate Oxidative Stress-Induced Chondrocyte Apoptosis. Cell Stress Chaperones 2019, 24, 195–202. [Google Scholar] [CrossRef]
- Asada, S.; Fukuda, K.; Nishisaka, F.; Matsukawa, M.; Hamanisi, C. Hydrogen Peroxide Induces Apoptosis of Chondrocytes; Involvement of Calcium Ion and Extracellular Signal-Regulated Protein Kinase. Inflamm. Res. 2001, 50, 19–23. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Martel-Pelletier, J.; Abramson, S.B. Osteoarthritis, an Inflammatory Disease: Potential Implication for the Selection of New Therapeutic Targets. Arthritis Rheum. 2001, 44, 1237–1247. [Google Scholar] [CrossRef]
- Scher, J.U.; Pillinger, M.H.; Abramson, S.B. Nitric Oxide Synthases and Osteoarthritis. Curr. Rheumatol. Rep. 2007, 9, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T. Cartilage Destruction by Matrix Degradation Products. Mod. Rheumatol. 2006, 16, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Kühn, K.; Lotz, M. Mechanisms of Sodium Nitroprusside-Induced Death in Human Chondrocytes. Rheumatol. Int. 2003, 23, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Del Carlo, M.; Loeser, R.F. Nitric Oxide-Mediated Chondrocyte Cell Death Requires the Generation of Additional Reactive Oxygen Species. Arthritis Rheum. 2002, 46, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Kühn, K.; D’Lima, D.D.; Hashimoto, S.; Lotz, M. Cell Death in Cartilage. Osteoarthr. Cartil. 2004, 12, 1–16. [Google Scholar] [CrossRef]
- Whiteman, M.; Armstrong, J.S.; Cheung, N.S.; Siau, J.-L.; Rose, P.; Schantz, J.-T.; Jones, D.P.; Halliwell, B. Peroxynitrite Mediates Calcium-Dependent Mitochondrial Dysfunction and Cell Death via Activation of Calpains. FASEB J. 2004, 18, 1395–1397. [Google Scholar] [CrossRef]
- Mirzamohammadi, F.; Papaioannou, G.; Kobayashi, T. MicroRNAs in Cartilage Development, Homeostasis, and Disease. Curr. Osteoporos. Rep. 2014, 12, 410–419. [Google Scholar] [CrossRef]
- Makki, M.S.; Haseeb, A.; Haqqi, T.M. MicroRNA-9 Promotion of Interleukin-6 Expression by Inhibiting Monocyte Chemoattractant Protein-Induced Protein 1 Expression in Interleukin-1β-Stimulated Human Chondrocytes. Arthritis Rheumatol. 2015, 67, 2117–2128. [Google Scholar] [CrossRef]
- Li, F.; Yao, J.; Hao, Q.; Duan, Z. MiRNA-103 Promotes Chondrocyte Apoptosis by down-Regulation of Sphingosine Kinase-1 and Ameliorates PI3K/AKT Pathway in Osteoarthritis. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Yan, S.; Wang, M.; Zhao, J.; Zhang, H.; Zhou, C.; Jin, L.; Zhang, Y.; Qiu, X.; Ma, B.; Fan, Q. MicroRNA-34a Affects Chondrocyte Apoptosis and Proliferation by Targeting the SIRT1/P53 Signaling Pathway during the Pathogenesis of Osteoarthritis. Int. J. Mol. Med. 2016, 38, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Abouheif, M.M.; Nakasa, T.; Shibuya, H.; Niimoto, T.; Kongcharoensombat, W.; Ochi, M. Silencing MicroRNA-34a Inhibits Chondrocyte Apoptosis in a Rat Osteoarthritis Model in Vitro. Rheumatology 2010, 49, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Z.; Li, B.; Sun, F.; Chen, A.; Gong, M. Identification of MicroRNA-363-3p as an Essential Regulator of Chondrocyte Apoptosis in Osteoarthritis by Targeting NRF1 through the P53-signaling Pathway. Mol. Med. Rep. 2020, 21, 1077–1088. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, T.; Cai, B.; Wang, X.; Feng, W.; Han, Y.; Li, D.; Li, S.; Liu, J. MicroRNA-495 Enhances Chondrocyte Apoptosis, Senescence and Promotes the Progression of Osteoarthritis by Targeting AKT1. Am. J. Transl. Res. 2019, 11, 2232–2244. [Google Scholar] [PubMed]
- Yang, D.-W.; Qian, G.-B.; Jiang, M.-J.; Wang, P.; Wang, K.-Z. Inhibition of MicroRNA-495 Suppresses Chondrocyte Apoptosis through Activation of the NF-ΚB Signaling Pathway by Regulating CCL4 in Osteoarthritis. Gene Ther. 2019, 26, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zang, X.; Hou, H.; Sun, H.; Wang, L.; Zhang, T.; Tan, Y.; Liu, W.; Ye, P.; Gao, L.; et al. Bax Targeted by MiR-29a Regulates Chondrocyte Apoptosis in Osteoarthritis. BioMed Res. Int. 2019, 2019, e1434538. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, J.; Jing, W.; Yan, S.; Wang, X.; Xiao, C.; Ma, B. Role of MiR-146a in Human Chondrocyte Apoptosis in Response to Mechanical Pressure Injury in Vitro. Int. J. Mol. Med. 2014, 34, 451–463. [Google Scholar] [CrossRef]
- Makki, M.S.; Haqqi, T.M. MiR-139 Modulates MCPIP1/IL-6 Expression and Induces Apoptosis in Human OA Chondrocytes. Exp. Mol. Med. 2015, 47, e189. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, Y.; Chen, J.; Huang, K.; Ji, B.; Chen, Z.; Wang, Q.; Ma, J.; Shen, S.; Zhang, J. MiR-10a-5p Promotes Chondrocyte Apoptosis in Osteoarthritis by Targeting HOXA1. Mol. Ther. Nucleic Acids 2019, 14, 398–409. [Google Scholar] [CrossRef]
- Fu, M.; Huang, G.; Zhang, Z.; Liu, J.; Zhang, Z.; Huang, Z.; Yu, B.; Meng, F. Expression Profile of Long Noncoding RNAs in Cartilage from Knee Osteoarthritis Patients. Osteoarthr. Cartil. 2015, 23, 423–432. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, X.; Dai, L.; Hu, X.; Zhu, J.; Li, L.; Zhou, C.; Ao, Y. Long Noncoding RNA Related to Cartilage Injury Promotes Chondrocyte Extracellular Matrix Degradation in Osteoarthritis. Arthritis Rheum. 2014, 66, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Razmara, E.; Bitaraf, A.; Yousefi, H.; Nguyen, T.H.; Garshasbi, M.; Cho, W.C.-S.; Babashah, S. Non-Coding RNAs in Cartilage Development: An Updated Review. Int. J. Mol. Sci. 2019, 20, 4475. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Jiang, T.; Wang, W.; Yu, J.; Wang, Y.; Wu, X.; He, Y. Long Non-Coding HCG18 Promotes Intervertebral Disc Degeneration by Sponging MiR-146a-5p and Regulating TRAF6 Expression. Sci. Rep. 2017, 7, 13234. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Ahn, C.; Chun, C.-H.; Jin, E.-J. A Long Non-Coding RNA, GAS5, Plays a Critical Role in the Regulation of MiR-21 during Osteoarthritis. J. Orthop. Res. 2014, 32, 1628–1635. [Google Scholar] [CrossRef]
- Xing, D.; Liang, J.; Li, Y.; Lu, J.; Jia, H.; Xu, L.; Ma, X. Identification of Long Noncoding RNA Associated with Osteoarthritis in Humans. Orthop. Surg. 2014, 6, 288–293. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, P.; Jiang, P.; Lv, Y.; Dong, C.; Dai, X.; Tan, L.; Wang, Z. Upregulation of LncRNA HOTAIR Contributes to IL-1β-Induced MMP Overexpression and Chondrocytes Apoptosis in Temporomandibular Joint Osteoarthritis. Gene 2016, 586, 248–253. [Google Scholar] [CrossRef]
- Watson, E.C.; Grant, Z.L.; Coultas, L. Endothelial Cell Apoptosis in Angiogenesis and Vessel Regression. Cell. Mol. Life Sci. 2017, 74, 4387–4403. [Google Scholar] [CrossRef]
- Slomp, J.; Gittenberger-de Groot, A.C.; Glukhova, M.A.; Conny van Munsteren, J.; Kockx, M.M.; Schwartz, S.M.; Koteliansky, V.E. Differentiation, Dedifferentiation, and Apoptosis of Smooth Muscle Cells during the Development of the Human Ductus Arteriosus. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1003–1009. [Google Scholar] [CrossRef]
- Cho, A.; Courtman, D.W.; Langille, B.L. Apoptosis (Programmed Cell Death) in Arteries of the Neonatal Lamb. Circ. Res. 1995, 76, 168–175. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.K.; Shi, J.; Yu, P.L.; Zhang, G.Q. The Role of Endoplasmic Reticulum Stress-Related Apoptosis in Vascular Endothelium Pathogenesis. Biomed. Environ. Sci. 2018, 31, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Chen, Z.; Liu, M.; Peng, J.; Wu, P. Apoptosis and Calcification of Vascular Endothelial Cell under Hyperhomocysteinemia. Med. Oncol. 2015, 32, 403. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular Calcification: Pathobiological Mechanisms and Clinical Implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef]
- van Varik, B.J.; Rennenberg, R.J.M.W.; Reutelingsperger, C.P.; Kroon, A.A.; de Leeuw, P.W.; Schurgers, L.J. Mechanisms of Arterial Remodeling: Lessons from Genetic Diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef]
- Patel, J.J.; Bourne, L.E.; Davies, B.K.; Arnett, T.R.; MacRae, V.E.; Wheeler-Jones, C.P.; Orriss, I.R. Differing Calcification Processes in Cultured Vascular Smooth Muscle Cells and Osteoblasts. Exp. Cell Res. 2019, 380, 100–113. [Google Scholar] [CrossRef]
- Voelkl, J.; Cejka, D.; Alesutan, I. An Overview of the Mechanisms in Vascular Calcification during Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2019, 28, 289–296. [Google Scholar] [CrossRef]
- Lee, H.Y.; Oh, B.H. Aging and Arterial Stiffness. Circ. J. 2010, 74, 2257–2262. [Google Scholar] [CrossRef]
- Xu, J.; Shi, G.-P. Vascular Wall Extracellular Matrix Proteins and Vascular Diseases. Biochim. Biophys. Acta 2014, 1842, 2106–2119. [Google Scholar] [CrossRef]
- Watson, K.E.; Parhami, F.; Shin, V.; Demer, L.L. Fibronectin and Collagen I Matrixes Promote Calcification of Vascular Cells in Vitro, Whereas Collagen IV Matrix Is Inhibitory. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1964–1971. [Google Scholar] [CrossRef]
- Tyson, J.; Bundy, K.; Roach, C.; Douglas, H.; Ventura, V.; Segars, M.F.; Schwartz, O.; Simpson, C.L. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering 2020, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Rutkovskiy, A.; Lund, M.; Siamansour, T.S.; Reine, T.M.; Kolset, S.O.; Sand, K.L.; Ignatieva, E.; Gordeev, M.L.; Stensløkken, K.-O.; Valen, G.; et al. Mechanical Stress Alters the Expression of Calcification-Related Genes in Vascular Interstitial and Endothelial Cells. Interact. Cardiovasc. Thorac. Surg. 2019, 28, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Ni, L.; Zhang, C.; Hu, X.; Wu, X. Vascular Calcification: New Insights into Endothelial Cells. Microvasc. Res. 2020, 134, 104105. [Google Scholar] [CrossRef] [PubMed]
- Mantella, L.-E.; Quan, A.; Verma, S. Variability in Vascular Smooth Muscle Cell Stretch-Induced Responses in 2D Culture. Vasc. Cell 2015, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Herencia, C.; Rodríguez-Ortiz, M.E.; Muñoz-Castañeda, J.R.; Martinez-Moreno, J.M.; Canalejo, R.; Montes de Oca, A.; Díaz-Tocados, J.M.; Peralbo-Santaella, E.; Marín, C.; Canalejo, A.; et al. Angiotensin II Prevents Calcification in Vascular Smooth Muscle Cells by Enhancing Magnesium Influx. Eur. J. Clin. Investig. 2015, 45, 1129–1144. [Google Scholar] [CrossRef]
- Pescatore, L.A.; Gamarra, L.F.; Liberman, M. Multifaceted Mechanisms of Vascular Calcification in Aging. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1307–1316. [Google Scholar] [CrossRef]
- Wang, S.-S.; Wang, C.; Chen, H. MicroRNAs Are Critical in Regulating Smooth Muscle Cell Mineralization and Apoptosis during Vascular Calcification. J. Cell Mol. Med. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, X.; Shen, Y.; Chen, L.; Xu, C.; Zhao, H.; Wu, Y.; Zhang, Q.; Zhong, J.; Tang, Z.; et al. MicroRNA-32 Promotes Calcification in Vascular Smooth Muscle Cells: Implications as a Novel Marker for Coronary Artery Calcification. PLoS ONE 2017, 12, e0174138. [Google Scholar] [CrossRef]
- Fujita, H.; Yamamoto, M.; Ogino, T.; Kobuchi, H.; Ohmoto, N.; Aoyama, E.; Oka, T.; Nakanishi, T.; Inoue, K.; Sasaki, J. Necrotic and Apoptotic Cells Serve as Nuclei for Calcification on Osteoblastic Differentiation of Human Mesenchymal Stem Cells in Vitro. Cell Biochem. Funct. 2014, 32, 77–86. [Google Scholar] [CrossRef]
- Proudfoot, D.; Shanahan, C.M. Biology of Calcification in Vascular Cells: Intima versus Media. Herz 2001, 26, 245–251. [Google Scholar] [CrossRef]
- Duan, X.; Zhou, Y.; Teng, X.; Tang, C.; Qi, Y. Endoplasmic Reticulum Stress-Mediated Apoptosis Is Activated in Vascular Calcification. Biochem. Biophys. Res. Commun. 2009, 387, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, H.; Liu, S.; Gao, J.; Xia, J. Glycemic Variability Is Associated with Vascular Calcification by the Markers of Endoplasmic Reticulum Stress-Related Apoptosis, Wnt1, Galectin-3 and BMP-2. Diabetol. Metab. Syndr. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, H.; Tao, H.; Wu, N.; Yu, L.; Zhang, D.; Lu, X.; Zhu, J.; Lu, Z.; Zhu, Q. Metformin Inhibits Vascular Calcification in Female Rat Aortic Smooth Muscle Cells via the AMPK-ENOS-NO Pathway. Endocrinology 2013, 154, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-Q.; Sun, X.-J.; Wang, Y.; Zhu, Y.; Han, X.-Q.; Liu, N.-F. Restoring Mitochondrial Biogenesis with Metformin Attenuates β-GP-Induced Phenotypic Transformation of VSMCs into an Osteogenic Phenotype via Inhibition of PDK4/Oxidative Stress-Mediated Apoptosis. Mol. Cell. Endocrinol. 2019, 479, 39–53. [Google Scholar] [CrossRef]
- Lu, Y.; Bian, Y.; Wang, Y.; Bai, R.; Wang, J.; Xiao, C. Globular Adiponectin Reduces Vascular Calcification via Inhibition of ER-Stress-Mediated Smooth Muscle Cell Apoptosis. Int. J. Clin. Exp. Pathol. 2015, 8, 2545–2554. [Google Scholar]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Mallat, Z.; Tedgui, A. Apoptosis in the Vasculature: Mechanisms and Functional Importance. Br. J. Pharmacol. 2000, 130, 947–962. [Google Scholar] [CrossRef]
- Ghosh, S.; Luo, D.; He, W.; Chen, J.; Su, X.; Huang, H. Diabetes and Calcification: The Potential Role of Anti-Diabetic Drugs on Vascular Calcification Regression. Pharmacol. Res. 2020, 158, 104861. [Google Scholar] [CrossRef]
- Ziyad, A.-A. Arterial Calcification: A Tumor Necrosis Factor-Alpha Mediated Vascular Wnt-Opathy. Transl. Res. 2008, 151, 233–239. [Google Scholar] [CrossRef]
- Lee, K.-M.; Kang, H.-A.; Park, M.; Lee, H.-Y.; Choi, H.-R.; Yun, C.-H.; Oh, J.-W.; Kang, H.-S. Interleukin-24 Attenuates β-Glycerophosphate-Induced Calcification of Vascular Smooth Muscle Cells by Inhibiting Apoptosis, the Expression of Calcification and Osteoblastic Markers, and the Wnt/β-Catenin Pathway. Biochem. Biophys. Res. Commun. 2012, 428, 50–55. [Google Scholar] [CrossRef]
- Makarović, S.; Makarović, Z.; Steiner, R.; Mihaljević, I.; Milas-Ahić, J. Osteoprotegerin and Vascular Calcification: Clinical and Prognostic Relevance. Coll. Antropol. 2015, 39, 461–468. [Google Scholar] [PubMed]
- Scatena, M.; Almeida, M.; Chaisson, M.L.; Fausto, N.; Nicosia, R.F.; Giachelli, C.M. NF-KappaB Mediates Alphavbeta3 Integrin-Induced Endothelial Cell Survival. J. Cell Biol. 1998, 141, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Clemmons, D.R. Blocking Ligand Occupancy of the AlphaVbeta3 Integrin Inhibits Insulin-like Growth Factor I Signaling in Vascular Smooth Muscle Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11217–11222. [Google Scholar] [CrossRef] [PubMed]
- Strömblad, S.; Becker, J.C.; Yebra, M.; Brooks, P.C.; Cheresh, D.A. Suppression of P53 Activity and P21WAF1/CIP1 Expression by Vascular Cell Integrin AlphaVbeta3 during Angiogenesis. J. Clin. Investig. 1996, 98, 426–433. [Google Scholar] [CrossRef]
- Carmeliet, P.; Lampugnani, M.G.; Moons, L.; Breviario, F.; Compernolle, V.; Bono, F.; Balconi, G.; Spagnuolo, R.; Oosthuyse, B.; Dewerchin, M.; et al. Targeted Deficiency or Cytosolic Truncation of the VE-Cadherin Gene in Mice Impairs VEGF-Mediated Endothelial Survival and Angiogenesis. Cell 1999, 98, 147–157. [Google Scholar] [CrossRef]
- Zanetti, A.; Lampugnani, M.G.; Balconi, G.; Breviario, F.; Corada, M.; Lanfrancone, L.; Dejana, E. Vascular Endothelial Growth Factor Induces SHC Association with Vascular Endothelial Cadherin: A Potential Feedback Mechanism to Control Vascular Endothelial Growth Factor Receptor-2 Signaling. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 617–622. [Google Scholar] [CrossRef]
- Jones, P.L.; Crack, J.; Rabinovitch, M. Regulation of Tenascin-C, a Vascular Smooth Muscle Cell Survival Factor That Interacts with the Alpha v Beta 3 Integrin to Promote Epidermal Growth Factor Receptor Phosphorylation and Growth. J. Cell Biol. 1997, 139, 279–293. [Google Scholar] [CrossRef]
- Rüegg, C.; Yilmaz, A.; Bieler, G.; Bamat, J.; Chaubert, P.; Lejeune, F.J. Evidence for the Involvement of Endothelial Cell Integrin AlphaVbeta3 in the Disruption of the Tumor Vasculature Induced by TNF and IFN-Gamma. Nat. Med. 1998, 4, 408–414. [Google Scholar] [CrossRef]
- Pollman, M.J.; Naumovski, L.; Gibbons, G.H. Vascular Cell Apoptosis: Cell Type-Specific Modulation by Transforming Growth Factor-Beta1 in Endothelial Cells versus Smooth Muscle Cells. Circulation 1999, 99, 2019–2026. [Google Scholar] [CrossRef]
- Almeida, E.A.; Ilić, D.; Han, Q.; Hauck, C.R.; Jin, F.; Kawakatsu, H.; Schlaepfer, D.D.; Damsky, C.H. Matrix Survival Signaling: From Fibronectin via Focal Adhesion Kinase to c-Jun NH(2)-Terminal Kinase. J. Cell Biol. 2000, 149, 741–754. [Google Scholar] [CrossRef]
- Cance, W.G.; Golubovskaya, V.M. Focal Adhesion Kinase versus P53: Apoptosis or Survival? Sci. Signal 2008, 1, pe22. [Google Scholar] [CrossRef] [PubMed]
- Koyama, H.; Raines, E.W.; Bornfeldt, K.E.; Roberts, J.M.; Ross, R. Fibrillar Collagen Inhibits Arterial Smooth Muscle Proliferation through Regulation of Cdk2 Inhibitors. Cell 1996, 87, 1069–1078. [Google Scholar] [CrossRef]
- Harrington, E.O.; Smeglin, A.; Newton, J.; Ballard, G.; Rounds, S. Protein Tyrosine Phosphatase-Dependent Proteolysis of Focal Adhesion Complexes in Endothelial Cell Apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L342–L353. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Freyberg, M.A.; Schrimpf, G.; Friedl, P. Apoptosis Induced by Lack of Hemodynamic Forces Is a General Endothelial Feature Even Occuring in Immortalized Cell Lines. Endothelium 1999, 6, 325–334. [Google Scholar] [CrossRef]
- Zeng, Y.; Qiao, Y.; Zhang, Y.; Liu, X.; Wang, Y.; Hu, J. Effects of Fluid Shear Stress on Apoptosis of Cultured Human Umbilical Vein Endothelial Cells Induced by LPS. Cell Biol. Int. 2005, 29, 932–935. [Google Scholar] [CrossRef]
- Dimmeler, S.; Haendeler, J.; Rippmann, V.; Nehls, M.; Zeiher, A.M. Shear Stress Inhibits Apoptosis of Human Endothelial Cells. FEBS Lett. 1996, 399, 71–74. [Google Scholar] [CrossRef]
- Dimmeler, S.; Haendeler, J.; Nehls, M.; Zeiher, A.M. Suppression of Apoptosis by Nitric Oxide via Inhibition of Interleukin-1beta-Converting Enzyme (ICE)-like and Cysteine Protease Protein (CPP)-32-like Proteases. J. Exp. Med. 1997, 185, 601–607. [Google Scholar] [CrossRef]
- Siasos, G.; Tousoulis, D.; Siasou, Z.; Stefanadis, C.; Papavassiliou, A.G. Shear Stress, Protein Kinases and Atherosclerosis. Curr. Med. Chem. 2007, 14, 1567–1572. [Google Scholar] [CrossRef]
- Nicotera, P.; Bernassola, F.; Melino, G. Nitric Oxide (NO), a Signaling Molecule with a Killer Soul. Cell Death Differ. 1999, 6, 931–933. [Google Scholar] [CrossRef]
- Sagoo, P.; Chan, G.; Larkin, D.F.P.; George, A.J.T. Inflammatory Cytokines Induce Apoptosis of Corneal Endothelium through Nitric Oxide. Invest. Ophthalmol. Vis. Sci. 2004, 45, 3964–3973. [Google Scholar] [CrossRef]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased Expression of Matrix Metalloproteinases and Matrix Degrading Activity in Vulnerable Regions of Human Atherosclerotic Plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Mannello, F.; Luchetti, F.; Falcieri, E.; Papa, S. Multiple Roles of Matrix Metalloproteinases during Apoptosis. Apoptosis 2005, 10, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.J.; Seaton, T.; Mitchell, J.; Howe, A.; Blackburn, K.; Burkhart, W.; Moyer, M.; Patel, I.; Waitt, G.M.; Becherer, J.D.; et al. The Tumor Necrosis Factor-Alpha Converting Enzyme (TACE): A Unique Metalloproteinase with Highly Defined Substrate Selectivity. Biochemistry 2002, 41, 9462–9469. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R. Apoptosis in the Cardiovascular System. Heart 2002, 87, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Stoneman, V.E.A.; Bennett, M.R. Role of Fas/Fas-L in Vascular Cell Apoptosis. J. Cardiovasc. Pharmacol. 2009, 53, 100–108. [Google Scholar] [CrossRef]
- Boyle, J.J.; Weissberg, P.L.; Bennett, M.R. Tumor Necrosis Factor-Alpha Promotes Macrophage-Induced Vascular Smooth Muscle Cell Apoptosis by Direct and Autocrine Mechanisms. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Aravani, D.; Foote, K.; Figg, N.; Finigan, A.; Uryga, A.; Clarke, M.; Bennett, M. Cytokine Regulation of Apoptosis-Induced Apoptosis and Apoptosis-Induced Cell Proliferation in Vascular Smooth Muscle Cells. Apoptosis 2020, 25, 648–662. [Google Scholar] [CrossRef]
- Lindner, H.; Holler, E.; Ertl, B.; Multhoff, G.; Schreglmann, M.; Klauke, I.; Schultz-Hector, S.; Eissner, G. Peripheral Blood Mononuclear Cells Induce Programmed Cell Death in Human Endothelial Cells and May Prevent Repair: Role of Cytokines. Blood 1997, 89, 1931–1938. [Google Scholar] [CrossRef]
- Karsan, A.; Yee, E.; Harlan, J.M. Endothelial Cell Death Induced by Tumor Necrosis Factor-Alpha Is Inhibited by the Bcl-2 Family Member, A1. J. Biol. Chem. 1996, 271, 27201–27204. [Google Scholar] [CrossRef]
- Van Antwerp, D.J.; Martin, S.J.; Verma, I.M.; Green, D.R. Inhibition of TNF-Induced Apoptosis by NF-Kappa B. Trends Cell Biol. 1998, 8, 107–111. [Google Scholar] [CrossRef]
- Badrichani, A.Z.; Stroka, D.M.; Bilbao, G.; Curiel, D.T.; Bach, F.H.; Ferran, C. Bcl-2 and Bcl-XL Serve an Anti-Inflammatory Function in Endothelial Cells through Inhibition of NF-KappaB. J. Clin. Investig. 1999, 103, 543–553. [Google Scholar] [CrossRef]
- Forde, H.; Harper, E.; Davenport, C.; Rochfort, K.D.; Wallace, R.; Murphy, R.P.; Smith, D.; Cummins, P.M. The Beneficial Pleiotropic Effects of Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) within the Vasculature: A Review of the Evidence. Atherosclerosis 2016, 247, 87–96. [Google Scholar] [CrossRef]
- Michowitz, Y.; Goldstein, E.; Roth, A.; Afek, A.; Abashidze, A.; Ben Gal, Y.; Keren, G.; George, J. The Involvement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) in Atherosclerosis. J. Am. Coll. Cardiol. 2005, 45, 1018–1024. [Google Scholar] [CrossRef]
- Sato, K.; Niessner, A.; Kopecky, S.L.; Frye, R.L.; Goronzy, J.J.; Weyand, C.M. TRAIL-Expressing T Cells Induce Apoptosis of Vascular Smooth Muscle Cells in the Atherosclerotic Plaque. J. Exp. Med. 2006, 203, 239–250. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Shui, C.; Riggs, B.L.; Dunstan, C.R.; Spelsberg, T.C.; O’Brien, T.; Khosla, S. Effects of Immunosuppressants on Receptor Activator of NF-KappaB Ligand and Osteoprotegerin Production by Human Osteoblastic and Coronary Artery Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2001, 280, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S. Role of Mitochondrial Function in Cell Death and Body Metabolism. Front. Biosci. 2016, 21, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, S.; Peng, H.; Lv, Y.; Li, W.; Cheng, S.; Liu, J. Fibroblast Growth Factor 21 Attenuates Vascular Calcification by Alleviating Endoplasmic Reticulum Stress Mediated Apoptosis in Rats. Int. J. Biol. Sci. 2019, 15, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Olapoju, S.O.; Adejobi, O.I.; Le Thi, X. Fibroblast Growth Factor 21; Review on Its Participation in Vascular Calcification Pathology. Vascul. Pharmacol. 2020, 125, 106636. [Google Scholar] [CrossRef]
- García de la Cadena, S.; Massieu, L. Caspases and Their Role in Inflammation and Ischemic Neuronal Death. Focus on Caspase-12. Apoptosis 2016, 21, 763–777. [Google Scholar] [CrossRef]
- Cao, F.; Liu, X.; Cao, X.; Wang, S.; Fu, K.; Zhao, Y.; Shen, F.; Liu, J. Fibroblast Growth Factor 21 Plays an Inhibitory Role in Vascular Calcification in Vitro through OPG/RANKL System. Biochem. Biophys. Res. Commun. 2017, 491, 578–586. [Google Scholar] [CrossRef]
- Salvayre, R.; Auge, N.; Benoist, H.; Negre-Salvayre, A. Oxidized Low-Density Lipoprotein-Induced Apoptosis. Biochim. Biophys. Acta 2002, 1585, 213–221. [Google Scholar] [CrossRef]
- Sata, M.; Walsh, K. Oxidized LDL Activates Fas-Mediated Endothelial Cell Apoptosis. J. Clin. Investig. 1998, 102, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ishibashi, S.; Miyashita, T.; Osuga, J.; Yagyu, H.; Ohashi, K.; Yazaki, Y.; Yamada, N. Bcl-2 Protein Inhibits Oxysterol-Induced Apoptosis through Suppressing CPP32-Mediated Pathway. FEBS Lett. 1997, 411, 63–66. [Google Scholar] [CrossRef]
- Escargueil-Blanc, I.; Meilhac, O.; Pieraggi, M.T.; Arnal, J.F.; Salvayre, R.; Nègre-Salvayre, A. Oxidized LDLs Induce Massive Apoptosis of Cultured Human Endothelial Cells through a Calcium-Dependent Pathway. Prevention by Aurintricarboxylic Acid. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Wu, Q.; Muszynski, M.; Hansson, G.K.; Libby, P. Apoptosis of Vascular Smooth Muscle Cells Induced by in Vitro Stimulation with Interferon-Gamma, Tumor Necrosis Factor-Alpha, and Interleukin-1 Beta. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 19–27. [Google Scholar] [CrossRef]
- Bennett, M.R.; Boyle, J.J. Apoptosis of Vascular Smooth Muscle Cells in Atherosclerosis. Atherosclerosis 1998, 138, 3–9. [Google Scholar] [CrossRef]
- Mallat, Z.; Heymes, C.; Ohan, J.; Faggin, E.; Lesèche, G.; Tedgui, A. Expression of Interleukin-10 in Advanced Human Atherosclerotic Plaques: Relation to Inducible Nitric Oxide Synthase Expression and Cell Death. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 611–616. [Google Scholar] [CrossRef]
- Pollman, M.J.; Yamada, T.; Horiuchi, M.; Gibbons, G.H. Vasoactive Substances Regulate Vascular Smooth Muscle Cell Apoptosis. Countervailing Influences of Nitric Oxide and Angiotensin II. Circ. Res. 1996, 79, 748–756. [Google Scholar] [CrossRef]
- Haendeler, J.; Weiland, U.; Zeiher, A.M.; Dimmeler, S. Effects of Redox-Related Congeners of NO on Apoptosis and Caspase-3 Activity. Nitric Oxide 1997, 1, 282–293. [Google Scholar] [CrossRef]
- Kim, Y.M.; Bombeck, C.A.; Billiar, T.R. Nitric Oxide as a Bifunctional Regulator of Apoptosis. Circ. Res. 1999, 84, 253–256. [Google Scholar] [CrossRef]
- Yamada, T.; Akishita, M.; Pollman, M.J.; Gibbons, G.H.; Dzau, V.J.; Horiuchi, M. Angiotensin II Type 2 Receptor Mediates Vascular Smooth Muscle Cell Apoptosis and Antagonizes Angiotensin II Type 1 Receptor Action: An in Vitro Gene Transfer Study. Life Sci. 1998, 63, PL289–PL295. [Google Scholar] [CrossRef]
- Leeper, N.J.; Maegdefessel, L. Non-Coding RNAs: Key Regulators of Smooth Muscle Cell Fate in Vascular Disease. Cardiovasc. Res. 2018, 114, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, G.; Wei, T.; Yang, Z.; Tan, W.; Mo, Z.; Liu, J.; Li, D.; Wei, Y.; Zhang, L.; et al. MicroRNA-25 Protects Smooth Muscle Cells against Corticosterone-Induced Apoptosis. Oxid. Med. Cell Longev. 2019, 2019, 2691514. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.O.; Tan, K.M.; Chan, S.L.; Yee, K.S.; Bevort, M.; Ang, K.C.; Yu, V.C. MAP-1, a Novel Proapoptotic Protein Containing a BH3-like Motif That Associates with Bax through Its Bcl-2 Homology Domains. J. Biol. Chem. 2001, 276, 2802–2807. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, H.; Yue, Y.; Li, S.; Zhang, D.; He, R. H19 Knockdown Suppresses Proliferation and Induces Apoptosis by Regulating MiR-148b/WNT/β-Catenin in Ox-LDL -Stimulated Vascular Smooth Muscle Cells. J. Biomed. Sci. 2018, 25, 11. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, H.; Yue, Y.; Li, S.; Zhang, D.; He, R. TUG1 Knockdown Ameliorates Atherosclerosis via Up-Regulating the Expression of MiR-133a Target Gene FGF1. Cardiovasc. Pathol. 2018, 33, 6–15. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, X.; Cheng, J.; Zhang, K.; Ma, C. LncRNA TUG1 Regulates Proliferation and Apoptosis by Regulating MiR-148b/IGF2 Axis in Ox-LDL-Stimulated VSMC and HUVEC. Life Sci. 2020, 243, 117287. [Google Scholar] [CrossRef]
- Peng, Y.; Meng, K.; Jiang, L.; Zhong, Y.; Yang, Y.; Lan, Y.; Zeng, Q.; Cheng, L. Thymic Stromal Lymphopoietin-Induced HOTAIR Activation Promotes Endothelial Cell Proliferation and Migration in Atherosclerosis. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mineral | Short Name | Chemical Formula | Mineralization Site | References |
|---|---|---|---|---|
| Amorphous calcium phosphate | APC | Ca9(PO4)6 | Atherosclerotic plaque, aortic valves, and brain | [21,22,23] |
| Brushite or dicalcium phosphate dihydrate | DCPD | CaHPO4·2(H2O) | Heart valves and kidney stones | [24,25] |
| Calcium carbonate | CaCO | Ca10(PO4)6(OH)2−2x(CO3)x or Ca10−x(PO4)6−x(CO3)x(OH)2−x | Tendons | [26] |
| Calcium oxalate | CaOx | CaC2O4 | Breast, renal stones, and vascular | [27,28] |
| Calcium pyrophosphate dihydrate | CPPD | Ca2P2O7 | Knee joint, meniscus, and tendons | [29,30,31,32] |
| Hydroxyapatite | HA | (Ca10(PO4)6OH2) | Aortic valve, brain, cardiovascular tissue, kidney, skin, and tendons | [23,33,34,35,36,37] |
| Tricalcium phosphate | TCMP | Ca3(PO4)2 | Breast and tendons | [26,38] |
| Octacalcium phosphate | OCP | Ca8(HPO4)2(PO4)4·5H2O | Heart valves, knee joint compartments | [24,39,40] |
| Whitlockite | WH | Ca18Mg2(HPO4)2(PO4)12 | aorta, breast, cartilage, prostate, salivary glands | [41,42,43,44,45,46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boraldi, F.; Lofaro, F.D.; Quaglino, D. Apoptosis in the Extraosseous Calcification Process. Cells 2021, 10, 131. https://doi.org/10.3390/cells10010131
Boraldi F, Lofaro FD, Quaglino D. Apoptosis in the Extraosseous Calcification Process. Cells. 2021; 10(1):131. https://doi.org/10.3390/cells10010131
Chicago/Turabian StyleBoraldi, Federica, Francesco Demetrio Lofaro, and Daniela Quaglino. 2021. "Apoptosis in the Extraosseous Calcification Process" Cells 10, no. 1: 131. https://doi.org/10.3390/cells10010131
APA StyleBoraldi, F., Lofaro, F. D., & Quaglino, D. (2021). Apoptosis in the Extraosseous Calcification Process. Cells, 10(1), 131. https://doi.org/10.3390/cells10010131