Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lipids: Diet Incorporation, Biosynthesis and Storage
2.1. Triglycerides
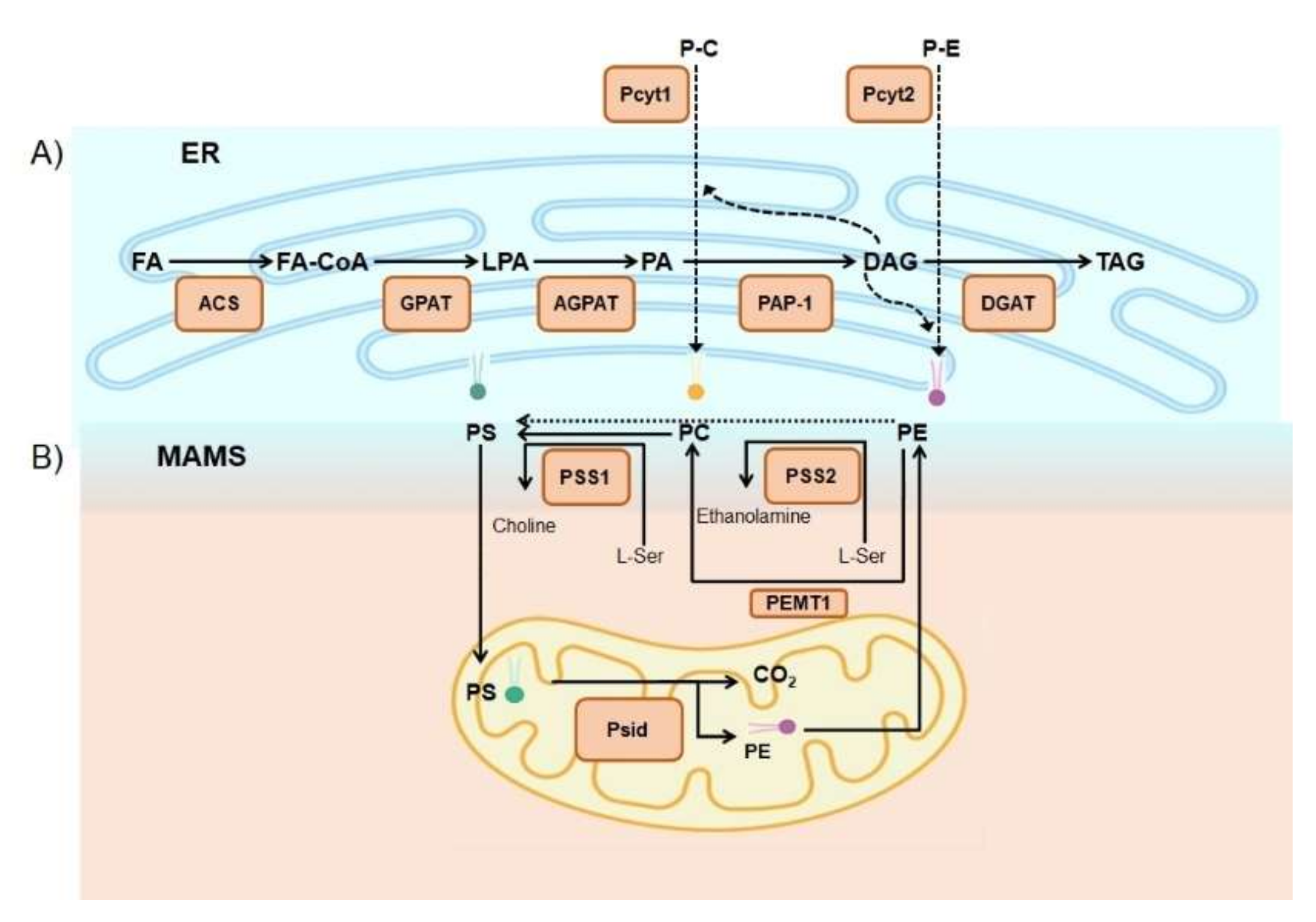
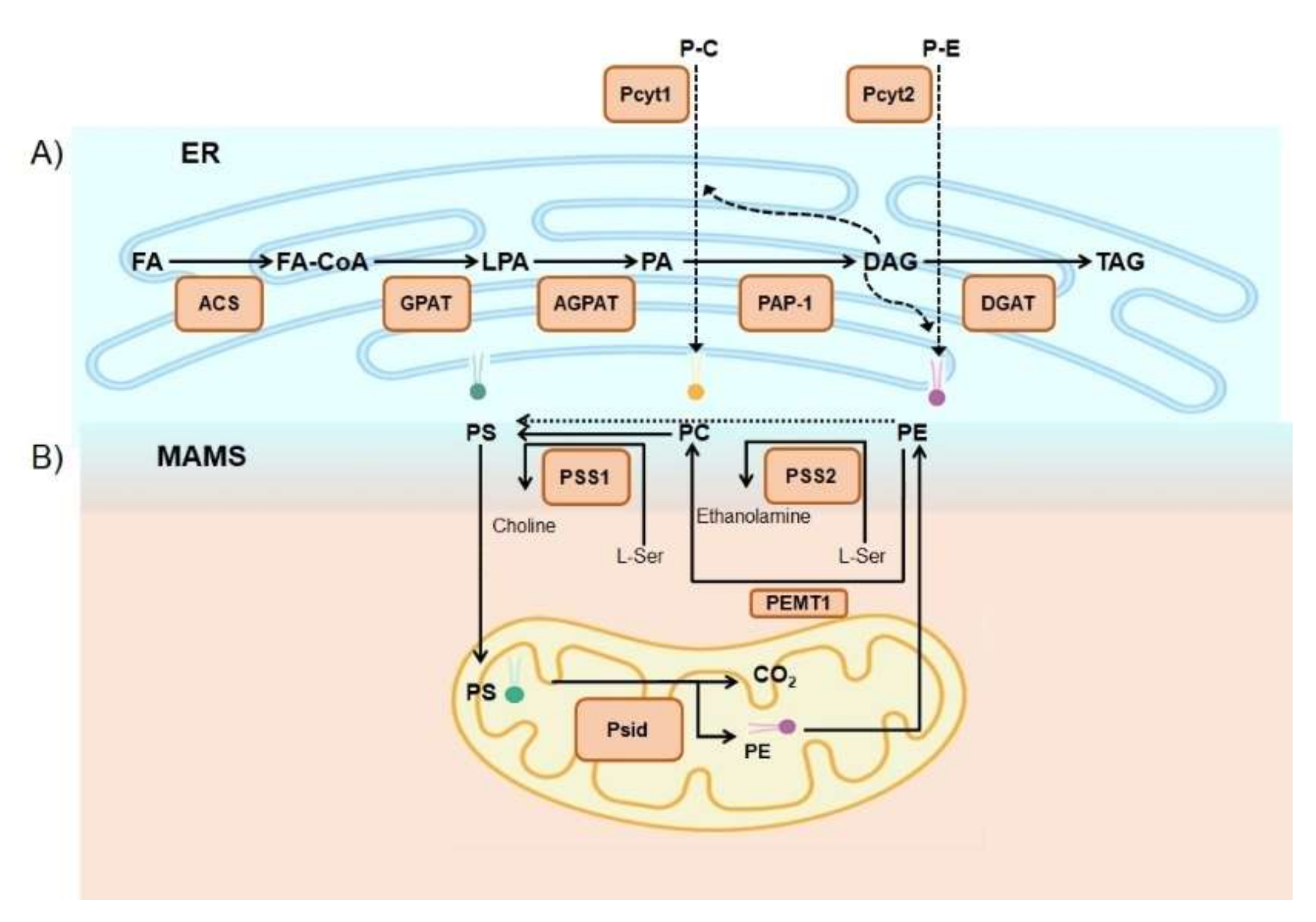
2.2. Phospholipids
2.3. Sterols
2.4. Eicosanoids
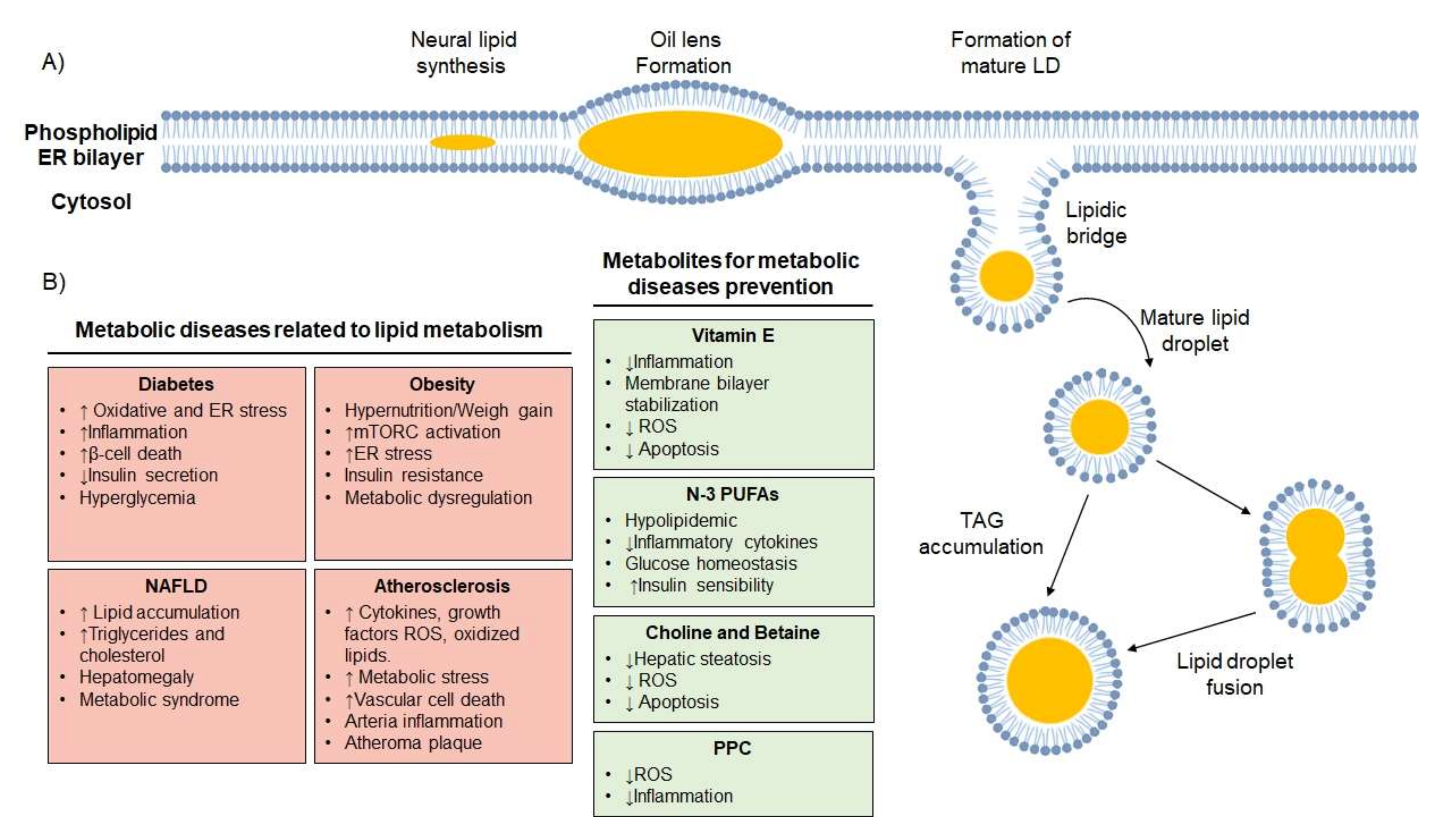
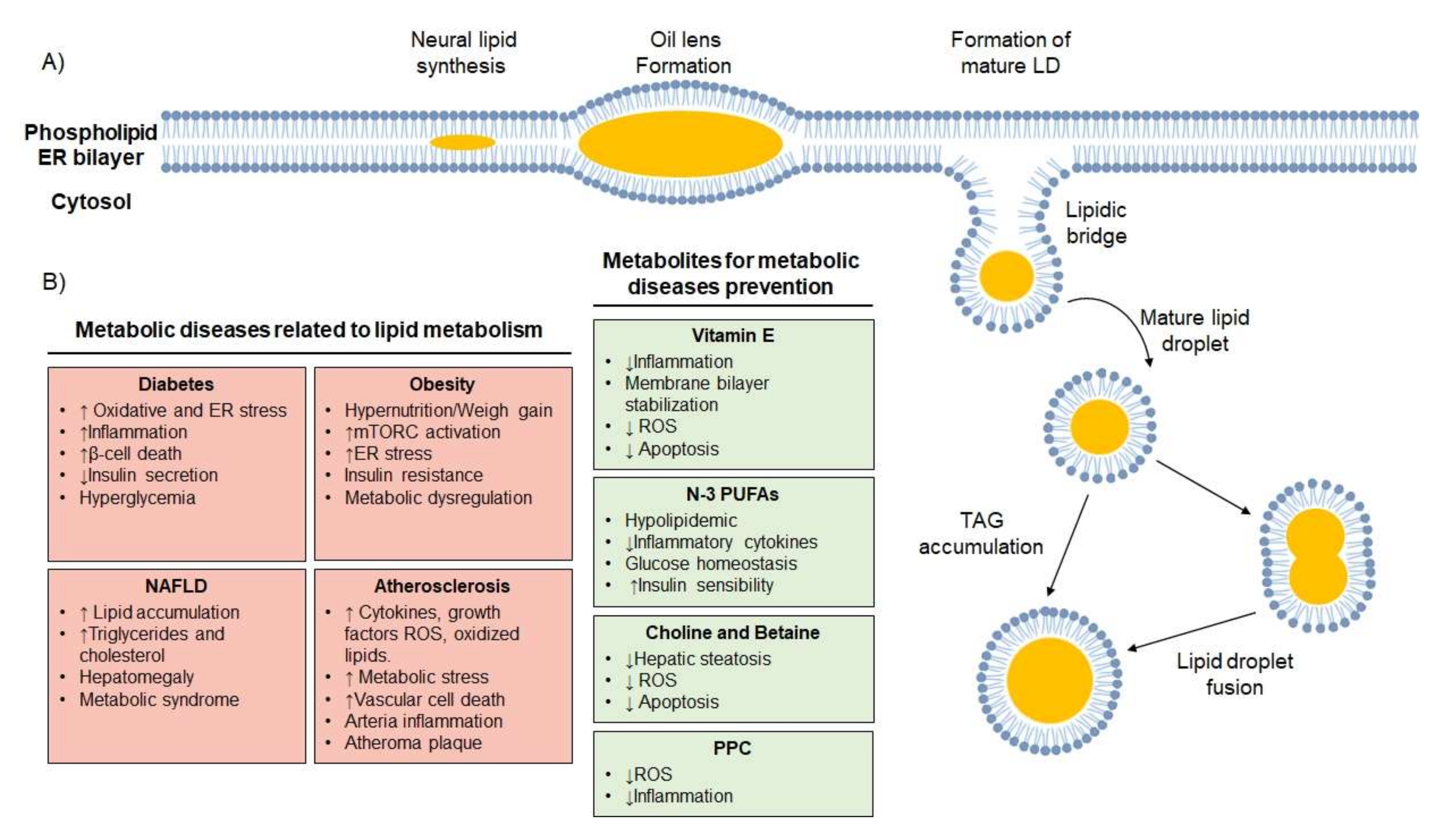
2.5. Lipid Droplets
3. Dietary Lipid Metabolism in Metabolic Diseases
3.1. TAG Associated with Lipid Disorders
3.2. Cholesterol Associated with Lipid Disorders
3.3. Eicosanoids Associated with Lipid Disorders
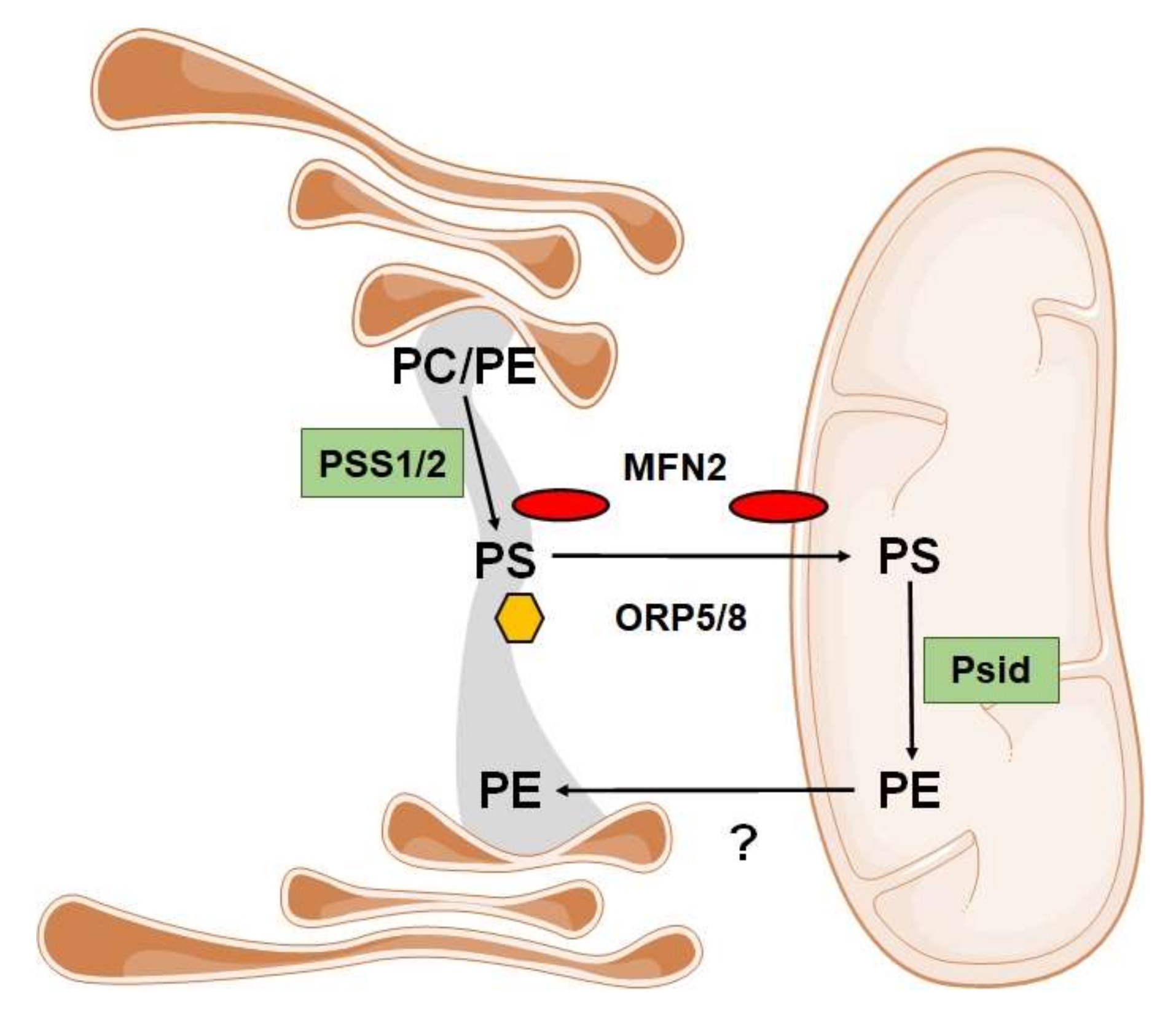
3.4. Phospholipids Associated with Lipid Disorders
4. Potential Role of Autophagy in the Modulation of Lipid Metabolism
4.1. Phospholipid Dependence of Autophagosome Formation
4.2. Metabolic Diseases Related to Defective Autophagy
5. Regulation of Lipid Stores and Metabolism by Lipophagy
Metabolic Diseases Related to Defective Lipophagy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cercato, C.; Fonseca, F.A. Cardiovascular risk and obesity. Diabetol. Metab. Syndr. 2019, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hurt, R.T.; Kulisek, C.; Buchanan, L.A.; McClave, S.A. The Obesity Epidemic: Challenges, Health Initiatives, and Implications for Gastroenterologists. Gastroenterol. Hepatol. 2010, 6, 780–792. [Google Scholar]
- Liu, A.G.; Ford, N.A.; Hu, F.B.; Zelman, K.M.; Mozaffarian, D.; Kris-Etherton, P.M. A healthy approach to dietary fats: Understanding the science and taking action to reduce consumer confusion. Nutr. J. 2017, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- What Are Lipids? Available online: https://med.libretexts.org/Courses/American_Public_University/APUS%3A_An_Introduction_to_Nutrition_(Byerley)/Text/04%3A_Lipids/4.02%3A_What_Are_Lipids%3F (accessed on 18 September 2020).
- Pol, A.; Gross, S.P.; Parton, R.G. Biogenesis of the multifunctional lipid droplet: Lipids, proteins, and sites. J. Cell Biol. 2014, 204, 635–646. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef]
- Fink, H.H.; Mikesky, A.E. Practical Applications in Sports Nutrition; Jones & Bartlett Publishers: Burlington, MA, USA, 2013; ISBN 978-1-4496-9005-2. [Google Scholar]
- Astrup, A.; Bradley, B.H.R.; Brenna, J.T.; Delplanque, B.; Ferry, M.; Torres-Gonzalez, M. Regular-Fat Dairy and Human Health: A Synopsis of Symposia Presented in Europe and North America (2014–2015). Nutrients 2016, 8, 463. [Google Scholar] [CrossRef] [Green Version]
- Pelech, S.L.; Vance, D.E. Regulation of phosphatidylcholine biosynthesis. Biochim. Biophys. Acta BBA Rev. Biomembr. 1984, 779, 217–251. [Google Scholar] [CrossRef]
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Phosphatidylcholine and the CDP–choline cycle. Biochim. Biophys. Acta BBA Mol. 2013, 1831, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Küllenberg, D.; Taylor, L.; Schneider, M.; Massing, U. Health effects of dietary phospholipids. Lipids Heal. Dis. 2012, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Blesso, C.N. Egg Phospholipids and Cardiovascular Health. Nutrients 2015, 7, 2731–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- E Vance, J.; E Vance, D. Phospholipid biosynthesis in mammalian cells. Biochem. Cell Biol. 2004, 82, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2014, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef] [PubMed]
- Galmes, R.; Houcine, A.; Van Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [PubMed]
- Van Der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine Deficiency in Mammalian Mitochondria Impairs Oxidative Phosphorylation and Alters Mitochondrial Morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E.; Tasseva, G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 543–554. [Google Scholar] [CrossRef]
- Malformation Syndromes Caused by Disorders of Cholesterol Synthesis. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2999931/ (accessed on 12 October 2020).
- SolimanHugo, G.A. Dietary Cholesterol and the Lack of Evidence in Cardiovascular Disease. Nutrients 2018, 10, 780. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Ali, A.; Khan, S.A.; Zahran, S.A.; Damanhouri, G.; Azhar, E.I.; Qadri, I. Unraveling the Complex Relationship Triad between Lipids, Obesity, and Inflammation. Mediat. Inflamm. 2014, 2014, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lin, H.; Gu, Y. Multiple roles of dihomo-γ-linolenic acid against proliferation diseases. Lipids Heal. Dis. 2012, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourdot, B.E.; Ahmed, I.; Holinstat, M. The emerging role of oxylipins in thrombosis and diabetes. Front. Pharmacol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Dumancas, G.G.; Murdianti, B.S.; Lucas, E.A. Arachidonic Acid: Dietary Sources and General Functions; Nova Science Publishers Incorporated: Hauppauge, NY, USA, 2013; ISBN 978-1-62257-481-0. [Google Scholar]
- Thiam, A.R.; Farese, R.V.F., Jr.; Walther, T.C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, A.R.; Brasaemle, D.L.; McAndrews-Hill, M.; Sztalryd, C.; Londos, C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins: TABLE 1. J. Lipid Res. 2010, 51, 468–471. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Wang, H.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.-X.; Graham, M.; Christiano, R.; Fröhlich, F.; et al. Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Kassan, A.; Herms, A.; Fernández-Vidal, A.; Bosch, M.; Bosch, R.; Schieber, N.L.; Reddy, B.J.N.; Fajardo, A.; Gelabert-Baldrich, M.; Tebar, F.; et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 2013, 203, 985–1001. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S. Lipid Droplets as Organelles. TAM Recept. Health Dis. 2018, 337, 83–110. [Google Scholar] [CrossRef]
- Soni, K.G.; Mardones, G.A.; Sougrat, R.; Smirnova, E.; Jackson, C.L.; Bonifacino, J.S. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 2009, 122, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Thiam, A.R.; Antonny, B.; Wang, J.; Delacotte, J.; Wilfling, F.; Walther, T.C.; Beck, R.; Rothman, J.E.; Pincet, F. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc. Natl. Acad. Sci. USA 2013, 110, 13244–13249. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Li, P. The size matters: Regulation of lipid storage by lipid droplet dynamics. Sci. China Life Sci. 2016, 60, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Gong, J.; Wu, H.; Xu, W.; Wu, L.; Xu, D.; Gao, J.; Wu, J.-W.; Yang, H.; Yang, M.; et al. Perilipin1 promotes unilocular lipid droplet formation through the activation of Fsp27 in adipocytes. Nat. Commun. 2013, 4, 1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennisi, E.M.; Arca, M.; Bertini, E.; Bruno, C.; Cassandrini, D.; D’Amico, A.; Garibaldi, M.; Gragnani, F.; Maggi, L.; Massa, R.; et al. Neutral Lipid Storage Diseases: Clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J. Rare Dis. 2017, 12, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuerschner, L.; Moessinger, C.; Thiele, C. Imaging of Lipid Biosynthesis: How a Neutral Lipid Enters Lipid Droplets. Traffic 2008, 9, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Azrad, M.; Turgeon, C.E.; Demark-Wahnefried, W. Current Evidence Linking Polyunsaturated Fatty Acids with Cancer Risk and Progression. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telle-Hansen, V.H.; Gaundal, L.; Myhrstad, M.C.W. Polyunsaturated Fatty Acids and Glycemic Control in Type 2 Diabetes. Nutrients 2019, 11, 1067. [Google Scholar] [CrossRef] [Green Version]
- De Lombardo, Y.B.; Chicco, A.G. Effects of dietary polyunsaturated n-3 fatty acids on dyslipidemia and insulin resistance in rodents and humans. A review. J. Nutr. Biochem. 2006, 17, 1–13. [Google Scholar] [CrossRef]
- Stupin, M.; Kibel, A.; Stupin, A.; Selthofer-Relatić, K.; Matić, A.; Mihalj, M.; Mihaljević, Z.; Jukić, I.; Drenjančević, I. The Physiological Effect of n-3 Polyunsaturated Fatty Acids (n-3 PUFAs) Intake and Exercise on Hemorheology, Microvascular Function, and Physical Performance in Health and Cardiovascular Diseases; Is There an Interaction of Exercise and Dietary n-3 PUFA Intake? Front. Physiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Risérus, U.; Willett, W.C.; Hu, F.B. Dietary fats and prevention of type 2 diabetes. Prog. Lipid Res. 2009, 48, 44–51. [Google Scholar] [CrossRef] [Green Version]
- De Caterina, R.; Madonna, R.; Bertolotto, A.; Schmidt, E.B. n-3 Fatty Acids in the Treatment of Diabetic Patients: Biological rationale and clinical data. Diabetes Care 2007, 30, 1012–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.B.; Manson, J.E.; Willett, W.C. Types of Dietary Fat and Risk of Coronary Heart Disease: A Critical Review. J. Am. Coll. Nutr. 2001, 20, 5–19. [Google Scholar] [CrossRef] [PubMed]
- A Jacobson, K.; Mouritsen, O.G.; Anderson, R.G.W. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef]
- Eckel, R.H.; Jakicic, J.M.; Ard, J.D.; De Jesus, J.M.; Houston Miller, N.; Hubbard, V.S.; Lee, I.M.; Lichtenstein, A.H.; Loria, C.M.; Millen, B.E.; et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk. Circulation 2014, 129, S76–S99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutungi, G.; Ratliff, J.; Puglisi, M.; Torres-Gonzalez, M.; Vaishnav, U.; Leite, J.O.; Quann, E.; Volek, J.S.; Fernandez, M.L. Dietary Cholesterol from Eggs Increases Plasma HDL Cholesterol in Overweight Men Consuming a Carbohydrate-Restricted Diet. J. Nutr. 2008, 138, 272–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuller, N.R.; Sainsbury, A.; Caterson, I.D.; Denyer, G.S.; Fong, M.; Gerofi, J.; Leung, C.; Lau, N.S.; Williams, K.H.; Januszewski, A.S.; et al. Effect of a high-egg diet on cardiometabolic risk factors in people with type 2 diabetes: The Diabetes and Egg (DIABEGG) Study—Randomized weight-loss and follow-up phase. Am. J. Clin. Nutr. 2018, 107, 921–931. [Google Scholar] [CrossRef]
- European Guidelines on Cardiovascular Disease Prevention in Clinical Practice: Executive Summary | European Heart Journal | Oxford Academic. Available online: https://academic.oup.com/eurheartj/article/28/19/2375/494218 (accessed on 12 October 2020).
- Ristic-Medic, D.; Vucic, V. Dietary Fats And Metabolic Syndrome. J. Nutr. Health Food Sci. 2013, 1, 1. [Google Scholar] [CrossRef]
- Brouwer, I.A.; Wanders, A.J.; Katan, M.B. Trans fatty acids and cardiovascular health: Research completed? Eur. J. Clin. Nutr. 2013, 67, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Factorial Study of the Effect of n–3 Fatty acid Supplementation and Atorvastatin on the Kinetics of HDL Apolipoproteins A-I and A-II in Men with Abdominal Obesity | The American Journal of Clinical Nutrition|Oxford Academic. Available online: https://academic.oup.com/ajcn/article/84/1/37/4633176 (accessed on 12 October 2020).
- Torrejon, C.; Jung, U.J.; Deckelbaum, R.J. n-3 Fatty acids and cardiovascular disease: Actions and molecular mechanisms. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Perona, J.S. Membrane lipid alterations in the metabolic syndrome and the role of dietary oils. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1690–1703. [Google Scholar] [CrossRef]
- Denisenko, Y.K.; Kytikova, O.Y.; Novgorodtseva, T.P.; Antonyuk, M.V.; Gvozdenko, T.A.; Kantur, T.A. Lipid-Induced Mechanisms of Metabolic Syndrome. J. Obes. 2020, 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Craig, C.; Liu, L.-F.; Perelman, D.; Allister, C.; Spielman, D.; Cushman, S.W. Adipose Cell Size and Regional Fat Deposition as Predictors of Metabolic Response to Overfeeding in Insulin-Resistant and Insulin-Sensitive Humans. Diabetes 2016, 65, 1245–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burhans, M.S.; Hagman, D.K.; Kuzma, J.N.; Schmidt, K.A.; Kratz, M. Contribution of Adipose Tissue Inflammation to the Development of Type 2 Diabetes Mellitus. Compr. Physiol. 2018, 9, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [Green Version]
- Caspar-Bauguil, S.; Fioroni, A.; Galinier, A.; Allenbach, S.; Pujol, M.C.; Salvayre, R.; Cartier, A.; Lemieux, I.; Richard, D.; Biron, S.; et al. Pro-inflammatory Phospholipid Arachidonic Acid/Eicosapentaenoic Acid Ratio of Dysmetabolic Severely Obese Women. Obes. Surg. 2012, 22, 935–944. [Google Scholar] [CrossRef]
- Hellmann, J.; Zhang, M.J.; Tang, Y.; Rane, M.; Bhatnagar, A.; Spite, M. Increased Saturated Fatty Acids in Obesity Alter Resolution of Inflammation in Part by Stimulating Prostaglandin Production. J. Immunol. 2013, 191, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Lima, T.; Kanunfre, C.; Pompéia, C.; Verlengia, R.; Curi, R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol. Vitr. 2002, 16, 741–747. [Google Scholar] [CrossRef]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [Green Version]
- De Jong, A.J.; Kloppenburg, M.; Toes, R.E.M.; Ioan-Facsinay, A. Fatty Acids, Lipid Mediators, and T-Cell Function. Front. Immunol. 2014, 5, 483. [Google Scholar] [CrossRef] [Green Version]
- Quinn, P.J. Is the Distribution of -Tocopherol in Membranes Consistent with Its Putative Functions? Biochem. Mosc. 2004, 69, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Noma, A.; Terao, J. Location of α-tocopherol and α-tocotrienol to heterogeneous cell membranes and inhibition of production of peroxidized cholesterol in mouse fibroblasts. SpringerPlus 2014, 3, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botchlett, R.; Woo, S.-L.; Liu, M.; Pei, Y.; Guo, X.; Li, H.; Wu, C. Nutritional approaches for managing obesity-associated metabolic diseases. J. Endocrinol. 2017, 233, R145–R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nan, Y.-M.; Wu, W.-J.; Fu, N.; Liang, B.-L.; Wang, R.-Q.; Li, L.-X.; Zhao, S.-X.; Zhao, J.-M.; Yu, J. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand. J. Gastroenterol. 2009, 44, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Giménez, M.S.; Oliveros, L.B.; Gomez, N.N. Nutritional Deficiencies and Phospholipid Metabolism. Int. J. Mol. Sci. 2011, 12, 2408–2433. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta BBA Bioenerg. 2014, 1837, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Zelber-Sagi, S.; Ratziu, V.; Oren, R. Nutrition and physical activity in NAFLD: An overview of the epidemiological evidence. World J. Gastroenterol. 2011, 17, 3377–3389. [Google Scholar] [CrossRef]
- Schiller, J.; Zschörnig, O.; Petković, M.; Müller, M.; Arnhold, J.; Arnold, K. Lipid analysis of human HDL and LDL by MALDI-TOF mass spectrometry and (31)P-NMR. J. Lipid Res. 2001, 42, 1501–1508. [Google Scholar]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Kartsoli, S.; E Kostara, C.; Tsimihodimos, V.; Bairaktari, E.T.; Christodoulou, D.K. Lipidomics in non-alcoholic fatty liver disease. World J. Hepatol. 2020, 12, 436–450. [Google Scholar] [CrossRef]
- Duric, M.; Sivanesan, S.; Bakovic, M. Phosphatidylcholine functional foods and nutraceuticals: A potential approach to prevent non-alcoholic fatty liver disease. Eur. J. Lipid Sci. Technol. 2012, 114, 389–398. [Google Scholar] [CrossRef]
- Cao, M.; Li, X.; Zhang, B.; Han, S.; Yang, Y.; Zhou, B.; Zhang, Y. The effect of polyene phosphatidyl choline intervention on nonalcoholic steatohepatitis and related mechanism. Am. J. Transl. Res. 2016, 8, 2325–2330. [Google Scholar] [PubMed]
- Mitsuhashi, S.; Nishino, I. Phospholipid synthetic defect and mitophagy in muscle disease. Autophagy 2011, 7, 1559–1561. [Google Scholar] [CrossRef]
- Zhang, J.; Han, J.; Wang, Y.; Wu, Y.; Song, X.; Ji, G. Neutral lipid storage disease with myopathy presenting asymmetrical muscle weakness: A case report. Int. J. Clin. Exp. Pathol. 2020, 13, 559. [Google Scholar] [PubMed]
- Flanagan, J.L.; A Simmons, P.; Vehige, J.; Willcox, M.D.; Garrett, Q. Role of carnitine in disease. Nutr. Metab. 2010, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Shantharam, P.; Srinivasarao, P. Activity of myelin membrane Na+/K+-ATPase and 5′-nucleotidase in relation to phospholipid acyl profiles, ganglioside composition and phosphoinositides in developing brains of undernourished rats. Biochim. Biophys. Acta BBA Biomembr. 1989, 982, 115–122. [Google Scholar] [CrossRef]
- Bazan, N.G.; Molina, M.F.; Gordon, W.C. Docosahexaenoic Acid Signalolipidomics in Nutrition: Significance in Aging, Neuroinflammation, Macular Degeneration, Alzheimer’s, and Other Neurodegenerative Diseases. Annu. Rev. Nutr. 2011, 31, 321–351. [Google Scholar] [CrossRef] [Green Version]
- Kamphuis, P.J.; Wurtman, R.J. Nutrition and Alzheimer’s disease: Pre-clinical concepts. Eur. J. Neurol. 2009, 16 (Suppl. 1), 12–18. [Google Scholar] [CrossRef]
- Levant, B.; Ozias, M.K.; Carlson, S.E. Specific Brain Regions of Female Rats Are Differentially Depleted of Docosahexaenoic Acid by Reproductive Activity and an (n-3) Fatty Acid-Deficient Diet. J. Nutr. 2007, 137, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Burdge, G.C.; Calder, P.C. Conversion of α-linolenic acid to longer-chain polyunsaturated fatty acids in human adults. Reprod. Nutr. Dev. 2005, 45, 581–597. [Google Scholar] [CrossRef]
- Sun, G.Y.; Shelat, P.B.; Jensen, M.B.; He, Y.; Sun, A.Y.; Simonyi, A. Phospholipases A2 and Inflammatory Responses in the Central Nervous System. Neuro Mol. Med. 2010, 12, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.-W.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Thiébaut, A.C.M.; Rotival, M.; Gauthier, E.; Lenoir, G.M.; Boutron-Ruault, M.-C.; Joulin, V.; Clavel-Chapelon, F.; Chajès, V. Correlation Between Serum Phospholipid Fatty Acids and Dietary Intakes Assessed a Few Years Earlier. Nutr. Cancer 2009, 61, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Poppitt, S.D.; Kilmartin, P.A.; Butler, P.; Keogh, G.F. Assessment of erythrocyte phospholipid fatty acid composition as a biomarker for dietary MUFA, PUFA or saturated fatty acid intake in a controlled cross-over intervention trial. Lipids Heal. Dis. 2005, 4, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, L.-T.; Glew, R.H.; Li, C.-C.; VanderJagt, D.J.; Broyles, J.S.; Ray, G.M.; Shah, V.O. Comparison of the fatty acid composition of the serum phospholipids of controls, prediabetics and adults with type 2 diabetes. J. Diabetes Mellit. 2012, 2, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nat. Cell Biol. 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, M.; Li, X.; Liu, J.; Li, H. Origin of the Autophagosome Membrane in Mammals. BioMed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E. Chapter 10 Monitoring Autophagy by Electron Microscopy in Mammalian Cells. Methods Enzymol. 2009, 452, 143–164. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nat. Cell Biol. 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Tito, S.; Hervás, J.H.; Van Vliet, A.R.; Tooze, S.A. The Golgi as an Assembly Line to the Autophagosome. Trends Biochem. Sci. 2020, 45, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; I Vaccaro, M.; Codogno, P.; Morel, E. ER –plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI 3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.W.; Yamamoto, H.; Oikawa, Y.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ohsumi, Y. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3350–3355. [Google Scholar] [CrossRef] [Green Version]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef] [Green Version]
- Rabouille, C. COPII vesicles and the expansion of the phagophore. eLife 2019, 8, e44944. [Google Scholar] [CrossRef]
- Watada, H.; Fujitani, Y. Minireview: Autophagy in Pancreatic β-Cells and Its Implication in Diabetes. Mol. Endocrinol. 2015, 29, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Moulis, M.; Vindis, C. Autophagy in Metabolic Age-Related Human Diseases. Cells 2018, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1–S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamışlıgil, G.S. Defective Hepatic Autophagy in Obesity Promotes ER Stress and Causes Insulin Resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Cao, Y.; Tong, T.; Shi, J.; Zhang, Y.; Yang, Y.; Liu, C. Autophagy in Atherosclerosis. Chin. Med J. 2015, 128, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, B.; Goikuria, H.; Vega, R.; Rodríguez-Antigüedad, A.; Medina, A.L.; Freijo, M.D.M.; Vandenbroeck, K.; Alloza, I. Autophagic Marker MAP1LC3B Expression Levels Are Associated with Carotid Atherosclerosis Symptomatology. PLoS ONE 2014, 9, e115176. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy Links Inflammasomes to Atherosclerotic Progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, I.; Aquila, S. The Role of Oxidative Stress and Autophagy in Atherosclerosis. Oxidative Med. Cell. Longev. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Brasaemle, D.L. DisseCCTing Phospholipid Function in Lipid Droplet Dynamics. Cell Metab. 2011, 14, 437–438. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Spandl, J.; Lohmann, D.; Kuerschner, L.; Moessinger, C.; Thiele, C. Ancient Ubiquitous Protein 1 (AUP1) Localizes to Lipid Droplets and Binds the E2 Ubiquitin Conjugase G2 (Ube2g2) via Its G2 Binding Region. J. Biol. Chem. 2011, 286, 5599–5606. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Li, M.; Chen, X.; Ni, H.; Lin, C.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.-M. Autophagy Reduces Acute Ethanol-Induced Hepatotoxicity and Steatosis in Mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovsan, J.; Blüher, M.; Tarnovscki, T.; Klöting, N.; Kirshtein, B.; Madar, L.; Shai, I.; Golan, R.; Harman-Boehm, I.; Schön, M.R.; et al. Altered Autophagy in Human Adipose Tissues in Obesity. J. Clin. Endocrinol. Metab. 2011, 96, E268–E277. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The role of lipids and lipoproteins in atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., De Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, I.; Rotter, V. Regulation of lipid metabolism by p53–Fighting two villains with one sword. Trends Endocrinol. Metab. 2012, 23, 567–575. [Google Scholar] [CrossRef]
- Broz, D.K.; Mello, S.S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cisa-Wieczorek, S.; Hernández-Alvarez, M.I. Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases. Cells 2020, 9, 2605. https://doi.org/10.3390/cells9122605
Cisa-Wieczorek S, Hernández-Alvarez MI. Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases. Cells. 2020; 9(12):2605. https://doi.org/10.3390/cells9122605
Chicago/Turabian StyleCisa-Wieczorek, Sabina, and María Isabel Hernández-Alvarez. 2020. "Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases" Cells 9, no. 12: 2605. https://doi.org/10.3390/cells9122605
APA StyleCisa-Wieczorek, S., & Hernández-Alvarez, M. I. (2020). Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases. Cells, 9(12), 2605. https://doi.org/10.3390/cells9122605