GADD45β Regulates Hepatic Gluconeogenesis via Modulating the Protein Stability of FoxO1

 , , ,
, , ,  , ,
, ,  and
and
Abstract
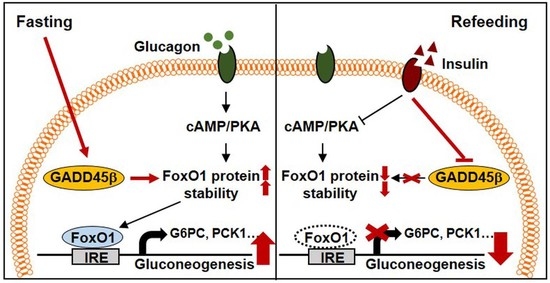
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Animal Experiments
2.2. Plasmids and Recombinant Adenoviruses
2.3. Culture of Primary Hepatocytes
2.4. Quantitative PCR
2.5. Glucose Production in Primary Hepatocytes
2.6. Seahorse Analysis
2.7. Western Blot Analysis
2.8. Cycloheximide Chase Assay
2.9. Luciferase Assay
2.10. Immunocytochemistry
2.11. Nuclear/Cytoplasmic Fractionation
2.12. Immunoprecipitation
2.13. Statistical Analysis
3. Results
3.1. Hepatic GADD45β Deficiency Suppresses Hepatic Gluconeogenesis
3.2. Hepatic GADD45β Regulates Glucose Production by Modulating Hepatic Gluconeogenesis
3.3. GADD45β Enhances FoxO1 Protein Stability Rather Than AKT-Mediated FoxO1 Phosphorylation and Fsk-Induced CREB Phosphorylation
3.4. GADD45β Promotes Transcriptional Activity through an Increase in FoxO1 Protein Stability
3.5. Hepatic GADD45β is Involved in the Insulin-Mediated Reduction of Hepatic Gluconeogenic Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Wu, Y.; Ding, Y.; Tanaka, Y.; Zhang, W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int. J. Med. Sci. 2014, 11, 1185–1200. [Google Scholar] [CrossRef]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Röder, P.V.; Wu, B.; Liu, Y.; Han, W. Pancreatic regulation of glucose homeostasis. Exp. Mol. Med. 2016, 48, e219. [Google Scholar] [CrossRef]
- Goldstein, I.; Hager, G.L. The three Ds of transcription activation by glucagon: Direct, delayed, and dynamic. Endocrinology 2018, 159, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C.; Copps, K.D.; Guo, S.; Li, Y.; Kollipara, R.; DePinho, R.A.; White, M.F. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008, 8, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Park, B.C.; Accili, D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J. Biol. Chem. 1999, 274, 15982–15985. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xu, W.; You, H.; Su, D.; Xing, J.; Li, M.; Li, L.; Liang, X. FoxO1 inhibits transcription and membrane trafficking of epithelial Na+ channel. J. Cell Sci. 2015, 128, 3621–3630. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Y.; Tian, M.; Huang, Q. Molecular mechanisms of FOXO1 in adipocyte differentiation. J. Mol. Endocrinol. 2019, 62, R239–R253. [Google Scholar] [CrossRef]
- Rena, G.; Prescott, A.R.; Guo, S.; Cohen, P.; Unterman, T.G. Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem. J. 2001, 354, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Daitoku, H.; Hatta, M.; Tanaka, K.; Fukamizu, A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc. Natl. Acad. Sci. USA 2003, 100, 11285–11290. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; Depinho, R.A.; Accili, D. Impaired Regulation of Hepatic Glucose Production in Mice Lacking the Forkhead Transcription Factor Foxo1 in Liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pan, Q.; Yan, H.; Zhang, K.; Guo, X.; Xu, Z.; Yang, W.; Qi, Y.; Guo, C.A.; Hornsby, C.; et al. Novel mechanism of Foxo1 phosphorylation in glucagon signaling in control of glucose homeostasis. Diabetes 2018, 67, 2167–2182. [Google Scholar] [CrossRef] [PubMed]
- Liebermann, D.A.; Hoffman, B. Gadd45 in stress signaling. J. Mol. Signal. 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.E.; de Vasconcellos, J.F.; Sarkar, D.; Libermann, T.A.; Fisher, P.B.; Zerbini, L.F. GADD45 proteins: Central players in tumorigenesis. Curr. Mol. Med. 2012, 12, 634–651. [Google Scholar] [CrossRef]
- Salvador, J.M.; Brown-Clay, J.D.; Fornace, A.J., Jr. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef]
- Weavers, H.; Wood, W.; Martin, P. Injury activates a dynamic cytoprotective network to confer stress resilience and drive repair. Curr. Biol. 2019, 29, 3851–3862. [Google Scholar] [CrossRef]
- Bauer, M.; Hamm, A.C.; Bonaus, M.; Jacob, A.; Jaekel, J.; Schorle, H.; Pankratz, M.J.; Katzenberger, J.D. Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol. Genomics 2004, 17, 230–244. [Google Scholar] [CrossRef]
- Fuhrmeister, J.; Zota, A.; Sijmonsma, T.P.; Seibert, O.; Cıngır, Ş.; Schmidt, K.; Vallon, N.; de Guia, R.M.; Niopek, K.; Diaz, M.B.; et al. Fasting-induced liver GADD45β restrains hepatic fatty acid uptake and improves metabolic health. EMBO Mol. Med. 2016, 8, 654–669. [Google Scholar] [CrossRef]
- Kim, J.H.; Qu, A.; Reddy, J.K.; Gao, B.; Gonzalez, F.J. Hepatic oxidative stress activates the Gadd45b gene by way of degradation of the transcriptional repressor STAT3. Hepatology 2014, 59, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Ogawa, W.; Ozaki, M.; Haga, S.; Matsumoto, M.; Furukawa, K.; Hashimoto, N.; Kido, Y.; Mori, T.; Sakaue, H.; et al. Role of STAT-3 in regulation of hepatic gluconeogenic genes and carbohydrate metabolism in vivo. Nat. Med. 2004, 10, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell. Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Ogawa, W.; Okamoto, Y.; Takashima, M.; Inoue, H.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; Kasuga, M. Role of hepatic STAT3 in the regulation of lipid metabolism. Kobe J. Med. Sci. 2008, 54, E200–E208. [Google Scholar] [PubMed]
- Oh, K.J.; Park, J.; Kim, S.S.; Oh, H.; Choi, C.S.; Koo, S.H. TCF7L2 modulates glucose homeostasis by regulating CREB- and FoxO1-dependent transcriptional pathway in the liver. PLoS Genet. 2012, 8, e1002986. [Google Scholar] [CrossRef] [PubMed]
- Dagher, Z.; Ruderman, N.; Tornheim, K.; Ido, Y. Acute regulation of fatty acid oxidation and AMP-activated protein kinase in human umbilical vein endothelial cells. Circ. Res. 2001, 88, 1276–1282. [Google Scholar] [CrossRef]
- Assifi, M.M.; Suchankova, G.; Constant, S.; Prentki, M.; Saha, A.K.; Ruderman, N.B. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E794–E800. [Google Scholar] [CrossRef]
- Green, M.H. Are Fatty Acids Gluconeogenic Precursors? J. Nutr. 2020, 150, 2235–2238. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Z.; Hu, Y.; Lu, Y.; Li, D.; Liu, J.; Liao, S.; Hu, M.; Wang, Y.; Zhang, D.; et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc. Natl. Acad. Sci. USA 2019, 116, 21732–21738. [Google Scholar] [CrossRef]
- Wagner, M.; Moore, D.D. Endoplasmic reticulum stress and glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 367–373. [Google Scholar] [CrossRef]
- Kimura, K.; Yamada, T.; Matsumoto, M.; Kido, Y.; Hosooka, T.; Asahara, S.; Matsuda, T.; Ota, T.; Watanabe, H.; Sai, Y.; et al. Endoplasmic reticulum stress inhibits STAT3-dependent suppression of hepatic gluconeogenesis via dephosphorylation and deacetylation. Diabetes 2012, 61, 61–73. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Lee, D.S.; An, T.H.; Park, T.-J.; Lee, E.-W.; Han, B.S.; Kim, W.K.; Lee, C.-H.; Lee, S.C.; Oh, K.-J.; et al. GADD45β Regulates Hepatic Gluconeogenesis via Modulating the Protein Stability of FoxO1. Biomedicines 2021, 9, 50. https://doi.org/10.3390/biomedicines9010050
Kim H, Lee DS, An TH, Park T-J, Lee E-W, Han BS, Kim WK, Lee C-H, Lee SC, Oh K-J, et al. GADD45β Regulates Hepatic Gluconeogenesis via Modulating the Protein Stability of FoxO1. Biomedicines. 2021; 9(1):50. https://doi.org/10.3390/biomedicines9010050
Chicago/Turabian StyleKim, Hyunmi, Da Som Lee, Tae Hyeon An, Tae-Jun Park, Eun-Woo Lee, Baek Soo Han, Won Kon Kim, Chul-Ho Lee, Sang Chul Lee, Kyoung-Jin Oh, and et al. 2021. "GADD45β Regulates Hepatic Gluconeogenesis via Modulating the Protein Stability of FoxO1" Biomedicines 9, no. 1: 50. https://doi.org/10.3390/biomedicines9010050
APA StyleKim, H., Lee, D. S., An, T. H., Park, T.-J., Lee, E.-W., Han, B. S., Kim, W. K., Lee, C.-H., Lee, S. C., Oh, K.-J., & Bae, K.-H. (2021). GADD45β Regulates Hepatic Gluconeogenesis via Modulating the Protein Stability of FoxO1. Biomedicines, 9(1), 50. https://doi.org/10.3390/biomedicines9010050